

Abstract
Coxiella burnetii, the causative agent of Q fever, is an intracellular pathogen that relies on a Type IV Dot/Icm Secretion System to establish a replicative niche. A cohort of effectors are translocated through this system into the host cell to manipulate host processes and allow the establishment of a unique lysosome-derived vacuole for replication. The method presented here involves the combination of two well-established techniques: specific gene silencing using siRNA and measurement of effector translocation using a FRET-based substrate that relies on β-lactamase activity. Applying these two approaches, we can begin to understand the role of host factors in bacterial secretion system function and effector translocation. In this study we examined the role of Rab5A and Rab7A, both important regulators of the endocytic trafficking pathway. We demonstrate that silencing the expression of either protein results in a decrease in effector translocation efficiency. These methods can be easily modified to examine other intracellular and extracellular pathogens that also utilize secretion systems. In this way, a global picture of host factors involved in bacterial effector translocation may be revealed.
Keywords: Infection, Issue 113, Host-pathogen interactions, bacterial secretion systems, β-lactamase translocation assay, BlaM substrate, fluorescence, microbiology, intracellular bacterial pathogens, Coxiella burnetii, gene silencing, siRNA, molecular biology
Introduction
Coxiella burnetii is a unique intracellular pathogen that causes the zoonotic human infection Q fever. This disease is associated with a broad spectrum of clinical presentations extending from asymptomatic seroconversion to life-threatening chronic infection that often manifests as endocarditis years after exposure1. Human infection occurs primarily through the inhalation of contaminated aerosols with ruminants the major reservoir for infection, in particular, dairy cows, sheep and goats. Although Coxiella infection in these animals is typically subclinical, the infection may trigger abortion and the considerable bacterial load within the birthing fluid and placenta can contaminate the local environment1. An example of the enormous burden such contamination can have on both public health and the agriculture industry was recently observed in the Q fever outbreak that occurred in the Netherlands2. Between 2007 and 2010, over 4,000 human cases of Q fever were diagnosed and this outbreak was linked to significant contamination of goat farms3. Additionally, Coxiella is a potential biological weapon, as classified by the US Centers for Disease Control and Prevention, due to the environmental stability of the bacteria and low infectious dose necessary to cause severe morbidity and mortality4.
Coxiella exists in two phases: Phase I organisms, isolated from natural sources, are extremely virulent and Phase II organisms are highly attenuated in vivo. For example, after several in vitro passages of Coxiella burnetii Nine Mile Phase I organisms, Phase II bacteria were produced that contain an irreversible chromosomal deletion resulting in truncated lipopolysaccharide (LPS)5. This strain, C. burnetii NMII, is phenotypically similar to Phase I in tissue culture models and provides a safer model for researchers to study Coxiella pathogenesis in laboratories5. In recent years several breakthroughs have rapidly advanced the field of Coxiella genetics. Most notably, the development of axenic media (acidified citrate cysteine medium - ACCM-2) has allowed the cell-free growth of Coxiella in both liquid and on solid media6,7. This resulted in direct improvements of genetic tools available for Coxiella including an inducible gene expression system, shuttle vectors and random transposon systems8-11. Most recently, two methods for targeted gene inactivation have also been developed, paving the way for examining specific virulence gene candidates12.
Following internalization by alveolar macrophages, Coxiella replicates to high numbers within a membrane-bound compartment termed the Coxiella-containing vacuole (CCV). The CCV requires host endocytic trafficking through early and late endosomes until it matures into a lysosome-derived organelle13. Throughout this process, the CCV acquires host factors that either appear transiently or remain associated with the vacuole, including, but not limited to, Rab5, Rab7, CD63 and LAMP-113-15. Replication of Coxiella within host cells is entirely dependent on a fully functional Dot/Icm Type IVB Secretion System (T4SS)8,16,17. This secretion system is a multi-protein structure ancestrally related to conjugation systems and spans both bacterial and vacuolar membranes to deliver bacterial proteins, termed effectors, into the host cytoplasm18. The Coxiella T4SS is functionally very similar to the well characterized Type IVB Dot/Icm Secretion System of Legionella pneumophila19,20. Interestingly, activation of the T4SS and subsequent effector translocation does not occur until Coxiella reaches the acidic lysosome-derived organelle, approximately 8 hr post-infection17,21. To date, over 130 Dot/Icm effectors have been identified9,17,22-24. Many of these effectors likely play important roles during replication of Coxiella within host cells; however, only a few effectors have been functionally characterized25-29.
In this study we utilize a fluorescence based translocation assay that relies on cleavage of the CCF2-AM FRET substrate (hereafter referred to as the BlaM substrate) via β-lactamase activity within the host cell cytoplasm (Figure 1). The gene of interest is fused to TEM-1 β-lactamase (BlaM) on a reporter plasmid that provides constitutive expression. The BlaM substrate is composed of two fluorophores (coumarin and fluorescein) that form a FRET pair. Excitation of the coumarin results in FRET of the fluorescein and green fluorescence emission in the absence of effector translocation; however, if the BlaM-effector fusion protein is translocated into the host cytoplasm, the resultant β-lactamase activity cleaves the β-lactam ring of the BlaM substrate, separating the FRET pair producing blue fluorescence emission following excitation. This translocation assay has been well proven as an approach to identify effector proteins from a range of different intracellular and extracellular bacteria, including C. burnetii, L. pneumophila, L. longbeachae, Chlamydia trachomatis, enteropathogenic E. coli, Salmonella and Brucella17,30-35.
To determine the role of specific host factors on Coxiella effector translocation we utilize a well-established method for gene silencing known as RNA interference, in particular small interfering RNA (siRNA). Originally identified in Caenorhabditis elegans, RNA interference is a conserved endogenous cellular process used for innate defense against viruses as well as gene regulation36,37. After binding of sequence-specific siRNA, degradation of mRNA occurs through RISC (RNA-induced silencing complex) resulting in specific gene silencing or knockdown38. In this study, siRNA was used to target two host proteins, Rab5A and Rab7A, which are important regulators of the endocytic pathway. The impact of silencing Rab5A and Rab7A on effector translocation was ascertained using C. burnetii pBlaM-CBU0077. CBU0077 was selected as it was previously shown to be translocated by the Dot/Icm secretion system of Coxiella17.
Utilizing both siRNA gene silencing and the fluorescencebased translocation assay described here, we are beginning to establish a role for host factors in the translocation of effector proteins by Coxiella. This approach can be applied to a wide range of both intracellular and extracellular bacteria that possess similar secretion systems responsible for the translocation of effector proteins.
