

Abstract
Membrane proteins serve important functions in cells such as nutrient transport, motility, signaling, survival and virulence, yet constitute only ~1% percent of known structures. There are two types of membrane proteins, α-helical and β-barrel. While α-helical membrane proteins can be found in nearly all cellular membranes, β-barrel membrane proteins can only be found in the outer membranes of mitochondria, chloroplasts, and Gram-negative bacteria. One common bottleneck in structural studies of membrane proteins in general is getting enough pure sample for analysis. In hopes of assisting those interested in solving the structure of their favorite β-barrel outer membrane protein (OMP), general protocols are presented for the production of target β-barrel OMPs at levels useful for structure determination by either X-ray crystallography and/or NMR spectroscopy. Here, we outline construct design for both native expression and for expression into inclusion bodies, purification using an affinity tag, and crystallization using detergent screening, bicelle, and lipidic cubic phase techniques. These protocols have been tested and found to work for most OMPs from Gram-negative bacteria; however, there are some targets, particularly for mitochondria and chloroplasts that may require other methods for expression and purification. As such, the methods here should be applicable for most projects that involve OMPs from Gram-negative bacteria, yet the expression levels and amount of purified sample will vary depending on the target OMP.
Keywords: Chemistry, Issue 113, β-barrel, membrane protein, protein crystallization, structural biology, membranes, protein refolding, protein purification
Introduction
β-barrel OMPs can only be found in the outer membranes of mitochondria, chloroplasts, and Gram-negative bacteria1-3. While they serve similar roles as α-helical proteins, they have a very different fold consisting of a central membrane-embedded β-barrel domain ranging from 8-26 anti-parallel β-strands with each strand being intimately connected to the two neighboring strands (Figures 1 and 2). The first and last strands of the β-barrel domain then interact with one another, almost exclusively in an anti-parallel fashion (except for mitochondrial VDAC), to close and seal the β-barrel domain from the surrounding membrane. All β-barrel OMPs have extracellular loops of varying sequence and length which play an important role in ligand interactions and/or protein-protein contacts, with these loops sometimes being as large as 75 residues, such as found in Neisserial transferrin binding protein A (TbpA)4. β-barrel OMPs can also have N-terminal or C-terminal periplasmic extensions which serve as additional domains for the protein's functional purpose (e.g., BamA5-7, FimD8,9, FadL10). While many types of β-barrel OMPs exist11, two of the more common types are described below as examples for those less familiar with the field, (1) TonB-dependent transporters and (2) autotransporters.
TonB-dependent transporters (e.g., FepA, TbpA, BtuB, Cir, etc.) are essential for nutrient import and contain an N-terminal plug domain consisting of ~150 residues that is found tucked inside a C-terminal 22-stranded β-barrel domain embedded into the outer membrane12 (Figure 3). While this plug domain prevents substrate from freely passing through the barrel domain, substrate binding induces a conformational change within the plug domain that leads to pore formation (either by plug rearrangement or by partial/full ejection of the plug) which can then facilitate substrate transport across the outer membrane into the periplasm. TonB-dependent transporters are especially important for the survival of some pathogenic strains of Gram-negative bacteria such as Neisseria meningitidis that have evolved specialized transporters that hijack nutrients such as iron directly from human host proteins4,13,14.
Autotransporters belong to the type V secretion system of Gram-negative bacteria and are β-barrel OMPs that consist of a β-barrel domain (typically 12-strands as with EstA and EspP) and a passenger domain that is either secreted or presented at the surface of the cell15,16 (Figure 3). These β-barrel OMPs often serve important roles in cell survival and virulence with the passenger domain serving either as a protease, adhesin, and/or other effector that mediates pathogenesis.
Structural methods such as X-ray crystallography, NMR spectroscopy, and electron microscopy (EM) allow us to determine models for the OMPs at atomic resolution which can in turn be used to decipher exactly how they function within the outer membrane. This invaluable information may then be used for drug and vaccine development if applicable. For example, transferrin binding protein A (TbpA) is found on the surface of Neisseria and is required for pathogenesis because it directly binds human transferrin and then extracts and imports the iron for its own survival. Without TbpA, Neisseria cannot scavenge iron from the human host and are rendered non-pathogenic. After the crystal structure of human transferrin bound to TbpA4 was solved, it became much clearer how the two proteins associated, what regions of TbpA mediated the interaction, what residues were important for iron extraction by TbpA, and how one might develop therapeutics against Neisseria targeting TbpA. Therefore, given the importance of β-barrel OMPs in Gram-negative bacteria for survival and pathogenesis, as well as in mitochondria and chloroplast function, and the need for additional structural information about this unique class of membrane proteins and the systems in which they function, general protocols are presented with the overall goal of expressing and purifying target OMPs at high levels for characterization by structural methods.
Protocol
1. Cloning and Expression
Note: To enable structural studies, sufficient quantities of highly purified protein must be prepared, and this generally starts with the cloning and overexpression of the target β-barrel outer membrane protein (OMP) in E. coli (Figure 4). To date, all β-barrel OMP structures, including those structures for mitochondrial VDAC, have been derived from bacterially expressed protein11. Here, general protocols are presented for cloning and expressing β-barrel OMPs for (1) native expression directly into bacterial membranes and (2) expression into inclusions bodies for in vitro refolding17.
- Designing an Expression Construct
- Obtain or purchase the codon optimized target OMP gene.
- Purchase or obtain an T7 expression vector (i) containing a periplasmic localization signal sequence, an N-terminal 6x-Histidine tag, and a TEV protease site4,18,19 for in vivo expression to the membrane or (ii) without a periplasmic localization signal sequence for expression to inclusion bodies for in vitro refolding. Note: An N-terminal protease cleavable 6x or 10x-Histidine tag would provide an easy method for purification. As with soluble proteins, vary the choice of affinity tag (Histidine, Strep, GST, etc.), the position of the affinity tag, and the inclusion of a protease site (TEV, enterokinase, thrombin, etc.) for subsequent tag removal.
- PCR amplify the target sequence with appropriate primers and subclone into the expression vector. Use ligation independent cloning (LIC)20,21 or conventional cloning techniques (restriction enzymes/ligation) for subcloning. However, LIC can facilitate high throughput, allowing for a vast number of various constructs (truncations, variety of tags, variety of promoters) to be cloned in parallel more easily.
- In Vivo Membrane-targeted Expression
- Use sequencing analysis to verify the target β-barrel OMP is cloned downstream and in frame with a signal sequence (i.e., pelB, OmpA) in the expression construct. Note: The signal sequence directs the nascent chain to the Sec translocon for secretion to the periplasm and subsequent assembly into the outer membrane.
- Transform the construct into an expression strain of bacteria for expression by pipetting 1.0 μl of plasmid into 50 μl of BL21(DE3) chemically competent cells and mix gently by pipetting up and down. Incubate on ice for 30 min.
- Heat pulse at 42 ºC for 30 sec using a water bath and then place back on ice for 1 min.
- Add 1 ml of pre-warmed SOC media and shake at 37 ºC for 1 hr at 1,000 rpm using a benchtop shaker incubator.
- Plate 100 μl of cells onto LB agar plates containing an appropriate antibiotic and incubate overnight at 37 ºC inverted.
