

Abstract
Site-directed mutagenesis has long been used as a method to interrogate protein structure, function and evolution. Recent advances in massively-parallel sequencing technology have opened up the possibility of assessing the functional or fitness effects of large numbers of mutations simultaneously. Here, we present a protocol for experimentally determining the effects of all possible single amino acid mutations in a protein of interest utilizing high-throughput sequencing technology, using the 263 amino acid antibiotic resistance enzyme TEM-1 β-lactamase as an example. In this approach, a whole-protein saturation mutagenesis library is constructed by site-directed mutagenic PCR, randomizing each position individually to all possible amino acids. The library is then transformed into bacteria, and selected for the ability to confer resistance to β-lactam antibiotics. The fitness effect of each mutation is then determined by deep sequencing of the library before and after selection. Importantly, this protocol introduces methods which maximize sequencing read depth and permit the simultaneous selection of the entire mutation library, by mixing adjacent positions into groups of length accommodated by high-throughput sequencing read length and utilizing orthogonal primers to barcode each group. Representative results using this protocol are provided by assessing the fitness effects of all single amino acid mutations in TEM-1 at a clinically relevant dosage of ampicillin. The method should be easily extendable to other proteins for which a high-throughput selection assay is in place.
Keywords: Molecular Biology, Issue 113, mutagenesis, saturation mutagenesis, next generation sequencing, high throughput sequencing, TEM-1 beta-lactamase, antibiotic resistance, orthogonal primers
Introduction
Mutagenesis has long been employed in the laboratory to study the properties of biological systems and their evolution, and to produce mutant proteins or organisms with enhanced or novel functions. While early approaches relied on methods which produce random mutations in organisms, the advent of recombinant DNA technology enabled researchers to introduce select changes to DNA in a site-specific manner, i.e., site-directed mutagenesis1,2. With current techniques, typically using mutagenic oligonucleotides in a polymerase chain reaction (PCR), it is relatively facile to create and assess small numbers of mutations (e.g., point mutations) in a given gene3,4. It is far more difficult however when the goal approaches, for example, the creation and assessment of all possible single-site (or higher-order) mutations.
While much has been learned from early studies attempting to assess large numbers of mutations in genes, the techniques used were often laborious, for example requiring the assessment of each mutation independently using nonsense suppressor strains5-7, or were limited in their quantitative ability due to the low sequencing depth of Sanger sequencing8. The techniques used in these studies have largely been supplanted by methods utilizing high-throughput sequencing technology9-12. These conceptually simple approaches entail creating a library comprising a large number of mutations, subjecting the library to a screen or selection for function, and then deep-sequencing (i.e., on the order of >106 sequencing reads) the library obtained before and after selection. In this way, the phenotypic or fitness effects of a large number of mutations, represented as the change in population frequency of each mutant, can be assessed simultaneously and more quantitatively.
We previously introduced a simple approach for assessing libraries of all possible single amino acid mutations in proteins (i.e., whole-protein saturation mutagenesis libraries), applicable to genes with a length longer than the sequencing read length11,13: First, each amino acid position is randomized by site-directed mutagenic PCR. During this process, the gene is split into groups composed of contiguous positions with a total length accommodated by the sequencing platform. The mutagenic PCR products for each group are then combined, and each group independently subjected to selection and high-throughput sequencing. By maintaining a correspondence between the location of mutations in the sequence and the sequencing read length, this approach has the advantage of maximizing sequencing depth: while one could simply sequence such libraries in short windows without splitting into groups (e.g., by a standard shotgun sequencing approach), most reads obtained would be wild-type and thus the majority of sequencing throughput wasted (e.g., for a whole-protein saturation mutagenesis library of a 500 amino acid protein sequenced in 100 amino acid (300 bp) windows, at minimum 80% of reads will be the wild-type sequence).
Here, a protocol is presented which utilizes high-throughput sequencing for the functional assessment of whole-protein saturation mutagenesis libraries, using the above approach (outlined in Figure 1). Importantly, we introduce the usage of orthogonal primers in the library cloning process to barcode each sequence group, which allows them to be multiplexed into one library, subjected simultaneously to screening or selection, and then de-multiplexed for deep sequencing. Since the sequence groups are not subjected to selection independently, this reduces the workload and ensures that each mutation experiences the same level of selection. TEM-1 β-lactamase, an enzyme which confers high-level resistance to β-lactam antibiotics (e.g., ampicillin) in bacteria is used as a model system14-16. A protocol is described for the assessment of a whole-protein saturation mutagenesis library of TEM-1 in E. coli under selection at an approximate serum level for a clinical dose of ampicillin (50 µg/ml)17,18.
Protocol
Note: See Figure 1 for outline of protocol. Several steps and reagents in the protocol require safety measures (indicated with "CAUTION"). Consult material safety data sheets before use. All protocol steps are performed at RT unless other indicated.
1. Prepare Culture Media and Plates
Prepare and sterilize by autoclaving 1 L purified water, 100 ml Super Optimal Broth (SOB; Table 1), 1 L Luria-Bertani broth (LB; Table 2) and 1 L LB-agar (Table 3). Prepare separately and sterilize three culture flasks each containing 1 L LB. Note: Throughout the protocol "water" refers to autoclave-sterilized purified water; SOB, LB and LB-agar refer to the autoclave-sterilized solutions.
Prepare a 12 mg/ml stock of Tet by dissolving 0.12 g tetracycline hydrochloride in 10 ml of 70% ethanol. Sterilize using a 0.2 µm filter and store at 4 °C protected from light.
Cool LB-agar to 50 °C and then add 1 ml of Tet stock (final concentration of 12 µg/ml Tet). Pour into petri plates and cool at RT protected from light. Store at 4 °C protected from light.
2. Construction of the Whole-gene Saturation Mutagenesis Library
Note: Primers; completed PCRs, restriction digests and ligations; and purified DNA samples can be stored at -20 °C.