Protocol
Note: All procedures involving the growth or manipulation of Coxiella burnetii RSA439 NMII should be performed in a Physical Containment Level 2 Laboratory and within a biological safety cabinet in compliance with local guidelines. A schematic diagram of the reverse transfection and translocation assay workflow described below is shown in Figure 2.
1. Preparation of C. burnetii Culture Expressing CBU0077 Fused to β-lactamase (pBlaM-CBU0077) (DAY 1)
- Prepare 1X ACCM-26:
- Combine the components listed in Table 1. Adjust pH to 4.75 with 6 M NaOH and filter sterilize (do not autoclave). ACCM-2 is stable for approximately three weeks stored at 4 °C.
- Generate C. burnetii pBlaM-CBU0077 stocks.
- Infect HeLa 229 cells with the C. burnetii strain carrying pBlaM-CBU0077 and passage until large vacuoles are observed in the majority of cells.
- Grow the C. burnetii strain carrying pBlaM-CBU0077 in 20 ml ACCM-2 containing 3 µg/ml chloramphenicol in a 75 cm2 tissue culture flask with vented cap for 7 days at 37 °C in 5 % CO2, 2.5 % O2.
- Centrifuge the C. burnetii pBlaM-CBU0077 culture at 15,000 × g for 15 min at RT.
- Re-suspend the pellet in 20 ml DMEM + 10 % fetal calf serum (FCS) + 3 µg/ml chloramphenicol (maintenance media). Remove media from a 50 % confluent 75 cm2 flask of HeLa 229 cells. Add re-suspended C. burnetii to HeLa 229 cells. Incubate for 2 days at 37 °C in 5 % CO2 to allow infection of HeLa 229 cells to establish.
- Split cells into 175 cm2 tissue culture flask.
- Remove media from the 75 cm2 flask and wash the monolayer twice with 10 ml pre-warmed PBS.
- Add 1 ml of 0.05 % trypsin-EDTA solution and place cells at 37 °C + 5 % CO2 for 3 - 5 min to detach cells.
- Re-suspend cells in 10 ml of maintenance media. Add 2 ml of re-suspended cells into a 175 cm2 flask containing 38 ml of maintenance media.
- Incubate at 37 °C in 5 % CO2 for a further 3 - 5 days until 100 % confluency is reached and large vacuoles are observed.
- Collect supernatant from the 175 cm2 flask, add 10 ml dH2O to cover the infected HeLa 229 cells and incubate for 15 min to lyse infected cells.
- Combine supernatant and lysed cells and pellet at 15,000 × g for 15 min at RT.
- Re-suspend pellet in 10 ml dH2O. Lyse cells further by repeatedly passing the re-suspension through a 23G needle attached to a 10 ml syringe at least 20 times. Pellet at 500 × g for 5 min to remove cellular debris.
- Remove and pellet supernatant at 15,000 × g for 15 min at RT to collect C. burnetii.
- Re-suspend C. burnetii in DMEM + 10 % FCS and quantify number of bacteria as per 4. Dilute to 1 × 109 genomes/ml using DMEM + 10 % FCS + 10 % dimethyl sulfoxide (DMSO), aliquot 50 µl stocks into microfuge tubes and store at -80 °C.
Inoculate 10 ml of pre-warmed ACCM-2 containing 3 µg/ml chloramphenicol with 20 µl of C. burnetii pBlaM-CBU0077 strain17 (from stocks generated in 1.2 stored at -80 °C) in a 25 cm2 tissue culture flask with vented cap. Grow bacterial culture for 7 days at 37 °C in 5 % CO2, 2.5 % O2.
2. Reverse Transfection of siRNA and Seeding of HeLa 229 Cells (DAY 4)
- When using the experimental siRNA for the first time prepare the siRNA as follows:
- Briefly centrifuge the lyophilized siRNA to ensure that the siRNA pellet is collected at the bottom of the tube.
- Re-suspend the pelleted siRNA to a final stock concentration of 20 µM by pipetting the appropriate volume of 1x siRNA buffer (Table 2).
- Pipette the solution up and down 3 - 5 times to mix and place on an orbital mixer for 30 min at RT, inverting the tube every 5 - 10 min.
- Briefly centrifuge the tube to ensure the siRNA is collected at the bottom of the tube.
- Aliquot the siRNA into 50 µl aliquots in microfuge tubes and store at -20 °C until required.
- To generate 1 µM working concentration of siRNA, pipette 950 µl of 1X siRNA buffer into a 50 µl aliquot stock (20 µM) when necessary. Note: This 1 µM working concentration can be freeze-thawed multiple times for repeated transfections.
Place DMEM + 10 % FCS, 0.05 % trypsin-EDTA and PBS in a 37 °C water bath (or equivalent) for 30 min to warm prior to use.
- Preparation of transfection components: Note: The following protocol has been optimized for HeLa 229 cells in a 96-well microplate with black walls and flat, transparent bottom. Optimization of the amount of transfection reagent, concentration of siRNA and seeding density of cells may be necessary for different cell lines.
- For each well, use 0.1 µl transfection reagent, 15.9 µl reduced-serum medium (minimal essential media used for transfections, refer to Table of Materials) and 4 µl of 1 µM siRNA (resulting in a final siRNA concentration of 40 nM). Use separate microfuge tubes for OTP, PLK1, Rab5A and Rab7A. Measure each assay condition in triplicate. An example of a 96-well plate layout is shown in Figure 3. Note: This protocol incorporates the use of two controls: OTP and PLK1. OTP is composed of four pooled siRNA that have been optimized to reduce off-target effects and thus OTP is used as a negative, non-targeting control. Scrambled siRNA could also be used as a negative control. Loss of PLK1 expression results in extensive cell death in a number of cell lines by inducing pro-apoptotic pathways and therefore PLK1 siRNA is used as positive control for transfection where cell death correlates to transfection efficiency.
- For each experimental siRNA transfection, combine 0.4 µl transfection reagent and 63.6 µl reduced-serum medium in an individual microfuge tube. Vortex all microfuge tubes and allow components to complex for 5 min at RT.
- Add 16µl of each experimental siRNA to the corresponding microfuge tubes containing the transfection complex. Vortex and incubate for 20 min at RT. Note: It is important not to exceed 30 min, as transfection efficiency will decrease.