- Perform small-scale expression tests by inoculating a 5 ml LB+antibiotic culture with a single colony. Repeat for 5-10 colonies. Note: Because of the potential toxicity of a β-barrel OMP and the reliance on basal expression levels, colony-to-colony variability is sometimes observed. Therefore, screening multiple colonies is recommended for each construct being tested. For small-scale expression tests, rather than conventional 5 ml cultures, growing 25 ml cultures in 125 ml baffled Erlenmeyer flasks is suggested. These conditions often better reflect what will happen in a large-scale growth. Additionally, it is a good idea to also screen various types of culture media (TB, LB, 2xYT, M9, etc.), since varying levels of success have been reported depending on the target protein.
- Grow at 37 ºC with shaking to an OD600 of ~0.6.
- Induce expression of the target OMP by adding 5 μl of 1 M isopropyl β-D-1-thiogalactopyranoside (IPTG) to each culture tube and allow to grow an additional 1-2 hr.
- Compare expression levels for all the colonies by centrifuging 1 ml of each culture (OD600 of ~0.6) for 1 min at 15,000 x g using a microcentrifuge.
- Remove the supernatant and resuspend the cells in 200 μl of 1x SDS-PAGE loading buffer. Heat at 100 ºC for 5 min and then centrifuge again at 15,000 x g for 5 min.
- Analyze the samples using SDS-PAGE by pipetting 20 μl into each well of a 10% gel. Run the gel for 35 min at constant 200 V. Note: It would be beneficial at this point to be able to determine if the protein target is being properly folded or not. This can often be accomplished by doing small-scale purifications or by assaying for heat modifiability.
- Select the colony showing the best expression and perform large-scale expression using 12-24 L of medium. Note: Conventional methods typically grow the cultures at 37 ºC with shaking to an OD600 of ~0.6 and then induce with an appropriate inducer (i.e., 0.1-1 mM for IPTG or ~0.2% for arabinose) for an additional 2-4 hr. If desired, prior to the induction, reduce the temperature to as low as 20 °C and extend the induction time.
- For leaky expression method, grow a 25 ml inoculate culture at 37°C in LB medium containing the appropriate antibiotic(s) until OD600 reaches ~0.6. Then, add 1 ml of inoculate to twelve 1 L flasks of TB medium plus antibiotic(s) and grow at 20 °C to saturation (~3 days).
- Harvest the cells by centrifugation at 6,000 x g for 10 min.
- Proceed to purification step Section 2.1 or freeze the cell pellet in liquid nitrogen for long-term storage at -80 ºC.
- Expression to Inclusion Bodies for In Vitro Refolding
- Use sequencing analysis to verify the expression construct does not contain a signal sequence. The absence of a signal sequence will direct the target protein to accumulate in the cytosol as inclusion bodies.
- Transform the expression construct into an expression strain of bacteria for expression. For inclusion body expression, it is best to control expression by induction (i.e., IPTG, arabinose, etc.) to minimize possible toxicity effects.
- Plate 100 μl of transformed cells onto LB agar plates containing an appropriate antibiotic and incubate overnight at 37 ºC inverted.
- Perform small-scale expression tests by inoculating a 5 ml LB+antibiotic culture with a single colony.
- Repeat step 1.3.4 for 2-4 additional colonies.
- Grow at 37 ºC with shaking to an OD600 of ~0.6.
- Induce with an appropriate inducer for 1-2 hr.
- Compare expression levels for all the colonies by analysis using SDS-PAGE (see Step 1.2.6). Select the colony showing the best expression and perform large-scale expression using 12-24 L of medium.
- Grow the cells at 37 °C to an OD600 of 0.6-0.8 and induce at 37 °C for 3-5 hr. While expression to inclusion bodies is more robust than other types of expression, as with all protein expression experiments, expression can be improved by varying growth medium, time of induction, temperature of induction, and concentration of the inducing agent.
- Harvest the cells by centrifugation at 6,000 x g for 10 min.
- Proceed to purification step Section 2.2 or freeze the cell pellet in liquid nitrogen for long-term storage at -80 ºC.
2. Purification
- Isolation from Membrane Fractions Note: In contrast to soluble proteins, integral membrane proteins are embedded into the lipid bilayer and therefore require detergents to extract them for further purification and analysis (Figure 5). Following expression, the first step in the purification of a β-barrel OMP is extraction from the membrane fraction.
- Resuspend cells in Lysis buffer (50 mM Tris-HCl, pH 7.4, 200 mM NaCl, 10 mM MgCl2, 50 µg/ml AEBSF, 5 µg/ml DNase I) at a ratio of 5 ml/g cell paste.
- Lyse the cells using a French press or cell homogenizer. Spin the lysed cells at 15,000 x g for 30 min at 4 ºC to remove unlysed cells and cell debris.
- Transfer the supernatant to a clean tube and centrifuge again at high speed (200,000 x g) for 1 hr at 4 ºC. The resulting pellet is the membrane fraction which contains the protein of interest.
- Using a dounce homogenizer, resuspend the membrane fraction, first transferring the membranes and then adding Solubilization buffer (50 mM KH2PO4 pH 7.5, 200 mM NaCl, 20 mM imidazole, pH 8.0) (50 ml per 20 g cells) at 2x concentration, without detergent.
- Pour the resuspended membranes into a small beaker and add detergent (i.e., n-dodecyl-β-D-maltoside (DDM), lauryldimethylamine-N-oxide (LDAO), n-octyl-β-D-glucopyranoside (OG), Triton X-100, etc.) slowly to a final concentration of ~10x critical micelle concentration (CMC).
- Fill with water until a final concentration of 1x Solubilization buffer. Stir for 0.5-16 hr at 4 ºC, depending on how easily the target protein is extracted from the membranes. Note: Detergent is used to slowly solubilize the membranes to extract the target protein. There are 3 types of detergents that can be used here: ionic (SDS, deoxycholic acid), non-ionic (Elugent, TWEEN, Triton X-100, OG, DDM), and zwitterionic (LDAO, CHAPS). A protein-detergent complex that is stable and monodisperse during the purification step will greatly aid in the success of membrane protein crystallization. More information about detergents, their properties, CMC information, and their use in the purification of membrane proteins and other applications can be found on commercial supplier websites.
- Centrifuge the solubilized sample at 300,000 x g for 1 hr at 4 ºC. The supernatant now contains the detergent-solubilized target β-barrel OMP.
- Proceed with Section 2.3 as outlined below.
- Refolding from Inclusion Bodies Note: For in vitro refolding, the target β-barrel OMP is expressed directly into inclusion bodies. One advantage here is that these proteins can be produced at high levels. However the disadvantage is that refolding is often inefficient and varies from one refolding experiment to the next. Still, there are many examples of proteins that have been successfully refolded for structural studies. Following expression into inclusion bodies, one must now isolate the inclusion bodies for refolding experiments.
- Resuspend cells in Lysis buffer (50 mM Tris-HCl, pH 7.4, 200 mM NaCl, 10 mM MgCl2, 50 µg/ml AEBSF, 5 µg/ml DNase I, 4 mM 2-mercaptoethanol (BME)) (5 ml per g cell paste). Lyse using either a French press, sonication, or cell homogenizer.
- Pellet the inclusion bodies by a low speed spin at 6,000 x g for 10 min at 4 ºC.
- Wash the inclusion bodies by resuspending in 25 ml of 1.0 M urea using a dounce homogenizer. Pellet again by a low speed spin at 6,000 x g for 10 min at 4 ºC.
- Repeat step 2.2.3 as necessary.
- Resuspend the washed inclusion bodies to a final concentration of 10 mg/ml using buffer containing 8.0 M urea (or 6.0 M guanidinium hydrochloride (GdCl)) plus detergent (i.e., DDM or LDAO) at 2x CMC or higher. Note: If desired, for membrane protein targets containing a Histidine tag, immobilized metal-affinity chromatography (IMAC) purification can be performed using denaturing conditions (in 8.0 M urea or 6.0 M GdCl) as an initial step similar to that outlined below in Section 2.3.