- Designing mutagenesis primers
- To mutagenize each amino acid position to all possible amino acids, design a pair of complementary mutagenesis primers (sense/forward and antisense/reverse) for each amino acid position with the following guidelines:
- Replace the codon corresponding to the amino acid to be mutagenized by NNS (where N is a mixture of all four nucleotide bases and S is a mixture of cytosine (C) and guanine (G)) and center in the primer, flanked by approximately 15 nucleotides on each side.
- Ensure that the 5' and 3' ends terminate in C or G and that the melting temperature (Tm) is approximately 70 °C3. Use the computational script NNS_PrimerDesign.m (See Supplementary Code File) to design NNS mutagenesis primers according to these guidelines.
- Order primers from a commercial source. For ease of use, have them synthesized in 96-well plate format and pre-diluted in water to 50 µM, with one set of plates containing the sense mutagenesis primers and another the antisense primers.
- Fill a pipette basin with water and use a multichannel pipette to transfer 95 µl to 263 wells over three 96-well plates. Dilute the primers 20-fold to 2.5 µM by using a multichannel pipette to transfer 5 µl from the 96-well plates containing the sense mutagenesis primers to the cognate wells of the plates containing water.
- Repeat protocol step 2.1.3 to dilute the antisense mutagenesis primers.
- Synthesis of NNS sub-libraries for each amino acid position by two-step PCR site-directed mutagenesis
- Perform the first-round mutagenic PCRs. For each mutagenesis primer, prepare a 25 µl PCR reaction using pBR322_AvrII plasmid as template and primers AatII_F or AvrII_R (sense mutagenesis primers paired with primer AvrII_R, and antisense mutagenesis primers paired with primer AatII_F; total of 526 PCRs). See Table of Materials for AatII_F and AvrII_R sequences.
- Prepare a PCR "master mix" by adding the reagents from Table 4 to a 15 ml conical tube. Transfer to a pipette basin. Use a multichannel pipette to transfer 15 µl to 263 wells over three 96-well PCR plates. Use a multichannel pipette to transfer 10 µl from the 96-well plates containing the diluted sense mutagenesis primers to the cognate wells in the PCR plates.
- Cover each PCR plate with a 96-well plate seal. Centrifuge at 200 x g for 2 min.
- Transfer the PCR plates to thermocycler and run the following program: 98 °C for 30 sec; 20 cycles: 98 °C for 10 sec, 55 °C for 20 sec, 72 °C for 1 min; 72 °C for 2 min; hold at 4 °C.
- Repeat protocol steps 2.2.1.1 - 2.2.1.3 for the 96-well plates containing the diluted antisense mutagenesis primers.
- Perform the second-round mutagenic PCRs. For each amino acid position, prepare a 25 µl PCR reaction using primers AatII_F and AvrII_R, and the mixed and diluted first-round mutagenic PCR products as a template (total of 263 PCRs).
- Fill a pipette basin with water and use a multichannel pipette to transfer 198 µl to 263 wells over three 96-well plates.
- Combine and dilute 100-fold the mutagenic PCR products for each amino acid position by using a multichannel pipette to first transfer 1 µl from the 96-well PCR plates containing the PCR products resulting from the sense mutagenesis primers to the cognate wells of the plates containing water. Then repeat the transfer for the PCR products resulting from the antisense mutagenesis primers.
- Prepare a PCR "master mix" by adding the reagents from Table 5 to a 15 ml conical tube. Transfer to a pipette basin. Use a multichannel pipette to transfer 24 µl to 263 wells over three 96-well PCR plates. Use a multichannel pipette to transfer 1 µl from the 96-well plates containing the mixed and diluted first-round mutagenic PCR products to the cognate wells in the PCR plates.
- Cover each PCR plate with a 96-well plate seal. Centrifuge at approximately 200 x g for 2 min. Transfer plates to thermocycler and run the same program as in protocol step 2.2.1.3.
- Analyze results of the second-round mutagenic PCRs by gel electrophoresis. Ensure that all products are of the correct size and absent of contaminating products.
- Add 2 ml of 2x gel loading dye to a pipette basin and then use a multichannel pipette to transfer 6 µl to 263 wells over three 96-well plates. Use a multichannel pipette to transfer 6 µl from the 96-well PCR plates containing the second-round mutagenic PCR products to the cognate wells of the 96-well plates containing dye.
- Prepare a 1.5% agarose gel with 0.2 µg/ml ethidium bromide (CAUTION).
- Load DNA ladder in first and last lanes of each row. Then use a multichannel pipette to load 10 µl of samples from protocol step 2.2.3.1.
- Run the gel at 100 V for 40 min and image on a UV transilluminator.
- Repeat protocol steps 2.2.3.2 - 2.2.3.4 until all samples are analyzed.
- Accurately measure the concentration of each NNS sub-library PCR product using a dsDNA quantitation reagent.
- Transfer approximately 15 ml EB buffer to a pipette basin. Use a multichannel pipette to transfer 49 µl to 263 wells over three 96-well black-walled, clear bottom assay plates.
- Use a multichannel pipette to transfer 1 µl of each second-step PCR product (protocol step 2.2.2) to the cognate wells of the 96-well assay plates.
- Prepare a DNA concentration standard curve by diluting lambda phage DNA to 2 ng/µl in 300 µl EB buffer and then make ten two-fold dilutions (for total of 11 concentrations). Transfer 50 µl to first eleven columns of a row of one of the 96-well assay plates from the previous step which contains no sample; to the twelfth column add 50 µl of EB buffer (reagent blank).
- Prepare dsDNA quantitation reagent by adding 75 µl of reagent (see Table of Materials) to a 15 ml conical tube, then add 15 ml of EB buffer. Mix by inverting tube and then transfer to a pipette basin. Protect reagent from light.
- Use a multichannel pipette to transfer 50 µl of prepared dsDNA quantitation reagent to each well of the assay plates. Mix by pipetting up-and-down. Incubate plates at RT for 5 min protected from light.
- Measure fluorescence of each sample using a microplate reader and standard fluorescein wavelengths (excitation 485 nm, emission 520 nm; 0.1 sec).