During the 20 min incubation (2.3.1.2) add 20 µl of the transfection reagent + reduced-serum medium + siRNA mix into the appropriate wells of a sterile black, flat clear bottom 96-well tray.
- As soon as the 20 min incubation period is complete, seed HeLa 229 cells at a density of 3.92 × 103 cells/well.
- Harvest HeLa 229 cells from a confluent or sub-confluent 75 cm2 tissue culture flask in pre-warmed DMEM + 10% FCS and re-suspend cells to a final density of 4.9 × 104 cells/ml as per standard methods. To ensure transfection efficiency is not compromised, prepare cells during the 20 min incubation period (2.3.1.2).
- Wash the monolayer of HeLa 229 cells twice with 10 ml of pre-warmed PBS.
- Add 1 ml of 0.05 % trypsin-EDTA solution and place HeLa 229 cells at 37 °C + 5 % CO2 for 3 - 5 min to detach cells.
- Collect cells in 9 ml of pre-warmed DMEM + 10 % FCS. Count cells using a haemocytometer and prepare cells to a final density of 4.9 × 104 cells/ml using DMEM + 10 % FCS.
- Pipette 80 µl of HeLa 229 cells at density of 4.9 × 104 cells/ml into each well that contains the transfection reagent + reduced-serum medium + siRNA mix. This corresponds to a cell density of 3.92 × 103 cells/well. Note: Background subtraction is required for the BlaM substrate.
- Do not add cells or siRNA to "blank" wells (Figure 3). Instead, add 80 µl of DMEM + 10 % FCS to these wells set up in triplicate.
- Incubate for 24 hr at 37 °C + 5 % CO2.
3. Change Media (DAY 5)
After 24 hr, remove media using a multichannel adapter attached to a vacuum source or manually with a multi-channel pipette, and replace with 100 µl of fresh prewarmed DMEM + 10% FCS.
Incubate for a further 48 hr at 37 °C + 5 % CO2.
At this point, check the viability of cells visually using a standard light microscope. If the reverse transfection has been successful, significant cell death will be observed in the wells containing PLK1 siRNA compared to OTP siRNA.
4. Quantification of Coxiella burnetii pBlaM-CBU0077 Strain using qPCR (DAY 7)
After 7 days of growth in ACCM-2 at 37 °C in 5 % CO2, 2.5% O2 (1.3), centrifuge the 10 ml C. burnetii pBlaM-CBU0077 culture at 15,000 × g for 15 min at RT.
Re-suspend the bacterial pellet in 10 ml of pre-warmed DMEM + 10 % FCS. Prepare 1:10 and 1:100 dilutions of the culture (sample) using dH2O.
- Set up the components required for the qPCR. Note: gDNA extraction can be performed in advance and stored at -20 °C until required.
- Preparation of standards:
- Extract gDNA from Coxiella burnetii RSA439 NMII wild type strain grown in ACCM-2 at 37 °C in 5 % CO2, 2.5 % O2 for 7 days using a gDNA extraction kit, as per manufacturer's instructions.
- Measure the gDNA concentration (g/µl) using a Nanodrop (or equivalent) at an absorbance of 260 nm.
- Calculate number of genome copies per µl using the following formula below, and convert to number of genomes/ml.
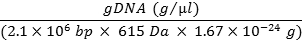
- Adjust the number of genomes/ml to 109/ml (in a final volume of 100 µl) in a new microfuge tube using dH2O. This can be boiled for 10 min and stored at -20 °C until required.
- On the day of the infection, generate 10-fold serial dilutions of 108 genomes/ml to 105 genomes/ml from the 109 genomes/ml stock.
- Pipette 10 µl of 109 genomes/ml into 90 µl of dH2O to generate 108 genomes/ml. Vortex to mix. Pipette 10 µl of 108 genomes/ml into 90 µl of dH2O to generate 107 genomes/ml. Vortex and repeat until 105 genomes/ml.
- Set up qPCR reaction. Create a master mix for 25 wells to account for any pipetting errors. For each well, use 10 µl qPCR Master Mix; 2 µl of ompA primer mix (4.3.2.2) and 3 µl of dH2O. Note: OmpA is an outer-membrane protein of C. burnetii'39 and an 82-base-pair section within a highly conserved region was selected for use as a probe as described previously40.
- Set up each standard (dH2O, 105, 106, 107, 108, 109 genomes/ml) and sample (1:10 and 1:100 dilution) in a 96-well PCR tear away plate (non-skirted) in duplicate or triplicate, respectively. Add 5 µl of sample or standard into the appropriate wells.
- Using dH2O, dilute both the ompA forward primer (5'-CAGAGCCGGGAGTCAAGCT-3') and ompA reverse primer (5'-CTGAGTAGGAGATTTGAATCGC-3') to a final concentration of 4 µM and combine into the same microfuge tube. Note: The ompA primer mix can be prepared in advance and stored at -20 °C until required.
- Combine 250 µl qPCR Master Mix, 50 µl ompA primer mix, 75 µl dH2O and vortex to mix. Add 15 µl of this master mix to wells containing either standards or samples.
- Place 8-lid PCR strip caps onto the appropriate wells and pulse centrifuge the PCR tubes to ensure everything is collected in the bottom of the tubes.
- Set up the following program on a quantitative PCR machine: 50 °C for 2 min, 95 °C for 10 min, followed by 45 cycles of 95 °C for 1 sec and 60 °C for 20 sec (Figure 4A).
- Analyze the Ct (cycle threshold) values from the qPCR:
- Transfer the Ct values to a spreadsheet using the program's export function.
- Create a scatterplot of the standards 105 genomes/ml through to 109 genomes/ml (expressed as log values). Discard any obvious outliers amongst the triplicate samples. Generate a logarithmic trendline and determine the equation of the graph.
- Calculate the total number of genomes/ml in the sample by using the equation generated in 4.3.3.2. Do not forget to account for the initial dilution (1:10 and 1:100) of the samples.
5. Infection of Reverse Transfected Cells (DAY 7)
- After calculating the total genomes/ml in the C. burnetii pBlaM-CBU0077 culture to be used for infection, adjust re-suspended bacterial culture to infect cells at a MOI of 300 in a final volume of 50 µl/well using prewarmed DMEM + 10% FCS. Note: The expected density of the seeded HeLa 229 cells with a normal doubling time after 72 hr is 3.136 × 104 cells/well. The amount of bacteria required to infect at a MOI of 300 is 9.408 × 106 in 50 µl, which equates to 1.88 × 108 bacteria/ml.