- Perform the refolding reaction by slowly (overnight) removing the denaturant by dialysis in buffer lacking the denaturant. Note: It may take several changes of the dialysis buffer to accomplish this. Alternatively, if the inclusion bodies are first bound to an IMAC column, one can do the refolding reaction while still bound to the column by slowly exchanging buffers to remove the denaturant. In either case, remember that this is a membrane protein, therefore, detergent must be present to stabilize the target. Also, if the detergent being used is dialyzable, it must be added to the dialysis buffer(s) as well.
- Spin the refolded samples at 200,000 x g for 1 hr at 4 ºC. The supernatant contains the refolded detergent-solubilized target β-barrel OMP.
- Proceed with Section 2.3 as outlined below.
- Purification Using IMAC, Ion-Exchange, and Gel Filtration Note: Assuming the protein has been engineered with a Histidine tag, whether natively expressed or refolded using in vitro methods, then immobilized metal-affinity chromatography (IMAC) is performed for purification much like for Histidine tagged soluble proteins, except detergent must be kept in all subsequent buffers to keep the target β-barrel OMP solubilized and stable.
- Prepare a 1-5 ml IMAC column or use a prepacked column. Perform subsequent chromatography steps at 4 °C. Flush with water to remove any traces of preservatives. If using an automated purification system, install the column according to manufacturer's instructions.
- Prepare 500 ml of IMAC Buffer A (50 mM K2HPO4, pH 7.5, 200 mM NaCl, 0.1% DDM) and 250 ml of IMAC Buffer B (50 mM K2HPO4, pH 7.5, 200 mM NaCl, 0.1% DDM, and 1.0 M imidazole). Note: Other detergents that may be used include Cymal-6 (0.05%), Triton X-100 (0.03%), and LDAO (0.05%).
- Equilibrate the IMAC column using 10 column volumes of IMAC Buffer A.
- Add imidazole to the protein sample to a final concentration of 25 mM and mix. Load the sample onto the equilibrated IMAC column at 2 ml/min. Collect the flow through.
- Wash the IMAC column with 5 column volumes each of increasing concentrations of imidazole using Buffer B (i.e., 25 mM, 50 mM, 100 mM). Collect the washes in 2 ml fractions.
- Elute the sample with a final concentration of 250 mM for 5 column volumes. Collect the eluted sample in 2 ml fractions.
- Analyze the flow through, the wash fractions and the elution fractions using SDS-PAGE analysis (see Step 1.2.6) based on their absorbance at 280 nm. Pool the fractions containing the target β-barrel OMP as verified by SDS-PAGE analysis.
- Remove the 6x-Histidine tag by adding TEV protease to the pooled sample (according to manufacturer's protocol) and incubating overnight at 4 ºC with gentle rocking.
- Load the sample/protease solution onto an IMAC column again to separate the target β-barrel OMP (flow through) from the cleaved tags and any uncleaved sample. The flow through will contain the cleaved sample lacking the tag. Note: This would also remove any protease like TEV-His, which also has a non-cleavable 6x-Histidine tag itself. Otherwise, other methods will be needed to remove the protease after digestion.
- Optionally, perform ion-exchange chromatography to further purify the sample. Follow the manufacturer's instructions for the column to be used, being sure to include detergent in all buffers.
- To prepare for crystallization, load the sample onto a gel filtration column into a buffer containing the detergent to be used for crystallization, such as 25 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1% OG.
- Collect 1 ml fractions and analyze using SDS-PAGE analysis (seeStep 1.2.6) based on their absorbance at 280 nm. Pool fractions with absorbances at 280 nm as they contain the target protein. Concentrate to ~10 mg/ml. Note: If desired, proper folding may be assessed using a heat modifiability assay as outlined below.
- Heat Modifiability Assay Note: One method to monitor proper folding of purified β-barrel OMPs is to perform heat modifiability assays using a semi-native SDS-PAGE22. Here, unheated samples that are folded properly will typically migrate differently from those that are heat denatured. This property is unique to β-barrel OMPs and widely used to study this family of membrane proteins.
- Prior to preparing samples, assemble the gel apparatus. To begin, place the buffer tank into an ice bucket and surround completely with ice. Insert an in-house cast or commercial native gradient gel into the holder and place into the tank. Fill the tank completely with cold 1x MES Running Buffer (50 mM 2-[N-morpholino]ethanesulfonic acid, 50 mM Tris base, 1 mM EDTA, 0.1% SDS, pH 7.3).
- Pipette 0.25 µl of the sample (10 mg/ml) into two 1.5 ml microcentrifuge tubes. Label one as 'Boiled' and the other as 'RT'.
- Add 9.75 µl of sample buffer to each and mix by pipetting. To both samples, add 10 µl of 2x SDS loading buffer (100 mM Tris-HCl, pH 6.8, 2% SDS, 0.2% bromophenol blue, and 20% glycerol) and mix gently by pipetting.
- Boil the 'Boiled' sample at 95 ºC for 5 min while leaving the 'RT' sample at RT. Spin the 'Boiled' sample briefly.
- Load 20 μl of both samples onto the preassembled native gel. Run for 60 min at constant 150 V. Remove the gel and soak in staining solution to visualize the results.
3. Crystallization
Note: For crystallization of both soluble and membrane protein targets, it is standard protocol to maximize sample purity and stability (i.e., best detergent, ligands, cofactors, etc.). Current methodology for crystallizing membrane protein targets in general encompasses three major approaches that satisfy the amphiphilic requirements of bilayer-embedded proteins: (1) detergent, (2) bicelle, and (3) lipidic cubic phase (LCP) (Figure 6)23. Use of a nanoliter crystallization robot is strongly recommended when possible in order to increase the number of conditions that can be screened for a given sample volume, as well as, utilizing recent advances in tools aimed to aid structure determination (Figure 7).
- Crystallization using Detergents
- Obtain a detergent solubilized protein sample that is at ~10 mg/ml concentration. Optionally, add the additive 1,2,3-heptanetriol directly to the sample to a final concentration of 3% to reduce detergent micelle size.
- Filter the sample using a 0.22 µm centrifugal filter to remove particulates and precipitation.
- Using a crystallization robot, perform broad matrix crystallization screening using commercially available 96 well screens by either hanging or sitting drop vaporization method with drops composed of ~200 nl protein sample and ~200 nl crystallization buffer.
- Incubate the crystallization plates at ~21 °C. Inspect the plates weekly for crystal growth using a stereomicroscope.
- Optimize crystallization leads by varying components of the crystallization condition in a systematic fashion (increasing/decreasing salt or precipitant concentrations, buffer pH, incubation temperature, and/or protein concentration).
- Crystallization Using Bicelles Note: A more detailed demonstration of bicelle techniques has previously been described by Ujwal and Abramson24.
- Prepare or purchase a 35% bicelle mixture consisting of DMPC:CHAPSO at a ratio of 2.8:1 according to published protocols25. Other concentrations may also be assayed, as well as, other lipids and detergents, as necessary.
- Obtain a detergent solubilized protein sample that is at ~10 mg/ml concentration.
- Add the bicelle mixture, on ice, at a starting ratio of 4:1 v/v (protein:bicelle) (e.g., add 10 µl of bicelle mixture to 40 µl of protein and mix rapidly by pipetting).