- Subtract the fluorescence value of the reagent blank from all the samples. Generate a standard curve from the fluorescence measurements of lambda phage samples. Calculate the concentration of each sample using their respective fluorescence measurements and the standard curve.
- Cloning of NNS sub-libraries into selection vectors
- Mix 100 ng of each NNS sub-library PCR product into five NNS sub-library groups. Following manufacturers' instructions, clean up samples using a DNA purification kit and then measure concentration using a dsDNA quantitation reagent. Note: Each group is composed of approximately 53 contiguous amino acids positions spaced along the TEM-1 sequence (NNS sub-library groups 1 - 5 are comprised of positions 26 - 78, 79 - 132, 133 - 183, 184 - 236, and 237 - 290, respectively; numbering according to Ambler et al.19).
- Create cloning vectors for each NNS sub-library group.
- Prepare five 100 µl PCRs according to Table 6, using primers AvrII_F and AatII_OP1_R - AatII_OP5_R, and plasmids pBR322_OP1-5 as template (AatII_OP1_R paired with pBR322_OP1, etc.).
- Transfer to thermocycler and run the following program: 98 °C for 30 sec; 25 cycles: 98 °C for 10 sec, 55 °C for 20 sec, 72 °C for 1.5 min; 72 °C for 2 min; hold at 4 °C. See Table of Materials for sequences of AatII_R and AvrII_F.
- Prepare a 1% agarose gel with 0.2 µg/ml ethidium bromide (CAUTION).
- Add 20 µl of 6x gel loading dye to each PCR sample. Load the first lane of the gel with DNA ladder; load entire volume of each sample, skipping at least one well between samples.
- Run gel at 100 V for 50 min.
- Visualize gel using a long-wavelength UV illuminator (CAUTION). Excise slices containing the PCR product at ~3,500 bp; transfer to separate microfuge tubes. Gel slices can be stored at -20 °C.
- Following manufacturers' instructions, purify samples using a gel extraction kit and measure concentration using a dsDNA quantitation reagent.
- For both the NNS sub-library groups (protocol step 2.3.1) and cloning vectors (protocol step 2.3.2), set up restriction digests with AatII and AvrII enzymes according to Table 7. Incubate at 37 °C for 1 hr. Following manufacturers' instructions, clean up samples using a DNA purification kit and then measure concentration using a dsDNA quantitation reagent.
- Set up ligation reactions following Table 8 for each restriction-digested NNS sub-library group with cognate restriction-digested cloning vector (NNS sub-library group 1 with pBR322_OP1, etc.). Incubate at RT for 1 hr. Clean up reactions using a DNA purification kit according to manufacturer's instructions; elute DNA with 20 µl of water.
- Transform the entirety of the purified ligation reactions into library-efficient E. coli cells by electroporation.
- Thaw electrocompetent E. coli cells and then place cells and purified ligation reactions on ice.
- Transfer 10 µl thawed cells to each purified ligation reaction and then transfer to electroporation cuvette. Electroporate at 1.8 kV.
- Recover cells by resuspending in 1 ml SOB. Incubate for 1 hr at 37 °C.
- Resuspend 10 µl of each recovery culture in 990 µl LB; spread 100 µl on LB-agar plates containing 12 µg/ml Tet. Incubate plates O/N (~16 hr) at 37 °C.
- For each recovery culture, prepare a 250 ml culture flask with 50 ml LB and 50 µl Tet stock. Transfer to flask the remaining ~1 ml of recovery culture. Incubate O/N (~16 hr) at 37 °C with vigorous shaking (~200 rpm).
- Count the number of colonies on each plate. Calculate the number of successful transformants as
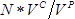 , where
, where  is the number of colonies,
is the number of colonies, 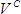 is the recovery culture volume (1,000 µl) and
is the recovery culture volume (1,000 µl) and 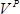 is volume of the recovery culture plated (1 µl). Note: To ensure complete coverage of all mutations, as a rule of thumb the number of successful transformants should be ≥100-fold over the number of expected mutations. Each NNS sub-library has ~53 positions, so the expected number of mutations is 53 positions × 32 codons/position ≈ 1.7 × 103; to give a library size ≥100-fold (≥1.7 × 105) there should be ≥170 colonies on each plate.
is volume of the recovery culture plated (1 µl). Note: To ensure complete coverage of all mutations, as a rule of thumb the number of successful transformants should be ≥100-fold over the number of expected mutations. Each NNS sub-library has ~53 positions, so the expected number of mutations is 53 positions × 32 codons/position ≈ 1.7 × 103; to give a library size ≥100-fold (≥1.7 × 105) there should be ≥170 colonies on each plate. - According to manufacturer's instructions, isolate plasmid DNA from cultures using a plasmid purification kit and then measure concentrations using a dsDNA quantitation reagent. Mix together 100 ng of each plasmid. This creates the final whole-protein saturation mutagenesis library.
3. Selection of the TEM-1 Whole-protein Saturation Mutagenesis Library for Antibiotic Resistance
- Preparation of the pre-selection culture.
- Dilute the plasmid from protocol step 2.3.7 to 0.5 ng/µl in water and transfer 20 µl to a microcentrifuge tube. Perform transformation, recovery, plating and O/N growth as previously described in protocol step 2.3.5, except transfer 1 µl of the SOB recovery culture to 999 µl LB.
- Count the number of colonies. To ensure complete coverage of all mutations there should be ≥100 colonies, indicating ≥106 successful transformants (100 × 263 positions × 32 codons/position ≈ 106).
- Measure the concentration of the 50 ml O/N culture.
- Prepare an LB blank by adding 1 ml LB to a spectrophotometer cuvette. Measure OD600 on a spectrophotometer.
- Dilute the O/N culture 10-fold by resuspending 100 µl in 900 µl LB. Measure the OD600. Subtract the OD600 reading of the blank and multiply by 10 to give the OD600 of the O/N culture.
- Pre-warm the three culture flasks from protocol step 1.1 for ~30 min at 37 °C. Dilute the O/N culture to OD600 = 0.1 and add 1 ml to one flask (final OD600 = 0.001). This is the "pre-selection culture".