- Using the value calculated from the qPCR (genomes/ml) (4.3.3.3), dilute the re-suspended bacteria using pre-warmed DMEM + 10 % FCS in a final volume of 2 ml to infect the reverse transfected cells.
Remove the existing media from the blank and reverse transfected cells.
Add 50 µl of the diluted bacteria (1.88 × 108 bacteria/ml) to the appropriate wells. Add 50 µl of DMEM + 10% FCS to both the blank and uninfected wells.
Incubate for 24 hr at 37 °C + 5 % CO2.
6. Addition of BlaM Substrate to Determine Level of Translocation (DAY 8)
- Prepare the solutions for the BlaM substrate 6x loading solution. Note: Solution A, B and C are provided within the BlaM substrate kit (Refer to Table of Materials). Solution B may form a precipitate at RT. Warm this solution to 37 °C prior to use and the precipitate should dissolve.
- When using the kit for the first time, add 185 µl of DMSO (supplied with the BlaM substrate kit) to the desiccated Solution A. Subsequently, store the re-suspended Solution A at -20 °C.
- Prepare 0.1 M Probenicid solution in advance and store in 1 ml aliquots at -20°C until required.
- Prepare 0.4 M NaOH (1.6 g in 100 ml dH2O).
- Prepare 100 mM Na phosphate buffer pH 8 (1.56 g NaH2PO4.2H2O and 1.41 g Na2HPO4 in 100 ml dH2O).
- Dissolve 1.25 g of Probenicid in 22 ml of 0.4 M NaOH by vigorous agitation.
- Add 22 ml of 100 mM Na phosphate buffer pH 8 (solution will turn cloudy) and stir to dissolve the precipitate that forms.
- Adjust the pH to 8 (if necessary) using 1 M NaOH/HCl.
- Stir vigorously and heat mildly (< 50 °C) until the solution is mostly clear.
- Filter (0.2 µm pore size) and aliquot in 1 ml volumes. Store aliquots at -20°C.
For each well, use 0.06 µl Solution A, 0.54 µl Solution B, 7.9 µl Solution C and 1.5 µl 0.1 M Probenicid. Create a master mix for the 30 wells by first combining 1.8 µl of Solution A and 16.2 µl of Solution B together in a microfuge tube. Mix well using a vortex. Add 237 µl of Solution C and 45 µl of 0.1 M Probenicid. Vortex to combine all components. Note: The 6x loading solution is stable for up to 4 hr (in the dark) but should be prepared as close as possible before use.
Add 10 µl of the 6x loading solution directly into all blank and reverse transfected wells without removing the existing media.
Incubate the plate at RT for 2 hr in the dark.
- To determine the level of translocation, quantify the amount of fluorescence using a microplate reader. Note: the following procedure is specific to the ClarioSTAR microplate reader. Equivalent microplate readers can also be utilized.
- Create a new fluorescence intensity protocol using endpoint as the reading mode.
- Adjust the basic parameters as follows:
- Select 96-well microplate.
- Under optic settings, select 2 multichromatics with the following user-defined settings: (1) Input 410-10 nm excitation and 520-10 nm emission. (2) Input 410-10 nm excitation and 450-10 nm emission. Ensure well multichromatics is selected.
- Select orbital averaging with a diameter of 3 mm.
- Under optic, select bottom optic.
- Under speed and precision, select precise. This corresponds to eight regions per well.
- Adjust the plate layout by selecting the appropriate wells as samples.
- Select start measurement.
- Remove the lid and place the tray into the plate reader. To avoid reaching maximum fluorescence values, ensure the gain value is adjusted. To do so, perform a gain adjustment on one of the infected wells that has been reverse transfected with OTP siRNA. This corresponds to the highest expected translocation.
- Select start measurement to measure all appropriate wells using bottom fluorescence at an excitation of 410 nm and emission of both 450 nm and 520 nm for blue and green fluorescence, respectively.
7. Analysis of Results:
- Calculate the ratio of 450 nm to 520 nm (blue to green) for all samples following blank reduction and express relative to the uninfected OTP sample. Note: the following analysis steps are provided for a simple spreadsheet program.
- Using the export function on the microplate reader, transfer the raw fluorescence values obtained for both 520 nm and 450 nm emission wavelengths (6.5) into a spreadsheet.
- Calculate the average of the "blank" readings (media only, no cells) for the 520 nm wavelength by adding the raw 520 nm fluorescence values and dividing by the number of wells (3). Repeat using the blank 450 nm wavelength values. These values represent the background fluorescence due to the 96-well plate and media.
- Subtract the background fluorescence. Using the values obtained in 7.1.2 subtract the average of the blank 520 nm wavelength from all sample 520 nm fluorescence values. Repeat for the 450 nm wavelength values.
- To obtain the 450:520 nm ratio use the blank corrected values (7.1.3). Divide each sample 450 nm fluorescence value by its corresponding 520 nm value.
- Calculate the average of the uninfected OTP ratio values by adding the 450:520 nm ratio values (from 7.1.4) and dividing by the number of wells (3).
- Calculate the ratio of the samples relative to uninfected OTP by dividing the 450:520 nm ratio calculated in 7.1.4 by the average of the uninfected OTP ratio value calculated in 7.1.5.
8. Addition of Nuclei Stain to Determine Cell Number after Translocation Assay (DAY 8)
Following quantification of fluorescence, remove media containing 6x loading solution from all wells using either a multi-channel adapter attached to a vacuum source or manually with a multi-channel pipette.
Add 50 µl of 4 % PFA (paraformaldehyde) solution in PBS to all appropriate wells to fix cells. Incubate at RT for 20 min.
Remove 4 % PFA PBS solution (as per 8.1) and wash cells twice with 75 µl of PBS.
Add 50 µl of a cell permeable nuclear stain, for example DAPI (4',6-diamidino-2-phenylindole), to each well. Acquire images with a fluorescent microscope and quantify the number of nuclei present in each well after siRNA treatment using appropriate image analysis software, such as ImageJ41.