- Incubate on ice 30-60 min. Note: The protein:bicelle solution should remain mostly clear but can often turn slightly opaque. However, if the protein:bicelle solution turns milky white, indicating precipitation, then other variables should be assayed (i.e., detergent, buffer, etc.). For new samples, doing small scale tests (8 µl protein and 2 µl bicelles) is recommended to verify stability prior to scaling up to prevent unnecessary sample loss.
- Using a crystallization robot, perform broad matrix crystallization screening using commercially available 96 well screens by either hanging or sitting drop vaporization method with drops composed of ~200 nl protein sample and ~200 nl crystallization buffer.
- Incubate the crystallization plates at ~21 °C. Inspect the plates weekly for crystal growth using a stereomicroscope.
- Optimize crystallization leads by varying components of the crystallization condition in a systematic fashion (increasing/decreasing salt or precipitant concentrations, buffer pH, incubation temperature, and/or protein concentration).
- Crystallization Using Lipidic Cubic Phase (LCP) Note: A more detailed demonstration of LCP techniques has previously been described by Liu and Cherezov26,27.
- Obtain a detergent solubilized protein sample that is at ~20 mg/ml concentration.
- Pre-warm the monoolein to ~40 ºC. Load 60 µl monoolein into one gas-tight 100 µl syringe and 40 µl protein sample into another.
- Using a mixing coupler, connect the syringes, being careful not to introduce air.
- Mix gently by applying alternating pressure on the syringe plungers pushing the mixture from one syringe to the other completely until the sample becomes translucent and homogenous.
- Using a crystallization robot designed for LCP methods, perform broad matrix crystallization screening using commercially available 96 well screens with sandwich-style crystallization plates (0.1 mm well thickness) with drops composed of 100 nl protein sample and 750 nl crystallization buffer. Note: Drop ratios may be adjusted if needed. Also, solid covers (i.e., glass or thick plastic) are recommended which support the drops nicely rather than thin films, which tend to allow the drops to move about the well as the plates are handled.
- Incubate the crystallization plates at ~21 °C. Inspect the plates weekly for crystal growth using a stereomicroscope.
- Optimize crystallization leads by varying components of the crystallization condition in a systematic fashion (increasing/decreasing salt or precipitant concentrations, buffer pH, incubation temperature, and/or protein concentration).
Representative Results
YiuR is a TonB dependent iron transporter that is a putative vaccine target against Yersinia pestis. It was originally identified using a microarray assay. Here, the steps that were taken to determine the structure of YiuR using X-ray crystallography are outlined (Figure 9). For cloning, the DNA sequence of YiuR (minus the N-terminal signal sequence) was PCR amplified from genomic DNA and subcloned into a vector containing an N-terminal pelB signal sequence and 10x-Histidine affinity tag followed by a TEV protease site. For expression, the YiuR-containing plasmid was transformed into BL21(DE3) competent cells and a single colony used to grow a 5 ml starter culture to OD600 ~0.5. Twelve flasks containing 750 ml of TB medium were then inoculated with 1 ml of the starter culture and allowed to grow for 3 d at 20 ºC (shaking at 220 rpm). The cells were then harvested, producing about 150-200 g of total cell paste, which was flash cooled in liquid nitrogen and stored at -80 ºC until use.
For purification of YiuR, 20 g of cells were resuspended in 120 ml of 1x PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4) by stirring at RT with DNase I and AEBSF being added. Lysis was performed by two passes through a homogenizer. The lysate was then centrifuged at 12,000 x g for 10 min (to spin down unbroken cells and inclusion bodies). The supernatant was centrifuged again at 235,000 x g for 60 min, with the pellet containing the membrane fraction (YiuR). The membranes were then resuspended in 100 ml of 1x PBS using a dounce homogenizer. YiuR was solubilized/extracted from membranes using Elugent at 5% final concentration, stirring overnight (up to 16 hr) at 4 ºC. The solubilized membranes were then centrifuged again at 371,000 x g for 60 min, with the supernatant containing the solubilized fraction including YiuR. To isolate YiuR, IMAC was performed using Buffer A (1x PBS, 0.1% DDM) and Buffer B (1x PBS, 1 M imidazole, 0.1% DDM), washed with imidazole concentrations from 30-50 mM and eluted with 250-500 mM. To remove the N-terminal 10x-Histidine tag, incubation with TEVHIS protease was performed overnight at 4 ºC during dialysis in 1x PBS buffer. The sample was then passed over an IMAC column again, the flow through concentrated, dialyzed, and then separated further using a 0-1.0 M NaCl gradient on an ion-exchange column in 50 mM Tris-HCl, pH 7.5. Fractions containing YiuR were then pooled, concentrated and ran over a gel filtration column using 25 mM Tris-HCl, pH 7.5, 200 mM NaCl, and 0.05% LDAO. The fractions containing YiuR were then pooled and concentrated to 10 mg/ml for crystallization.
Broad matrix screening was then performed and conditions determined that produced some initial diffracting crystals. Further optimization led to larger crystals which were used to collect native X-ray diffraction data. Crystals shown in Figure 8 are representative of crystals one may attain using the methods presented here. Crystals from detergent screening and bicelles can be as large as crystals obtained typically for soluble proteins; however, those from LCP are almost always much smaller. The data were good enough to be used for molecular replacement which led to structure determination to 2.65 Å resolution.
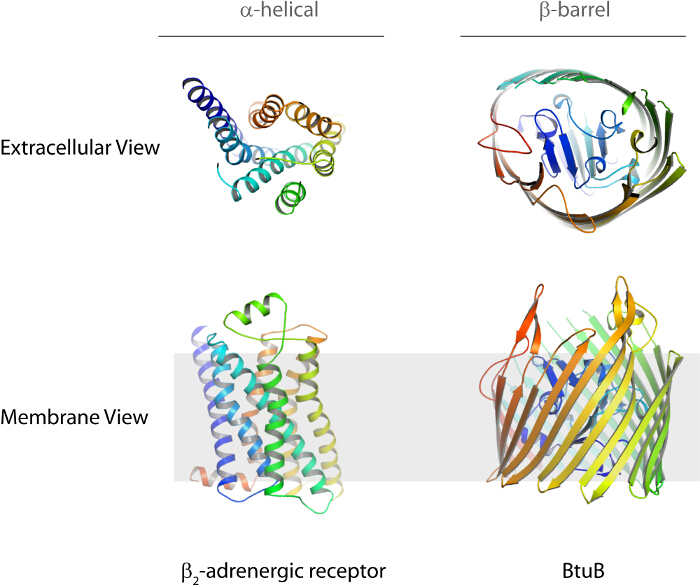 Figure 1.The two types of fully integrated membrane proteins are α-helical and β-barrel. Shown here are examples of each with the β2-adrenergic receptor (PDB code 2RH1, α-helical) and the TonB-dependent transporter BtuB (PDB code 1NQE, β-barrel). The top row shows the extracellular view while the bottom row shows the membrane view. Please click here to view a larger version of this figure.
Figure 1.The two types of fully integrated membrane proteins are α-helical and β-barrel. Shown here are examples of each with the β2-adrenergic receptor (PDB code 2RH1, α-helical) and the TonB-dependent transporter BtuB (PDB code 1NQE, β-barrel). The top row shows the extracellular view while the bottom row shows the membrane view. Please click here to view a larger version of this figure.