- Incubate the "pre-selection culture" at 37 °C with vigorous shaking (200 rpm). Periodically monitor growth by measuring OD600 as in protocol step 3.1.3 (it is not necessary to dilute culture 10-fold) until OD600 = 0.1 (~2.5 hr).
- Transfer 100 ml of the pre-selection culture to two 50 ml conical tubes. Centrifuge at 4,000 x g for 6 min at 4 °C. Remove most of supernatant and combine into a single 15 conical tube. Repeat centrifugation and remove all supernatant. Store at -20 °C.
- Selection for ampicillin resistance.
- While the pre-selection culture is incubating, prepare a 50 mg/ml stock of Amp in water by dissolving 0.5 g sodium ampicillin in 10 ml water. Sterilize using a 0.2 µm filter and store at 4 °C.
- To the other two flasks, add Tet to a final concentration of 12 µg/ml and a volume of the pre-selection culture such that the final OD600 = 0.001. To one flask, add 1 ml Amp, for a final concentration of 50 µg/ml - this is the "selection culture".
- Incubate the cultures at 37 °C with vigorous shaking (200 rpm). Monitor growth of the culture for which no ampicillin was added in the previous step, until OD600 = 0.1 (~2.5 hr). At this time, also measure the OD600 of the selection culture.
- Divide the OD600 of the selection culture into 0.1 and multiply by 100 ml. Transfer this volume (~400 ml) to 50 ml conical tubes and centrifuge at 4,000 x g for 6 min at 4 °C. Remove most of supernatant and combine into a single 15 conical tube. Repeat centrifugation and remove all supernatant.
- According to manufacturer's instructions, isolate plasmid DNA from the pre-selection (protocol step 3.1.6) and selection (protocol step 3.2.4) culture cell pellets and then measure concentrations using a dsDNA quantitation reagent.
4. High-throughput Sequencing to Determine the Fitness Effects of Mutations
- Preparation of samples for high-throughput sequencing
- Prepare 25 µl PCRs to de-multiplex the NNS sub-library groups with orthogonal primers
- Prepare a PCR master mix according to Table 9; transfer 23 µl to ten PCR tubes.
- Add 1 µl of 0.5 ng/µl purified plasmid DNA from the pre-selection culture to PCR tubes 1 - 5 and from the selection culture to tubes 6 - 10.
- Mix together 50 µl of 50 µM forward orthogonal primers OP1_F - OP5_F with the respective reverse orthogonal primers OP1_R - OP5_R. In the same order, transfer 1 µl to PCR tubes 1 - 5 and 6 - 10. See Table of Materials for sequences of OP1_F - OP5_F and OP1_R - OP5_R.
- Transfer PCR tubes to thermocycler. Run the following program: 98 °C for 30 sec; 20 cycles: 98 °C for 10 sec, 55 °C for 20 sec, 72 °C for 1.5 min; 72 °C for 2 min; hold at 4 °C.
- Prepare 25 µl PCRs to isolate each of the NNS sub-library groups
- Dilute 100-fold the ten PCRs from protocol step 4.1.1.4 by transferring 1 µl of each to separate PCR tubes and adding 99 µl of water. Mix, then pipette out 99 µl and discard.
- Mix together 50 µl of 50 µM forward primers Group1_F - Group5_F with the respective reverse primers Group1_R - Group5_R. In the same order, transfer 1 µl to PCR tubes 1 - 5 and 6 - 10. See Table of Materials for sequences of Group1_F - Group5_F and Group1_R - Group5_R.
- Prepare a PCR master mix according to Table 9, transfer 23 µl to each PCR tube. Transfer PCR tubes to thermocycler; run the same program as in protocol step 2.2.1.3.
- Carry out the final 25 µl PCRs to add indexing sequences
- Dilute 100-fold the ten PCRs from protocol step 4.1.2.3 by transferring 1 µl of each to separate PCR tubes and adding 99 µl of water. Mix, then pipette out 99 µl and discard.
- Prepare a PCR master mix according to Table 9, transfer 23 µl to each PCR tube.
- For tubes with template originating from NNS sub-library groups 1-5, transfer 0.5 µl per tube forward primers 501_F - 505_F respectively. For tubes with template resulting from the pre-selection and selection cultures, transfer 0.5 µl per tube reverse primers 701_R and 702_R, respectively. See Table of Materials for sequences of 501_F - 505_F, and 701_R and 702_R.
- Transfer PCR tubes to the thermocycler and run the program from step 2.2.1.3.
- Mix and purify samples
- Measure concentrations using a dsDNA quantitation reagent according to manufacturer's instructions. Mix 100 ng of each PCR product into a single microcentrifuge tube.
- Prepare a 2% agarose gel with 0.2 µg/ml ethidium bromide (CAUTION).
- Add 6x gel loading dye to the mixed PCR products sample. Load the first lane of the gel with DNA ladder; load entire volume of the sample.
- Run gel at 100 V for 50 min. Visualize gel using a long-wavelength UV illuminator (CAUTION). Excise slice containing the PCR product at ~360 bp; transfer to microfuge tube. Gel slice can be stored at -20 °C.
- Following manufacturers' instructions, purify sample using a gel extraction kit and measure concentration using a dsDNA quantitation reagent. This is the final sample for high-throughput sequencing.
- Sequence on a high-throughput sequencing platform (see Table of Materials for platform used in this protocol).
- Calculate sample concentration in nM as
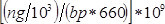 , where
, where 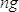 is the concentration of the sample in ng/µl and
is the concentration of the sample in ng/µl and 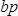 is the sequence length of the sample DNA (~360 bp). Dilute sample to 4 nM in EB buffer.
is the sequence length of the sample DNA (~360 bp). Dilute sample to 4 nM in EB buffer. - Follow manufacturer's instructions to denature sample and dilute to 9 pM in hybridization buffer.
- Load 600 µl of sample into reagent cartridge. Sequence following manufacturer's instructions and on-screen prompts.
- Analysis of sequencing data
- Download FLASh (Fast Length Adjustment of Short reads)20, place into folder along with fastq.gz files obtained from sequencing.