Representative Results
For this study, the C. burnetii pBlaM-CBU0077 strain was selected as CBU0077 has been previously shown to be a translocated effector of the Coxiella Dot/Icm secretion system17. Prior to infection, the total number of genomes/ml in the seven day C. burnetii pBlaM-CBU0077 culture was enumerated using qPCR. Figure 4 demonstrates an example of the cycle threshold (Ct) values expected from both standards and samples following qPCR. The Ct values (represented graphically in the Amplification Plot (Figure 4B) and numerically (Figure 4C)) were exported and analyzed as described in 4.3.3. Based on the representative data shown, there were 2.31 × 108 genomes/ml in the 10 ml re-suspended bacterial culture (Figure 4D). To generate the appropriate bacterial dilution (1.88 × 108 genomes/ml in 2 ml), 1.63 ml of resuspended bacteria was added to 370 µl of fresh, pre-warmed DMEM + 10% FCS. Cells reverse transfected with siRNA were subsequently infected with C. burnetii pBlaM-CBU0077 at an MOI of 300 for 24 hr.
After incubation of the reverse transfected infected cells with BlaM substrate quantification of effector translocation can be determined using a fluorescence plate reader. Following analysis as described in 7, all ratio values that have been normalized to uninfected OTP can also be normalized to infected OTP. The level of effector translocation after silencing of host genes can be grouped into three outcomes after comparison to infected OTP control: (1) No change; (2) Increased effector translocation; (3) Decreased effector translocation. Subsequent statistical analysis, for example a Student ttest, can be used to determine the significance of any observed difference to infected OTP control. The result of silencing either Rab5A or Rab7A on effector translocation from three independent experiments is shown in Figure 5A. A significant decrease in the level of BlaM-CBU0077 translocation occurred during silencing of both Rab5A and Rab7A compared to OTP control (p value = 0.0088 (Rab5A) and p value = 0.0059 (Rab7A), paired, Student ttest). The impact of siRNA treatment on cell viability was also established. Following the translocation assay, cells were fixed, stained with a nuclei stain and subsequently quantified. As shown in Figure 5C, although treatment with either Rab5A or Rab7A results in a reduction in cell viability compared to OTP control (12 % and 31 %, respectively), this reduction is not as severe as PLK1 (97 %), a gene known to cause cell death (p-value = 0.0102 (Rab5A); p-value = 0.0081 (Rab7A); p-value < 0.0001 (PLK1), paired, Student t-test). These results demonstrate how silencing of host genes using siRNA can negatively alter the levels of bacterial effector translocation.
 Figure 1. The Mechanism of Action of the BlaM Substrate During Infection.
C. burnetii is internalized by host cells and forms a lysosome-derived vacuole termed the Coxiella-containing vacuole. 24 hr post-infection, cells are loaded with the membrane permeable BlaM substrate that possesses two fluorophores forming a FRET pair (A). In the absence of β-lactamase i.e., no translocation of the BlaM-CBU0077 effector, excitation of the coumarin at 410 nm results in FRET to fluorescein, observed as a green fluorescence signal (520 nm) (B). In the event of a functional Dot/Icm secretion system, the BlaM-CBU0077 fusion protein will be translocated into the host cell and will cleave the BlaM substrate, via β-lactamase activity, causing the separation of the two dyes and resulting in FRET disruption and a blue fluorescence signal (450 nm) after excitation at 410 nm (C). Both the green and blue fluorescence signals can be detected using a fluorescence plate reader. Please click here to view a larger version of this figure.
Figure 1. The Mechanism of Action of the BlaM Substrate During Infection.
C. burnetii is internalized by host cells and forms a lysosome-derived vacuole termed the Coxiella-containing vacuole. 24 hr post-infection, cells are loaded with the membrane permeable BlaM substrate that possesses two fluorophores forming a FRET pair (A). In the absence of β-lactamase i.e., no translocation of the BlaM-CBU0077 effector, excitation of the coumarin at 410 nm results in FRET to fluorescein, observed as a green fluorescence signal (520 nm) (B). In the event of a functional Dot/Icm secretion system, the BlaM-CBU0077 fusion protein will be translocated into the host cell and will cleave the BlaM substrate, via β-lactamase activity, causing the separation of the two dyes and resulting in FRET disruption and a blue fluorescence signal (450 nm) after excitation at 410 nm (C). Both the green and blue fluorescence signals can be detected using a fluorescence plate reader. Please click here to view a larger version of this figure.
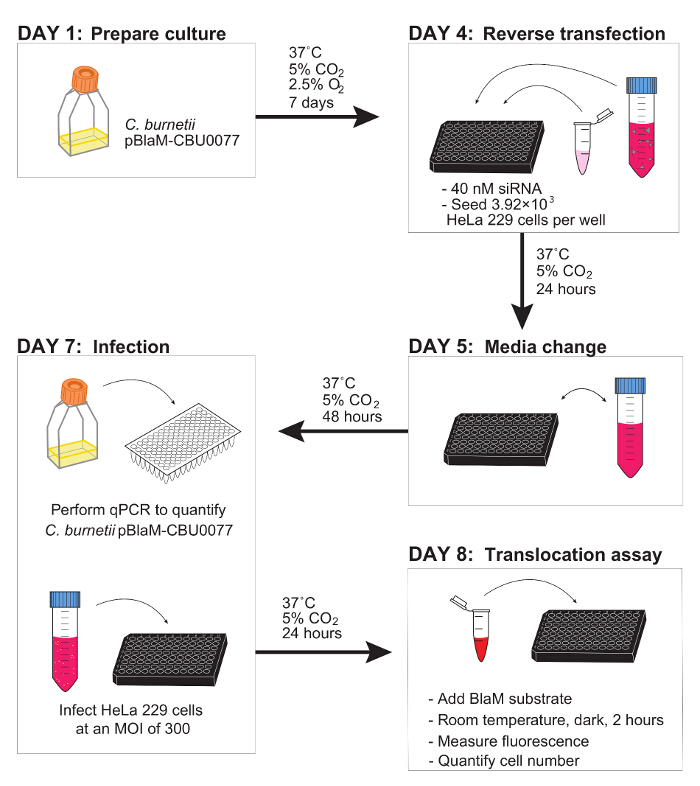 Figure 2. Schematic Overview of the Reverse Transfection and Translocation Assay Workflow.
Day 1: Prepare the C. burnetii pBlaM-CBU0077 culture. Day 4: Reverse transfect using 40 nM siRNA and seed HeLa 229 cells at a final density of 3.92 × 103 cells/well into a 96-well tray. Day 5: Change media. Day 7: Quantify the total genomes/ml of the C. burnetii pBlaM-CBU0077 culture using qPCR and subsequently infect reverse transfected cells with an MOI of 300. Day 8: Add 6x loading dye, measure fluorescence and quantify cell number. Please click here to view a larger version of this figure.