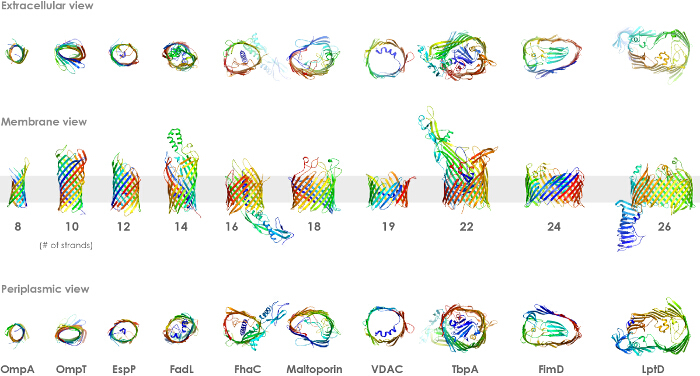 Figure 2.β-barrel OMPs serve many different functions and can have diverse structures. While α-helical membrane proteins can contain one or more transmembrane domains, β-barrel OMPs range from 8-26 strands and each strand intimately interacts with the neighboring strands. Outer residues, which interact with the membrane, are typically hydrophobic while the interior residues, which interact with solvent, are typically polar and hydrophilic. Top row shows the extracellular view, the middle row shows the membrane view, and the bottom row shows the periplasmic view. Please click here to view a larger version of this figure.
Figure 2.β-barrel OMPs serve many different functions and can have diverse structures. While α-helical membrane proteins can contain one or more transmembrane domains, β-barrel OMPs range from 8-26 strands and each strand intimately interacts with the neighboring strands. Outer residues, which interact with the membrane, are typically hydrophobic while the interior residues, which interact with solvent, are typically polar and hydrophilic. Top row shows the extracellular view, the middle row shows the membrane view, and the bottom row shows the periplasmic view. Please click here to view a larger version of this figure.
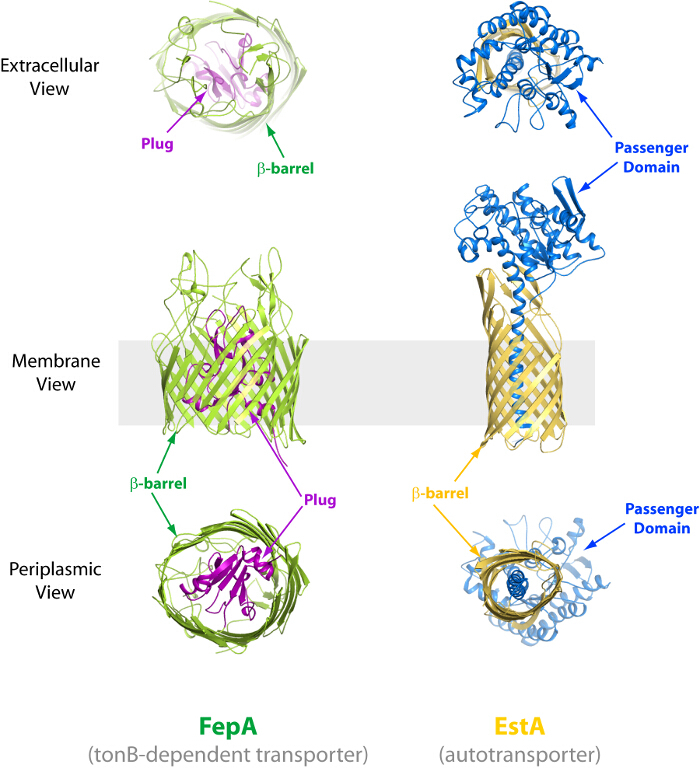 Figure 3.Examples of two types of common β-barrel OMPs. Left, the structure of the TonB-dependent transporter FepA (PDB code 1FEP), depicting the extracellular (top), membrane (middle) and periplasmic (bottom) views. The β-barrel domain is indicated in green while the plug domain is in magenta. Right, the structure of the autotransporter EstA (PDB code 3KVN), depicting the extracellular (top), membrane (middle) and periplasmic (bottom) views. The β-barrel domain is indicated in gold while the passenger domain is in blue. Please click here to view a larger version of this figure.
Figure 3.Examples of two types of common β-barrel OMPs. Left, the structure of the TonB-dependent transporter FepA (PDB code 1FEP), depicting the extracellular (top), membrane (middle) and periplasmic (bottom) views. The β-barrel domain is indicated in green while the plug domain is in magenta. Right, the structure of the autotransporter EstA (PDB code 3KVN), depicting the extracellular (top), membrane (middle) and periplasmic (bottom) views. The β-barrel domain is indicated in gold while the passenger domain is in blue. Please click here to view a larger version of this figure.
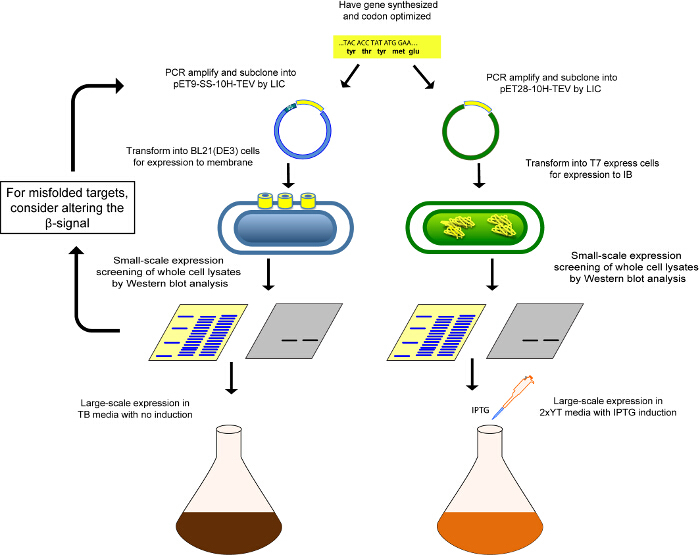 Figure 4.Schematic of cloning and expression pipeline for production of β-barrel OMPs. Schematic of the pipeline used to clone and express a target OMP by either native membrane expression (left) or by expression into inclusion bodies for refolding (right). Please click here to view a larger version of this figure.
Figure 4.Schematic of cloning and expression pipeline for production of β-barrel OMPs. Schematic of the pipeline used to clone and express a target OMP by either native membrane expression (left) or by expression into inclusion bodies for refolding (right). Please click here to view a larger version of this figure.
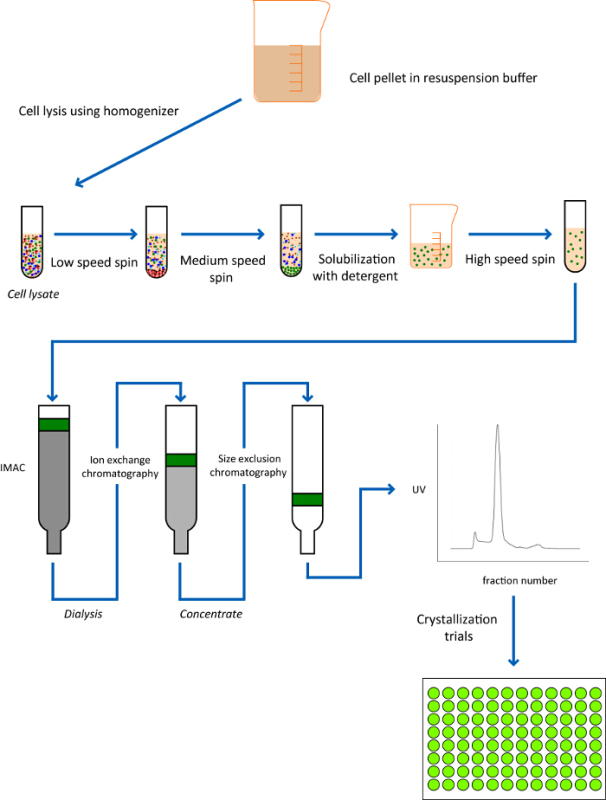 Figure 5.Schematic of purification pipeline for isolation of β-barrel OMPs. Schematic of the pipeline used for the extraction of OMPs that have been expressed directly into the OM. Here, the target OMP has to be extracted from the membrane by solubilization with an appropriate detergent and then purified by IMAC, ion exchange chromatography, and gel filtration. Please click here to view a larger version of this figure.