- Use FLASh to join the fastq.gz files corresponding to the paired-end reads for each pair of indices (forward and reverse reads for each NNS sub-library group for the pre-selection and selection cultures).
- Open Command Prompt and change directory to folder in protocol step 4.2.1. Join each pair of reads using command: flash <mates1.fastq.gz> <mates2.fastq.gz>, where mates1.fastq.gz and mates2.fastq.gz are the files containing the forward and reverse reads, respectively.
- After joining each pair of reads, place the out.extendedFrags.fastq output file into separate folders for results from the pre-selection or selection cultures. Rename the out.extendedFrags.fastq output file according to the NNS sub-library group to which it corresponds (i.e., 1.fastq, 2.fastq, etc.).
- Run the computational script NNS_DataAnalyzer.m (See Supplementary Code File) from each folder to compute the counts for each single amino acid mutation, and the counts for the wild-type, for each NNS sub-library group.
- Calculate the fitness effect
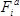 of each mutation
of each mutation  at each position
at each position  as the base ten logarithm of the ratio of counts obtained in the selection (
as the base ten logarithm of the ratio of counts obtained in the selection (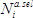 ) versus the pre-selection (
) versus the pre-selection (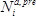 ) condition, relative to the wild-type:
) condition, relative to the wild-type: 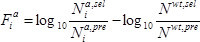 .
.
Representative Results
The plasmid map for the five modified pBR322 plasmids containing orthogonal priming sites (pBR322_OP1 - pBR322_OP5) is shown in Figure 2A. To test whether the orthogonal primers are specific, PCRs were performed using each pair of orthogonal primers individually, along with all five pBR322_OP1-5 plasmids, or with all plasmids minus the plasmid matching the orthogonal primer pair. The correct product was only obtained when the matching plasmid was included, and no product of any size was obtained in its absence (Figure 2B).
A representative experiment was performed following the protocol described in this text (Figure 1). Following processing (protocol section 4.2), 6.2 × 106 reads from the pre-selection condition and 6.3 × 106 reads from the 50 µg/ml ampicillin selection condition were obtained. The counts obtained for each amino acid mutation from the pre-selection condition display a characteristic log-normal distribution (Figure 3A)13. At least one count from sequencing of the pre-selection culture for 98.9% of mutations (58 had no counts), and greater than 100 counts for 91.2% (465 had less than 100 counts) were obtained. Figure 3B depicts the relative fitness effect () for each mutation at each position of TEM-1; the distribution of is shown in Figure 3C. Under selection at 50 µg/ml ampicillin, most mutations have a neutral or nearly-neutral fitness effect (≈ 0, corresponding to white pixels in Figure 3B and the large peak in Figure 3C). A small fraction of mutations at this concentration have substantial effects on fitness (<< 0, corresponding to blue pixels in Figure 3B and the left tail in Figure 3C); expectedly, these include mutations within the highly conserved active site residues (S70, K73, S130, D131, N132, K234 and G236)13,14. In contrast, few mutations considerably increase fitness over that of TEM-1 (>> 0, corresponding to red pixels in Figure 3B), as might be expected since TEM-1 is highly efficient at ampicillin hydrolysis (![]() ≈ 107 M-1s-1)14.
≈ 107 M-1s-1)14.
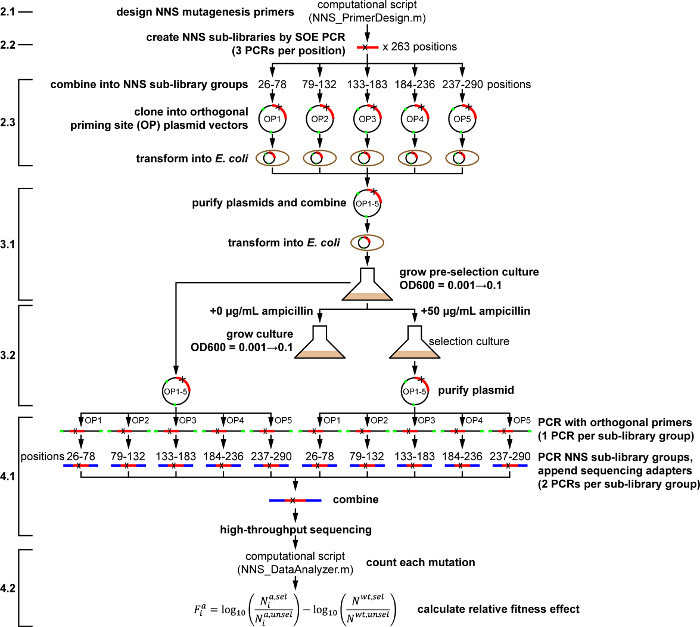 Figure 1. Outline of Whole-protein Saturation Mutagenesis Protocol for TEM-1 β-lactamase under Ampicillin Selection. Actions as described in the protocol are shown in bold. Numbered protocol steps are shown at left, for reference to the main text. Please click here to view a larger version of this figure.
Figure 1. Outline of Whole-protein Saturation Mutagenesis Protocol for TEM-1 β-lactamase under Ampicillin Selection. Actions as described in the protocol are shown in bold. Numbered protocol steps are shown at left, for reference to the main text. Please click here to view a larger version of this figure.