Figure 2. Schematic Overview of the Reverse Transfection and Translocation Assay Workflow.
Day 1: Prepare the C. burnetii pBlaM-CBU0077 culture. Day 4: Reverse transfect using 40 nM siRNA and seed HeLa 229 cells at a final density of 3.92 × 103 cells/well into a 96-well tray. Day 5: Change media. Day 7: Quantify the total genomes/ml of the C. burnetii pBlaM-CBU0077 culture using qPCR and subsequently infect reverse transfected cells with an MOI of 300. Day 8: Add 6x loading dye, measure fluorescence and quantify cell number. Please click here to view a larger version of this figure.
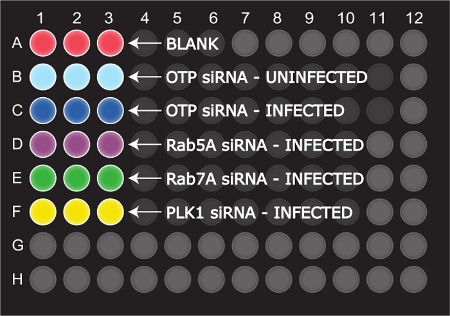 Figure 3. The 96-well Plate Layout Used to Set up the Reverse Transfection and Seeding of HeLa 229 Cells. The "blank" wells (shown in red) contain media only and do not need the addition of either siRNA or HeLa 229 cells. The OTP siRNA (shown in light blue for uninfected wells and dark blue for infected wells) is used as a non-targeting control, since it has been optimized to have fewer off-target effects. The maroon and green wells represent the Rab5A and Rab7A siRNA, respectively. PLK1 siRNA (shown in yellow) is used as a measure of transfection efficiency as loss of PLK1 expression can induce pro-apoptotic pathways and extensive cell death in a vast majority of cell lines. Please click here to view a larger version of this figure.
Figure 3. The 96-well Plate Layout Used to Set up the Reverse Transfection and Seeding of HeLa 229 Cells. The "blank" wells (shown in red) contain media only and do not need the addition of either siRNA or HeLa 229 cells. The OTP siRNA (shown in light blue for uninfected wells and dark blue for infected wells) is used as a non-targeting control, since it has been optimized to have fewer off-target effects. The maroon and green wells represent the Rab5A and Rab7A siRNA, respectively. PLK1 siRNA (shown in yellow) is used as a measure of transfection efficiency as loss of PLK1 expression can induce pro-apoptotic pathways and extensive cell death in a vast majority of cell lines. Please click here to view a larger version of this figure.
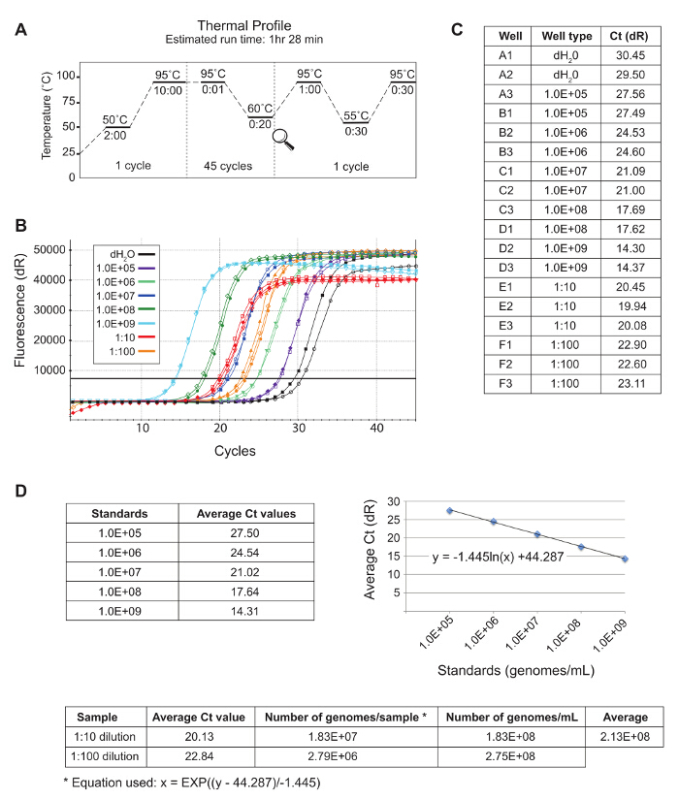 Figure 4. Representative qPCR Ct Values Used to Quantitate the Total Number of genomes/ml in C. burnetii pBlaM-CBU0077 Culture.(A) The cycle conditions used in the qPCR to enumerate the number of genomes/ml in Coxiella cultures. The Ct values for both standards and samples are represented in graphical (B) and numerical (C) formats. (D) The standard Ct values were averaged, graphed and a logarithmic trendline was calculated. Using the equation and the averages of the sample Ct values the total number of genomes/ml in the C. burnetii pBlaM-CBU0077 culture was determined to be 2.31 × 108. Please click here to view a larger version of this figure.
Figure 4. Representative qPCR Ct Values Used to Quantitate the Total Number of genomes/ml in C. burnetii pBlaM-CBU0077 Culture.(A) The cycle conditions used in the qPCR to enumerate the number of genomes/ml in Coxiella cultures. The Ct values for both standards and samples are represented in graphical (B) and numerical (C) formats. (D) The standard Ct values were averaged, graphed and a logarithmic trendline was calculated. Using the equation and the averages of the sample Ct values the total number of genomes/ml in the C. burnetii pBlaM-CBU0077 culture was determined to be 2.31 × 108. Please click here to view a larger version of this figure.