Figure 5.Schematic of purification pipeline for isolation of β-barrel OMPs. Schematic of the pipeline used for the extraction of OMPs that have been expressed directly into the OM. Here, the target OMP has to be extracted from the membrane by solubilization with an appropriate detergent and then purified by IMAC, ion exchange chromatography, and gel filtration. Please click here to view a larger version of this figure.
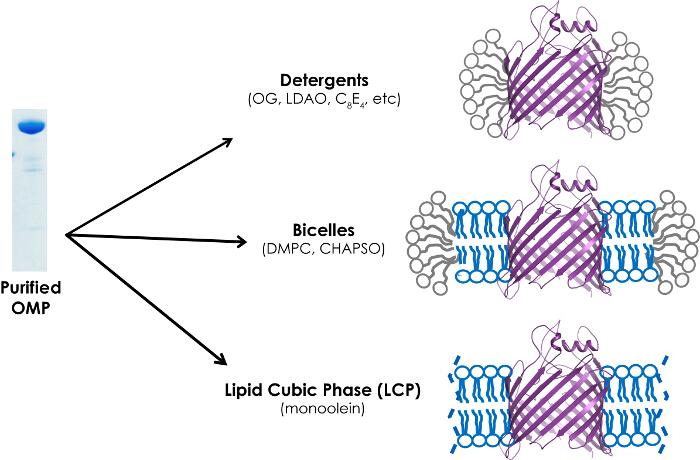 Figure 6.Schematic of crystallization pipeline for growing crystals of β-barrel OMPs. Three methods can be utilized for the crystallization of β-barrel OMPsincluding detergent screening, bicelles, and lipidic cubic phase (LCP). Please click here to view a larger version of this figure.
Figure 6.Schematic of crystallization pipeline for growing crystals of β-barrel OMPs. Three methods can be utilized for the crystallization of β-barrel OMPsincluding detergent screening, bicelles, and lipidic cubic phase (LCP). Please click here to view a larger version of this figure.
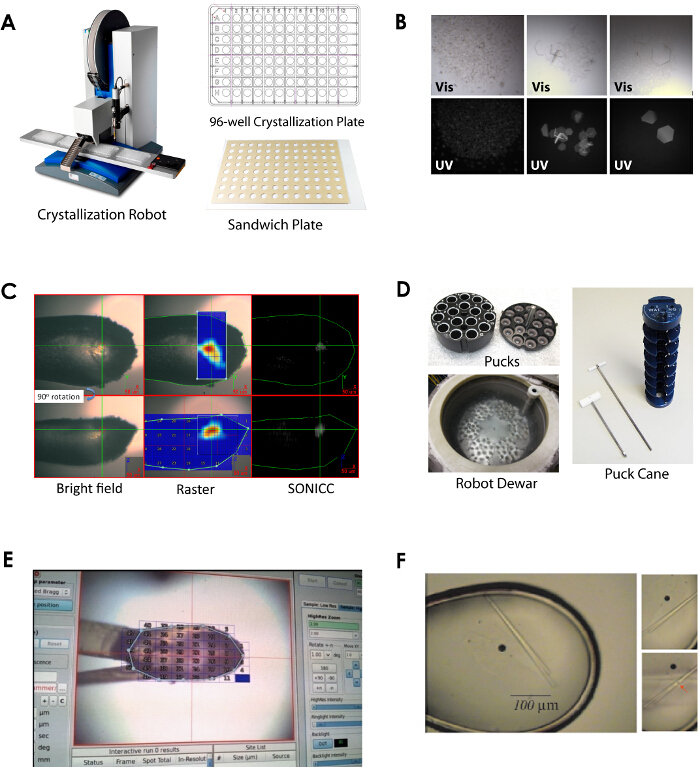 Figure 7.New tools in crystallography that have significantly contributed to structure determination of β-barrel OMPs. Examples include crystallization robots/LCP robots (A), UV microscopes (B), Second Order Nonlinear Imaging of Chiral Crystals (SONICC) visualization (C), robotic pucks/robots (D), rastering/vector/helical data collection methods (E), and microfocus beams (F). Please click here to view a larger version of this figure.
Figure 7.New tools in crystallography that have significantly contributed to structure determination of β-barrel OMPs. Examples include crystallization robots/LCP robots (A), UV microscopes (B), Second Order Nonlinear Imaging of Chiral Crystals (SONICC) visualization (C), robotic pucks/robots (D), rastering/vector/helical data collection methods (E), and microfocus beams (F). Please click here to view a larger version of this figure.
 Figure 8.Representative image of the crystals resulting from the protocol. Crystals from detergent screening and bicelles can be as large as crystals obtained typically for soluble proteins; however, those from LCP are almost always much smaller. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 8.Representative image of the crystals resulting from the protocol. Crystals from detergent screening and bicelles can be as large as crystals obtained typically for soluble proteins; however, those from LCP are almost always much smaller. Scale bar = 100 µm. Please click here to view a larger version of this figure.
 Figure 9.From constructs to crystals to structure determination for the putative vaccine target YiuR from Yersinia pestis. The overall pipeline used for structure determination of YiuR, illustrating the steps taken to clone, express, purify, crystallize, and solve the structure. Please click here to view a larger version of this figure.
Figure 9.From constructs to crystals to structure determination for the putative vaccine target YiuR from Yersinia pestis. The overall pipeline used for structure determination of YiuR, illustrating the steps taken to clone, express, purify, crystallize, and solve the structure. Please click here to view a larger version of this figure.
Discussion
β-barrel OMPs serve essential roles in Gram-negative bacteria, mitochondria and chloroplasts and are important targets for structural analysis that offer a wealth of information about essential molecular mechanisms at the outer membranes of these respective organelles. However, producing enough sample for structural analysis is not always straightforward and therefore, a general pipeline is presented for the production of sufficient quantities of target β-barrel OMPs for structure determination, explaining in detail the process from constructs to crystals. While these protocols have been tested and found to work for most OMPs from Gram-negative bacteria, there are limitations in that there are some targets, particularly for mitochondria and chloroplasts, which may require other methods for expression and purification. As such, the methods here should be applicable for most projects that involve OMPs from Gram-negative bacteria, yet the expression levels and amount of purified sample will vary depending on the target OMP.
There are two general approaches for the expression of β-barrel OMPs for structural studies, (1) in vivo expression directly into the outer membrane and (2) expression to inclusion bodies for in vitro refolding. For the in vivo expression approach, β-barrel OMPs are targeted to outer membrane of E. coli where they are natively folded and isolated directly from membranes. Expression to the membrane is the preferred approach since targets are more likely to be properly folded. While this approach typically yields lower levels of overall protein, it avoids the complications sometimes associated with in vitro refolding. Many β-barrel OMPs have been successfully expressed in this way for structure determination4,5,11. Success is often achieved using a system that relies on low-level constitutive expression from a T7 promoter, but other systems which rely on induction (i.e., IPTG, arabinose) for expression are also routinely used with success.