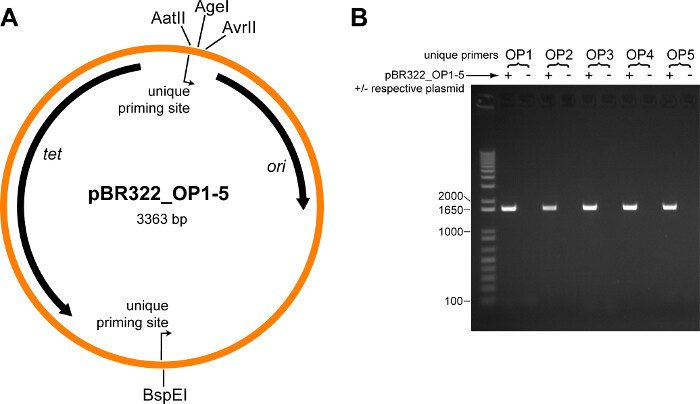 Figure 2. Validation of Orthogonal Priming Site Plasmid Vectors. (A) Plasmid map for the five modified pBR322 plasmids containing orthogonal priming sites (pBR322_OP1 - pBR322_OP5). Location and direction of the orthogonal priming sites are indicated. Locations of several restriction sites are labeled; the TEM-1 whole-protein saturation mutagenesis library (which includes the entire TEM-1 gene and promoter) is cloned in-between the AatII and AvrII restriction sites. Abbreviations: tet tetracycline resistance gene, ori origin of replication. (B) Each pair of orthogonal primers (OP1-OP5) was tested in a PCR containing all five pBR322_OP1-5 plasmids (+), or with all plasmids minus the respective plasmid (˗). Shown is an ethidium bromide stained agarose gel (1% w/v) loaded with each PCR reaction, size separated by electrophoresis. The expected size product is 1,628 bp; the first lane is a DNA ladder, sizes of relevant standards are indicated. Please click here to view a larger version of this figure.
Figure 2. Validation of Orthogonal Priming Site Plasmid Vectors. (A) Plasmid map for the five modified pBR322 plasmids containing orthogonal priming sites (pBR322_OP1 - pBR322_OP5). Location and direction of the orthogonal priming sites are indicated. Locations of several restriction sites are labeled; the TEM-1 whole-protein saturation mutagenesis library (which includes the entire TEM-1 gene and promoter) is cloned in-between the AatII and AvrII restriction sites. Abbreviations: tet tetracycline resistance gene, ori origin of replication. (B) Each pair of orthogonal primers (OP1-OP5) was tested in a PCR containing all five pBR322_OP1-5 plasmids (+), or with all plasmids minus the respective plasmid (˗). Shown is an ethidium bromide stained agarose gel (1% w/v) loaded with each PCR reaction, size separated by electrophoresis. The expected size product is 1,628 bp; the first lane is a DNA ladder, sizes of relevant standards are indicated. Please click here to view a larger version of this figure.
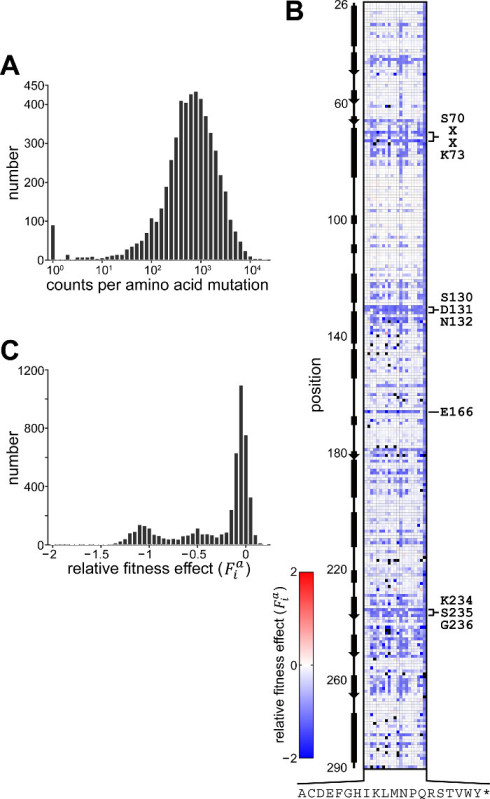 Figure 3. Results of TEM-1 β-lactamase Whole-protein Saturation Experiment. (A) Histogram showing the distribution of counts for each amino acid mutation obtained from high-throughput sequencing of the library from the pre-selection condition. For simplicity, mutations with zero counts (53 mutations) are shown as having a count of one. (B) Fitness effects of all single amino acid mutations in TEM-1 under selection at 50 µg/ml ampicillin. Shown is the data matrix containing the relative fitness effect () depicted colorimetrically with blue representing deleterious effect, red a positive effect and white no fitness effect relative to wild-type. Mutations for which no counts were obtained from the pre-selection culture are colored black. Rows depict positions along the primary sequence and columns indicate mutation to one of twenty amino acids or stop codon (indicated by one-letter code, * is stop codon); the secondary structure of TEM-1 is indicated at left and several highly conserved motifs within the active site are indicated at right. (C) Histogram showing the distribution of relative fitness effects. Shown are results for those mutations with >100 counts obtained from sequencing the pre-selection culture. Please click here to view a larger version of this figure.
Figure 3. Results of TEM-1 β-lactamase Whole-protein Saturation Experiment. (A) Histogram showing the distribution of counts for each amino acid mutation obtained from high-throughput sequencing of the library from the pre-selection condition. For simplicity, mutations with zero counts (53 mutations) are shown as having a count of one. (B) Fitness effects of all single amino acid mutations in TEM-1 under selection at 50 µg/ml ampicillin. Shown is the data matrix containing the relative fitness effect () depicted colorimetrically with blue representing deleterious effect, red a positive effect and white no fitness effect relative to wild-type. Mutations for which no counts were obtained from the pre-selection culture are colored black. Rows depict positions along the primary sequence and columns indicate mutation to one of twenty amino acids or stop codon (indicated by one-letter code, * is stop codon); the secondary structure of TEM-1 is indicated at left and several highly conserved motifs within the active site are indicated at right. (C) Histogram showing the distribution of relative fitness effects. Shown are results for those mutations with >100 counts obtained from sequencing the pre-selection culture. Please click here to view a larger version of this figure.
| Reagent | Mass or Volume | Comment |
| Typtone | 2 g | |
| Yeast extract | 0.5 g | |
| Sodium chloride | 0.06 g | |
| Potassium chloride | 0.02 g | |
| Magnesium sulfate | 0.24 g | |
| Purified water | 100 ml |
Table 1. Super Optimal Broth (SOB). Reagent names and quantities used in preparing 100 ml SOB (Protocol step 1.1).