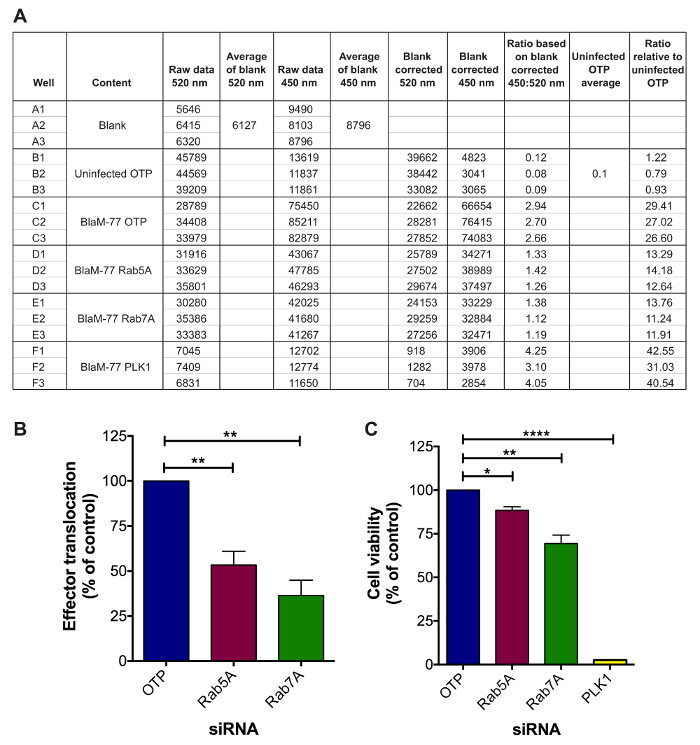 Figure 5. Translocation of the Dot/Icm Effector BlaM-CBU0077 during siRNA Silencing of Rab5A and Rab7A. HeLa 229 cells were reverse transfected with siRNA specific to Rab5A (maroon bars), Rab7A (green bars), OTP (blue bars) or PLK1 (yellow bars) and incubated for 72 hr before infection with the C. burnetii pBlaM-CBU0077 strain at an MOI of 300 for 24 hr. After the addition of the BlaM substrate, fluorescence was measured using a microplate reader. (A) The table demonstrates the raw fluorescence values obtained from a single experiment alongside the 450:520 nm ratio relative to uninfected OTP control after subtraction of the blank values (media only, no cells). The level of translocation (B) and cell viability (C) are presented as the percentage mean ± standard deviation relative to OTP reverse transfected infected cells from three independent experiments. * p-value < 0.05; **p-value < 0.01; **** p-value < 0.0001 (paired, Student t-test). Please click here to view a larger version of this figure.
Figure 5. Translocation of the Dot/Icm Effector BlaM-CBU0077 during siRNA Silencing of Rab5A and Rab7A. HeLa 229 cells were reverse transfected with siRNA specific to Rab5A (maroon bars), Rab7A (green bars), OTP (blue bars) or PLK1 (yellow bars) and incubated for 72 hr before infection with the C. burnetii pBlaM-CBU0077 strain at an MOI of 300 for 24 hr. After the addition of the BlaM substrate, fluorescence was measured using a microplate reader. (A) The table demonstrates the raw fluorescence values obtained from a single experiment alongside the 450:520 nm ratio relative to uninfected OTP control after subtraction of the blank values (media only, no cells). The level of translocation (B) and cell viability (C) are presented as the percentage mean ± standard deviation relative to OTP reverse transfected infected cells from three independent experiments. * p-value < 0.05; **p-value < 0.01; **** p-value < 0.0001 (paired, Student t-test). Please click here to view a larger version of this figure.
| Component | Concentration |
| citric acid | 13.4 mM |
| sodium citrate | 16.1 mM |
| potassium phosphate | 3.67 mM |
| magnesium chloride | 1 mM |
| calcium chloride | 0.02 mM |
| iron sulfate | 0.01 mM |
| sodium chloride | 125.4 mM |
| L-cysteine | 1.5 mM |
| neopeptone | 0.1 g/L |
| casamino acids | 2.5 g/L |
| methyl beta cyclodextrin | 1 g/L |
| RPMI | 125 ml/L |
Table 1. The Components Required to Make 1x ACCM-2.
| Target concentration | ||||
| 1µM | 5 µM | 10µM | 20µM | |
| Starting Moles | ||||
| 1 nmol | 1 ml | 200 µl | 100 µl | 50 µl |
| 2 nmol | 2 ml | 400 µl | 200 µl | 100 µl |
| 5 nmol | 5 ml | 1 ml | 500 µl | 250 µl |
| 10 nmol | 10 ml | 2 ml | 1 ml | 500 µl |
| 20 nmol | 20 ml | 4 ml | 2 ml | 1 ml |
Table 2. Volume of 1x siRNA Buffer Required for Hydrating Lyophilized siRNA to the Appropriate Concentration.
Discussion
Secretion systems, and the bacterial effector proteins these systems transport into the cytoplasm of host cells, are an important virulence component that many pathogenic bacteria utilize to establish an infection within unique replicative niches. The focus of many research groups has been to investigate the interplay between bacterial effectors and host proteins and the influence these effectors have on host cellular pathways. Very limited research, if any, has examined the potential for host proteins to be necessary for successful bacterial effector translocation.
In this study we used the obligate, intracellular pathogen Coxiella burnetii, the causative agent of zoonotic Q fever. After entry into host cells, the Coxiella-containing vacuole passively traffics through the canonical endocytic pathway until reaching an acidic lysosome-derived vacuole, where it replicates to very high numbers. Apart from taking advantage of the normal host trafficking pathway, the ability of Coxiella to establish a successful replicative niche also relies on a functional type IVB secretion system (T4SS) and subsequent effector translocation8,17. This secretion system is functionally analogous to the well-characterized Dot/Icm T4SS of Legionella. This was aptly demonstrated by complementing specific dot/icm mutants of Legionella with their respective Coxiella genes19,20. Although the T4SSs of both Legionella and Coxiella are essential for replication, the timing of when these systems are activated is quite different and reflects the divergent nature of their respective replicative niches. The Legionella T4SS is triggered immediately upon contact with host cells and the rapid translocation of effectors allows the bacteria to avoid the default endosomal-lysosomal pathway and instead create a unique ER-derived replicative vacuole42. In contrast, the T4SS of Coxiella is not activated until approximately 8 hr post-infection, after the bacteria reach the acidic lysosome-derived vacuole21.
Given that Coxiella passively transits through the endocytic pathway and the translocation system is not activated until it reaches the lysosome, a unique opportunity presents itself to use Coxiella as a tool to study endocytic maturation. The requirement for the CCV to become acidified before effectors are translocated indicates that a number of host proteins are important for the translocation of effector proteins into the host cytoplasm, in particular, but not limited to, proteins involved in the endocytic trafficking pathway. Utilizing siRNA in a specific targeted manner we can begin to elucidate the role of particular host proteins on effector translocation. In this study we focused on two proteins that regulate the eukaryotic endocytic trafficking pathway, and hence were predicted to effect translocation of the Coxiella effector CBU0077. Rab5A and Rab7A control membrane fusion with early and late endosomes, respectively. Silencing either of these proteins using siRNA resulted in a significant decrease in translocation efficiency of CBU0077 when compared to the control OTP siRNA (Figure 5B). The representative results shown here reflect our previous published findings21 and provide further validation to the importance of these human Rab proteins to Coxiella effector translocation. This technique has also been employed to characterize the impact of other host processes on Coxiella Dot/Icm effector translocation. The retromer complex was found to be important for intracellular replication of Coxiella. By silencing retromer components, using siRNA, and then conducting a translocation assay we could show that retromer is required to create the environment that induces Coxiella effector translocation 43.