Expression directly into the outer membrane requires the target protein to have an N-terminal signal sequence which directs the ribosome-nascent chain complex to the Sec translocon, for secretion of the nascent β-barrel OMP into the periplasm. The signal sequence is cleaved on the periplasmic side of the inner membrane, so it is important that the signal sequence precede any N-terminal tags added to the target. The nascent β-barrel OMP is then escorted by chaperones through the periplasm to the BAM complex for insertion into the outer membrane2,3.
For targets that cannot be overexpressed into the membrane, an alternative approach is expression into inclusion bodies for in vitro refolding28-30. This approach typically results in high level and robust expression of the target protein. However, it can be challenging to identify refolding conditions, as refolding can be inefficient. In addition, it can be difficult to assay when the target β-barrel OMP is properly folded. Still, there are many examples of proteins that have been successfully refolded for structural studies including OmpA31, Ail19, OmpF32, and VDAC33. For clones that do not express at all (natively or into inclusion bodies) or express but are not assembled properly into the membrane (natively), one could try mutating the β-barrel to more closely resemble that of E. coli34,35. The amino acid sequence in the β-signal of the barrel domain is important for recognition and assembly by the BAM complex and variations in the β-signal sequence can significantly affect both proper biogenesis and expression levels of the target β-barrel OMP11,34,35. Proper integration of the target β-barrel OMP can be monitored by screening for membrane fractionation and detergent extractability followed by heat modifiability assays.
Crystallization of membrane proteins in the presence of detergent micelles is the oldest technique and allows easy adaptation of traditional soluble protein crystallization equipment and strategies. By masking the membrane embedded regions with detergent molecules, concentrated protein can be treated in a similar fashion to soluble proteins (e.g., mixed with crystallization mother liquors and enclosed in a vapor diffusion apparatus that causes the slow dehydration of the protein drop). While simple in concept, detergent characteristics and properties add a significant layer of complexity on top of the normal crystallization challenges. Specifically, the molecular character of a detergent must be empirically tailored to a given target protein, including micelle size, head group polarity (anionic, cationic, non-ionic, or zwitterionic), and hydrocarbon chain length, as each of these affect the stability of the target membrane protein in solution. Drawbacks of this approach include the non-native chemical environment, potential obscuring of surface regions that could form crystal contacts, and detergent concentration issues.
Bicelles are a mixture of lipids and amphiphiles (e.g., detergents or short chain lipids) that assemble into individual particles that mimic a bilayer structure similar to the membranes found in cells36,37. The amphiphile masks the hydrophobic core of this bilayer where it is exposed at its edges in a similar fashion to the micelles in detergent crystallization. This provides a more native-like environment to help stabilize target proteins.
Membrane protein targets can also be crystallized with lipidic cubic phase (LCP) methods38. LCP is a mesophase formed by the mixing of lipid and water, in which a continuous bilayer is permeated by two non-intersecting networks of solvent channels. This three dimensional structure allows diffusion of lipid embedded membrane proteins in a largely native-like environment and allows crystal contacts to form between the hydrophobic and hydrophilic surfaces of the proteins and decreases the overall solvent content of crystals while enhancing their ordering. Since its introduction in the 1990s, LCP techniques have been critical in determining the structures of many elusive membrane protein targets, such as rhodopsins39,40 and GPCRs41-43. Use of LCP in high throughput screens requires special provisions (i.e., LCP-specific robot, gas-tight syringes, LCP dispensing tool, sandwich crystallization plates, etc.) as the thick monoolein commonly used for LCP cannot be handled by traditional nanoliter liquid handling robots.
Gel filtration chromatography is an extremely important step for crystallization of membrane proteins in general because it yields information on how stabilizing the chosen detergent is for the membrane protein target, which can be visualized by the chromatogram. By comparing the amount of sample in the void volume, retention times, and shapes of the peaks, the overall stability and monodispersity of the sample can be accessed. An ideal sample would have very little to no sample lost in the void volume and would have a single symmetric elution peak with a Gaussian distribution. The detergents C8E4 (0.8%), OG (1.0%), and LDAO (0.05%) are routinely used for crystallization of β-barrel OMPs with success and are good ones to start with. Ideally, small scale experiments are performed comparing several detergents or mixtures of detergents to determine which are most appropriate for crystallization. Those that are found to be most stabilizing are then used for large scale preparations of the target β-barrel OMP and for crystallization trials.
Once crystals are formed of the target β-barrel OMP, lead optimization (i.e., additive screening, cryo-screening, detergent additive screening, etc.) and other techniques similar to soluble protein targets can be followed with few differences. However, there are some recent advances that are extremely useful when working with membrane proteins. Specifically, membrane protein crystals can often be hard to detect within their mother liquor for a variety of reasons. Membrane proteins often form relatively small crystals and false positives can misdirect crystal optimization. LCP techniques especially present additional challenges, given the highly viscous, often opaque environments in which the crystals are grown. Strategies to address these issues include use of ultraviolet microscopes (UV) in conjunction with light microscopes, allowing naturally fluorescent protein crystals to be distinguished from non-fluorescing salt and detergent crystals (Figure 7). Challenges remain, however, in positive identification of protein crystals that form within fields of precipitant. Crystals can also be detected by exploitation of the frequency doubling effect of most chiral crystals when imaged by femtosecond scanning laser pulses, as implemented by SONICC technology44. This high resolution, high contrast technique can be used to distinguish submicron crystals from obscuring conditions.
Harvesting membrane proteins is done using standard crystallography techniques, particularly for detergent and bicelle crystallization. Looping is performed by hand using a low magnification microscope and a crystal-mounting loop (i.e., nylon fiber, wire, or polymer). Wicking of excess solvent and cryo protection prior to plunge freezing in cryogenic liquid are also standard procedures when harvesting β-barrel OMP crystals. However, harvesting of LCP grown crystals presents particular problems, especially if harvesting directly from sandwich plates, as crystals can be difficult to access and may not be easily observed through the microscope. Also, crystals grown in LCP must sometimes be harvested in bulk since the LCP mixture cannot be easily separated within the loop.
Data collection for β-barrel OMPs can be performed as with soluble protein crystals with only a few additional considerations. While the size of crystals grown by detergent and bicelle methods are often comparable to those grown with soluble proteins, crystals grown by the LCP method are almost always significantly smaller. In addition, because samples harvested from LCP matrix often contain multiple crystals which are difficult to observe, one must utilize synchrotron sources that have mini-beam and loop rastering capabilities which can systematically scan the entire loop to locate the positions of crystals based on diffraction (Figure 7). The small size of LCP crystals also makes them especially susceptible to radiation damage. Therefore data from multiple crystals are often merged in order to collect a complete dataset.
Once well diffracting crystals are obtained and a complete dataset is collected, structure determination for β-barrel OMPs can be accomplished using the same procedures as for soluble proteins, keeping in mind that membrane protein crystals generally exhibit a higher solvent content. As with all crystallography targets, whether soluble protein or membrane protein, each offers its own challenges and for that reason no single pipeline can directly apply to all targets. Therefore, it is the job of the primary researcher(s) to tailor these general protocols accordingly to ensure the success of his/her project.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We would like to thank Herve Celia of the CNRS for providing the UV images and Chris Dettmar and Garth Simpson in the Department of Chemistry at Purdue University for providing the SONICC images. We would like to acknowledge funding from the National Institute of Diabetes and Digestive and Kidney Diseases and the Intramural Research Program at the National Institutes of Health. Additionally, we would like to acknowledge additional funding from the National Institute of General Medical Sciences (A.M.S. and C.J.), National Institute of Allergy and Infectious Diseases (N.N. 1K22AI113078-01), and the Department of Biological Sciences at Purdue University (N.N.).