| Reagent | Mass or Volume | Comment |
| Typtone | 10 g | |
| Yeast extract | 5 g | |
| Sodium chloride | 10 g | |
| Purified water | 1 L |
Table 2. Luria-Bertani Broth (LB). Reagent names and quantities used in preparing 1 L LB (protocol step 1.1).
| Reagent | Mass or Volume | Comment |
| Typtone | 10 g | |
| Yeast extract | 5 g | |
| Sodium chloride | 10 g | |
| Agar | 15 g | |
| Purified water | 1 L |
Table 3. LB-agar. Reagent names and quantities used in preparing LB-agar (protocol step 1.1).
| Reagent | Volume | Comment |
| 5x PCR buffer | 1,450 µl | |
| PCR additive | 1,450 µl | |
| 2 mM dNTPs | 725 µl | 2 mM each nucleotide |
| 50 µM AatII_F or AvrII_R primer | 145 µl | |
| 1 ng/µl pBR322_AvrII plasmid | 145 µl | |
| 2 units/µl DNA polymerase | 72.5 µl | |
| water | 363 µl |
Table 4. First-round Mutagenic PCR Master Mix. Reagent names and quantities for preparing the master mix for the first-round mutagenic PCR (protocol step 2.2.1). Total quantity is sufficient for 290 25 µL reactions.
| Reagent | Volume | Comment |
| 5x PCR buffer | 1,450 µl | |
| PCR additive | 1,450 µl | |
| 2 mM dNTPs | 725 µl | 2 mM each nucleotide |
| 50 µM AatII_F primer | 145 µl | |
| 50 µM AvrII_R primer | 145 µl | |
| 2 units/µl DNA polymerase | 72.5 µl | |
| water | 2,973 µl |
Table 5. Second-round Mutagenic PCR Master Mix. Reagent names and quantities for preparing the master mix for the second-round mutagenic PCR (protocol step 2.2.2). Total quantity is sufficient for 290 25 µl reactions.
| Reagent | Volume | Comment |
| 5x PCR buffer | 20 µl | |
| PCR additive | 20 µl | |
| 2 mM dNTPs | 10 µl | 2 mM each nucleotide |
| 50 µM AvrII_F primer | 2 µl | |
| 50 µM AatII_OP1_R - AatII_OP5_R primer | 2 µl | one primer per reaction, paired with respective plasmid |
| 1 ng/µl pBR322_OP1-5 plasmid | 2 µl | one plasmid per reaction |
| 2 units/µl DNA polymerase | 1 µl | |
| water | 43 µl |
Table 6. Cloning Vector PCR. Reagent names and quantities for preparing the PCR to make the cloning vectors (protocol step 2.3.2).
| Reagent | Volume | Comment |
| 10x restriction enzyme buffer | 5 µl | |
| 4 units/µl AvrII | 2.5 µl | |
| 20 units/µl AatII | 0.5 µl | |
| DNA to restriction digest | volume for 500 ng | |
| water | to 50 µl total volume |
Table 7. Restriction Digests. Reagent names and quantities for restriction digests of cloning vectors and NNS sub-library groups (protocol step 2.3.3).
| Reagent | Volume | Comment |
| 10x T4 DNA ligase buffer | 5 µl | |
| purified restriction-digested NNS sub-library group DNA | volume for 48 ng | |
| purified restriction-digested cloning vector DNA | volume for 52 ng | |
| 400 units/µl T4 DNA ligase | 1 µl | |
| water | to 20 µl total volume |
Table 8. Ligations. Reagent names and quantities for ligations of cloning vectors with restriction-digested NNS sub-library groups in a 1:3 vector:insert molar ratio (protocol step 2.3.4).
| Reagent | Volume | Comment |
| 5x PCR buffer | 55 µl | |
| PCR additive | 55 µl | |
| 2 mM dNTPs | 27.5 µl | 2 mM each nucleotide |
| 2 units/µl DNA polymerase | 2.75 µl | |
| water | 113 µl |
Table 9. PCR Reagents for Preparing Samples for High-throughput Sequencing. Reagent names and quantities for preparing PCR master mixes used for de-multiplexing with orthogonal primers (4.1.1), isolating NNS sub-library groups (protocol step 4.1.2) and adding indexing sequences (protocol step 4.1.3). Total quantity is sufficient for 11 25 µl reactions.
Discussion
Here a protocol is described for performing the functional assessment of whole-protein saturation mutagenesis libraries, using high-throughput sequencing technology. An important aspect of the method is the use of orthogonal primers during the cloning process. Briefly, each amino acid position is randomized by mutagenic PCR, and mixed together into groups of positions whose combined sequence length is accommodated by high-throughput sequencing. These groups are cloned into plasmid vectors containing pairs of orthogonal priming sites, mixed together and subjected to selection, then de-multiplexed using the orthogonal primers, and subsequently deep sequenced. Since mutations are confined within the sequencing read length limit, this approach maximizes the number of useful reads containing mutations for genes of size longer than the sequencing read length. In addition, this technique allows for the simultaneous or "one-batch" selection of the entire mutational library, reducing the workload as well as the possibility that mutations experience different levels of selection. Practically, the critical steps in the protocol largely concern organization: during the cloning process (protocol step 2.3) one must ensure the correct mixing of mutagenic PCR products into groups and their subsequent cloning into the correct orthogonal priming site vector; during the preparation of the sample for sequencing (protocol step 4.1), the correct orthogonal primers, as well as primers to isolate each of the NNS sub-library groups and add indexing sequences, must be used.