References
- Walther DM, Rapaport D, Tommassen J. Biogenesis of beta-barrel membrane proteins in bacteria and eukaryotes: evolutionary conservation and divergence. Cell Mol Life Sci. 2009;66:2789–2804. doi: 10.1007/s00018-009-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci DP, Silhavy TJ. The Bam machine: A molecular cooper. Biochim Biophys Acta. 2012;1818:1067–1084. doi: 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan CL, Silhavy TJ, Kahne D. beta-Barrel membrane protein assembly by the Bam complex. Ann Rev of Biochem. 2011;80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- Noinaj N, et al. Structural basis for iron piracy by pathogenic Neisseria. Nature. 2012;483:53–58. doi: 10.1038/nature10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, et al. Structural insight into the biogenesis of beta-barrel membrane proteins. Nature. 2013;501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatzeva-Topalova PZ, Walton TA, Sousa MC. Crystal structure of YaeT: conformational flexibility and substrate recognition. Structure. 2008;16:1873–1881. doi: 10.1016/j.str.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, et al. Structure and function of an essential component of the outer membrane protein assembly machine. Science. 2007;317:961–964. doi: 10.1126/science.1143993. [DOI] [PubMed] [Google Scholar]
- Geibel S, Procko E, Hultgren SJ, Baker D, Waksman G. Structural and energetic basis of folded-protein transport by the FimD usher. Nature. 2013;496:243–246. doi: 10.1038/nature12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan G, et al. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature. 2011;474:49–53. doi: 10.1038/nature10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg B, Black PN, Clemons WM, Jr, Rapoport TA. Crystal structure of the long-chain fatty acid transporter FadL. Science. 2004;304:1506–1509. doi: 10.1126/science.1097524. [DOI] [PubMed] [Google Scholar]
- Fairman JW, Noinaj N, Buchanan SK. The structural biology of beta-barrel membrane proteins: a summary of recent reports. Curr Op Struct Bio. 2011;21:523–531. doi: 10.1016/j.sbi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, Guillier M, Barnard TJ, Buchanan SK. TonB-dependent transporters: regulation, structure, and function. Ann Rev of Micro. 2010;64:43–60. doi: 10.1146/annurev.micro.112408.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siburt CJ, et al. Hijacking transferrin bound iron: protein-receptor interactions involved in iron transport in N. gonorrhoeae. Metallomics. 2009;1:249–255. doi: 10.1039/b902860a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen CN, Hollander A. TonB-Dependent Transporters Expressed by Neisseria gonorrhoeae. Front in Micro. 2011;2(117) doi: 10.3389/fmicb.2011.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautin N, Bernstein HD. Protein secretion in gram-negative bacteria via the autotransporter pathway. Ann Rev of Micro. 2007;61:89–112. doi: 10.1146/annurev.micro.61.080706.093233. [DOI] [PubMed] [Google Scholar]
- Leo JC, Grin I, Linke D. Type V secretion: mechanism(s) of autotransport through the bacterial outer membrane. Phil Trans of the Royal Soc of London. Series B, Biological Sciences. 2012;367:1088–1101. doi: 10.1098/rstb.2011.0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan SK, Yamashita S, Fleming KG. In: Comprehensive Biophysics. Engelman EH, Tamm LK, editors. Vol. 5. Academic Press; 2011. pp. 139–163. [Google Scholar]
- Buchanan SK, et al. Structure of colicin I receptor bound to the R-domain of colicin Ia: implications for protein import. EMBO J. 2007;26:2594–2604. doi: 10.1038/sj.emboj.7601693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, et al. Structural insights into Ail-mediated adhesion in Yersinia pestis. Structure. 2011;19:1672–1682. doi: 10.1016/j.str.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun RS, Serventi IM, Moss J. Rapid reliable ligation-independent cloning of PCR products using modified plasmid vectors. BioTechniques. 1992;13:515–518. [PubMed] [Google Scholar]
- Rosenbusch JP. Characterization of the major envelope protein from Escherichia coli. Regular arrangement on the peptidoglycan and unusual dodecyl sulfate binding. J Biol Chem. 1974;249:8019–8029. [PubMed] [Google Scholar]
- Barnard TJ, Wally JL, Buchanan SK, et al. Coligan JE, et al., editors. Crystallization of integral membrane proteins. Curr Prot in Prot Science. 2007. p. Unit 17 19. [DOI] [PubMed]
- Ujwal R, Abramson J. High-throughput crystallization of membrane proteins using the lipidic bicelle method. JoVE. 2012. p. e3383. [DOI] [PMC free article] [PubMed]
- Faham S, Ujwal R, Abramson J, Bowie JU. Practical Aspects of Membrane Proteins Crystallization in Bicelles. Curr Topics in Membranes. 2009;63:111–127. [Google Scholar]
- Li D, Boland C, Walsh K, Caffrey M. Use of a robot for high-throughput crystallization of membrane proteins in lipidic mesophases. JoVE. 2012. p. e4000. [DOI] [PMC free article] [PubMed]
- Liu W, Cherezov V. Crystallization of membrane proteins in lipidic mesophases. JoVE. 2011. [DOI] [PMC free article] [PubMed]
- Buchanan SK. Overexpression and refolding of an 80-kDa iron transporter from the outer membrane of Escherichia coli. Biochem Soc Trans. 1999;27:903–908. doi: 10.1042/bst0270903. [DOI] [PubMed] [Google Scholar]
- Buchanan SK. Beta-barrel proteins from bacterial outer membranes: structure, function and refolding. Curr Op in Struct Bio. 1999;9:455–461. doi: 10.1016/S0959-440X(99)80064-5. [DOI] [PubMed] [Google Scholar]
- Stanley AM, Fleming KG. The process of folding proteins into membranes: challenges and progress. Arch of Biochem and Biophysics. 2008;469:46–66. doi: 10.1016/j.abb.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Pautsch A, Schulz GE. Structure of the outer membrane protein A transmembrane domain. Nature Struct Bio. 1998;5:1013–1017. doi: 10.1038/2983. [DOI] [PubMed] [Google Scholar]
- Cowan SW, et al. The structure of OmpF porin in a tetragonal crystal form. Structure. 1995;3:1041–1050. doi: 10.1016/s0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- Ujwal R, et al. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. PNAS. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert V, et al. Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS Bio. 2006;4:e377. doi: 10.1371/journal.pbio.0040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramasivam N, Habeck M, Linke D. Is the C-terminal insertional signal in Gram-negative bacterial outer membrane proteins species-specific or not? BMC Genomics. 2012;13:510. doi: 10.1186/1471-2164-13-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agah S, Faham S. Crystallization of membrane proteins in bicelles. Methods in Mol Bio. 2012;914:3–16. doi: 10.1007/978-1-62703-023-6_1. [DOI] [PubMed] [Google Scholar]
- Ujwal R, Bowie JU. Crystallizing membrane proteins using lipidic bicelles. Methods. 2011;55:337–341. doi: 10.1016/j.ymeth.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V. Lipidic cubic phase technologies for membrane protein structural studies. Cur Op in Struct Bio. 2011;21:559–566. doi: 10.1016/j.sbi.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 A resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- Kato HE, et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012;482:369–374. doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JF, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupert LM, Simpson GJ. Screening of protein crystallization trials by second order nonlinear optical imaging of chiral crystals (SONICC) Methods. 2011;55:379–386. doi: 10.1016/j.ymeth.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]