The three main steps of the protocol - library construction, selection, and sequencing - can be modified in several aspects. During library construction one could introduce mutations using a variety of techniques, for example, by error-prone PCR, or by constructing the gene using oligonucleotides synthesized by doping in a small fraction of alternative nucleotides21. One could construct the library to include double or higher-order mutations within segments of the protein (i.e., NNS sub-library groups), or in alternate genotype backgrounds22. Importantly, all modifications to the library construction step however should satisfy the criterion that the correspondence between the location of mutations in the sequence and the sequencing read length is maintained. This criterion therefore excludes the application of the protocol towards comprehensive studies of multiple mutations across a protein. Modifications to the second part of the protocol include alternate selection conditions: different β-lactam types (or combinations) and concentrations, external stress conditions (e.g., temperature, nutrient levels), host type (e.g., different types of bacteria), or different sampling times (hours to days). For example, in previous work we examined the fitness effects of all single amino acid mutations in TEM-1 under different concentrations of ampicillin, and under the third-generation cephalosporin cefotaxime13. With regards to the third step of the protocol, we currently do not recommend deviating from the choice of the sequencing platform used here (see Table of Materials). While sequencing read lengths are indeed currently longer in other platforms, the number of reads obtainable is currently far lower; in general the accuracy to which the effect of a mutation can be determined is proportional to the number of reads obtained (see equation in protocol step 4.2.5).
Largely for simplicity, the protocol uses TEM-1 β-lactamase as a model system, however the methodology described here can be extended to other systems for which a high-throughput selection or screening assay is in place. Constructing such assays is however often non-trivial: First, a strategy for compartmentalization of gene (mutation) and protein together must be established, for example within a cell, liquid droplet (as in a microfluidics platform), or by phage display. Second, and most importantly, a quantitative connection between protein function and a selectable phenotype, or fitness, must be established. For enzymes involved in metabolism or antibiotic resistance, the ability of cells to grow in nutrient drop-out or antibiotic media is often a direct function of enzymatic activity. A more synthetic approach could be used in other systems, for example by linking protein-protein binding affinity to reporter gene (e.g., fluorescent protein) expression in bacteria or yeast11,23, or using a fluorogenic enzyme substrate in a microfluidics system24. Lastly, such an assay must be scalable, to address the size of a whole-protein mutagenesis library.
In summary, a high-throughput sequencing-based approach for the functional assessment of whole-protein saturation mutagenesis libraries is described here. Central to the approach is the construction of the mutagenesis library within segments along the gene, and the utilization of orthogonal primer barcodes to tag each segment for multiplexing and de-multiplexing the library. We anticipate that this protocol could be readily applied to other proteins for which an appropriate high-throughput selection or screen has been developed.
Disclosures
The authors declare they have no competing financial interests
Acknowledgments
R.R. acknowledges support from the National Institutes of Health (RO1EY018720-05), the Robert A. Welch Foundation (I-1366), and the Green Center for Systems Biology.
References
- Hutchison CA, 3rd, et al. Mutagenesis at a specific position in a DNA sequence. J Biol Chem. 1978;253(18):6551–6560. [PubMed] [Google Scholar]
- Mullis KB, Faloona FA. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335–350. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- Papworth C, Bauer JC, Braman J, Wright DA. Site-directed mutagenesis in one day with >80% efficiency. Strategies. 1996;9(3):3–4. [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16(15):7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222(1):67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- Markiewicz P, Kleina LG, Cruz C, Ehret S, Miller JH. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as 'spacers' which do not require a specific sequence. J Mol Biol. 1994;240(5):421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- Kleina LG, Miller JH. Genetic studies of the lac repressor. XIII. Extensive amino acid replacements generated by the use of natural and synthetic nonsense suppressors. J Mol Biol. 1990;212(2):295–318. doi: 10.1016/0022-2836(90)90126-7. [DOI] [PubMed] [Google Scholar]
- Huang W, Petrosino J, Hirsch M, Shenkin PS, Palzkill T. Amino acid sequence determinants of beta-lactamase structure and activity. J Mol Biol. 1996;258(4):688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- Fowler DM, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7(9):741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietpas RT, Jensen JD, Bolon DN. Experimental illumination of a fitness landscape. Proc Natl Acad Sci U S A. 2011;108(19):7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RN, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491(7422):138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, et al. Deep sequencing of systematic combinatorial libraries reveals beta-lactamase sequence constraints at high resolution. J Mol Biol. 2012;424(3-4):150–167. doi: 10.1016/j.jmb.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiffler MA, Hekstra DR, Ranganathan R. Evolvability as a Function of Purifying Selection in TEM-1 beta-Lactamase. Cell. 2015;160(5):882–892. doi: 10.1016/j.cell.2015.01.035. [DOI] [PubMed] [Google Scholar]
- Matagne A, Lamotte-Brasseur J, Frere JM. Catalytic properties of class A beta-lactamases: efficiency and diversity. Biochem J. 1998;330 (Pt2):581–598. doi: 10.1042/bj3300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salverda ML, De Visser JA, Barlow M. Natural evolution of TEM-1 beta-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol Rev. 2010;34(6):1015–1036. doi: 10.1111/j.1574-6976.2010.00222.x. [DOI] [PubMed] [Google Scholar]
- Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312(5770):111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- Stewart SM, Fisher M, Young JE, Lutz W. Ampicillin levels in sputum, serum, and saliva. Thorax. 1970;25(3):304–311. doi: 10.1136/thx.25.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachetto G, et al. Ampicillin and penicillin concentration in serum and pleural fluid of hospitalized children with community-acquired pneumonia. Pediatr Infect Dis J. 2004;23(7):625–629. doi: 10.1097/01.inf.0000128783.11218.c9. [DOI] [PubMed] [Google Scholar]
- Ambler RP, et al. A standard numbering scheme for the class A beta-lactamases. Biochem J. 1991;276(Pt 1):269–270. doi: 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies) Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed D, Young DL, Gamble CE, Miller CR, Fields S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013;19(11):1537–1551. doi: 10.1261/rna.040709.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank C, Hietpas RT, Jensen JD, Bolon DN. A systematic survey of an intragenic epistatic landscape. Mol Biol Evol. 2015;32(1):229–238. doi: 10.1093/molbev/msu301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove SL, Joung JK, Hochschild A. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature. 1997;386(6625):627–630. doi: 10.1038/386627a0. [DOI] [PubMed] [Google Scholar]
- Romero PA, Tran TM, Abate AR. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc Natl Acad Sci U S A. 2015;112(23):7159–7164. doi: 10.1073/pnas.1422285112. [DOI] [PMC free article] [PubMed] [Google Scholar]