Although the representative results shown here is a comparison between infected OTP control and siRNA targeting Rab5A and Rab7A, the protocol can be modified according to experimental needs. For example, the level of effector translocation can also be compared to cells transfected with scrambled siRNA or non-transfected cells.
The focus of this particular study was the obligate, intracellular pathogen C. burnetii and proteins involved in endocytic trafficking, however, the methods described within can be easily adapted to other intracellular and extracellular pathogens that utilize secretion systems for virulence. Indeed, the fluorescent-based translocation assay that relies on β-lactamase activity within the host cytoplasm used here, has already been extensively used across a range of pathogens30-32,35,44. Naturally, the infection conditions of the specific cell lines used must be adapted to reflect the infection requirements of specific bacteria. Additionally, a known endogenous effector fused to β-lactamase is paramount to the success of the methods described within. One important consideration for successful implementation of the protocol explained here is the impact silencing a particular host target could have on bacterial entry and subsequent replication. Here, effector translocation by Coxiella is measured 24 hr post infection. In this instance, and for other intracellular bacteria, the ability of the organism to gain entry into host cells is vital. Additionally, specific gene silencing could also influence bacterial replication therefore altering the level of translocation observed. Confirmation that siRNA silencing does not significantly impact bacterial entry and/or replication, and consequently translocation efficiency, could be achieved using either microscopy or numeration of bacteria. The translocation data can then be normalized for the number of infected cells per siRNA treatment. Bacterial replication has not confounded the data presented here as Coxiella undergoes little to no replication during the 24 hr incubation period.
This method requires efficient transfection of siRNA into host cells to ensure sufficient silencing of the gene of interest. With this in mind, choosing the correct transfection reagent is extremely important. Apart from the reagent and conditions used in this particular study, which has been optimized for HeLa 229 cells, there are numerous other reagents to select from. The knockdown efficiency using different transfection reagents can be quantified using RT-qPCR to ensure that both the most appropriate reagent is selected and that the siRNA chosen specifically targets the transcript of interest. Although not presented in this study, we have previously demonstrated the knockdown efficiency of both Rab5A and Rab7A using RT-qPCR21. Additionally, the use of antibodies against the target of interest and confirmation by western blot can also confirm siRNA silencing of specific targets.
Two other important considerations to take note of when using siRNA include the possibility of off-target effects and the cell viability of transfected cells. Off-target effects occur when unintended targets are also destroyed. This can lead to phenotypic changes and might result in false positives. In this study we utilized a pool of four siRNA duplexes to silence a single host target. If silencing a particular target causes a reduction in translocation efficiency, validation that this result is not due to off-target effects can be achieved by separating the four siRNA duplexes and repeating the translocation assay. At least two out of four siRNA duplexes should demonstrate a similar phenotype to the original siRNA pool. With this result, one can be highly confident that the observed translocation efficiency is not due to off-target effects. The validity of the translocation results produced is also dependent on cell viability. The application of a nuclei stain after the translocation assay enables enumeration of cell viability following siRNA silencing. In the case of PLK1, significant cell death can be easily observed without staining; however, using epifluorescence microscopy a more precise representation can be achieved. If by silencing a particular host target significant cell death is observed, for example 50 % compared to the control, it is unlikely that an accurate picture can be inferred as to the importance of that particular host protein to the translocation of effectors. As shown in this study, although silencing either Rab5A or Rab7A has a slight impact on cell viability (Figure 5C), this reduction is not pronounced enough to negate the validity of the translocation data observed. Another consideration with regards to cell viability following siRNA silencing is the observation that significant cell death often leads to an increase in the translocation ratio. This is aptly demonstrated by the translocation data reported for PLK1 (Figure 5A). The reduced cell number results in a proportional increase in the multiplicity of infection. This will increase the infection rate and hence the translocation ratio.
Although not presented in this study, visualization of effector translocation can also yield a complimentary quantitative result. Using an epifluorescent microscope fitted with a long-pass dichroic mirror to separate excitation and emission light can provide a visual observation of intracellular BlaM substrate fluorescence. A possible alternative to the β-lactamase reporter system is to utilize the adenylate-cyclase reporter system. Similar to the reporter system described here, the gene of interest is fused to a calmodulin-dependent adenylate cyclase gene (cyaA) derived from Bordetella pertussis. Effector translocation can be measured by quantification of intracellular cyclic AMP (cAMP) using an immunoassay ELISA kit. Since the activity of CyaA is entirely dependent on host cell calmodulin, which is only present in the host cytosol, effector translocation is confirmed by an increase in cAMP levels. Although this method has also been used successfully to measure effector translocation for a variety of bacteria45-47, quantification of intracellular cAMP relies on the lysis of host cells to extract cAMP, a process that is more timeconsuming compared to the method described within. Measurement of effector translocation using the FRET-based β-lactamase reporter system is more efficient since the BlaM substrate can be added directly to the cells and fluorescence can be measured within hours. Additionally, the method described here can be more easily adapted to generate high-throughput results, especially in a 96-well format.
Many bacterial pathogens depend on protein translocation systems to introduce effector proteins into host cells allowing modulation of host cell functions to benefit the pathogen. Type IV secretion systems are used by intracellular bacteria such as Legionella, Coxiella and Brucella and Type III secretion systems are utilized by a range of pathogens including Chlamydia, attaching and effacing E. coli and Salmonella. By comparing the effect of silencing expression of specific host proteins on effector translocation by a range of pathogens, an understanding of host factors necessary for effector translocation by particular types of secretion systems or specific to an individual pathogen could be elucidated. This provides a unique approach to study the intricacies of host-pathogen interactions.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by National Health and Medical Research Council (NHMRC) grants (Grant ID 1062383 and 1063646) awarded to HJN. EAL is supported by an Australian Postgraduate Award.
References
- Delsing CE, Warris A, Bleeker-Rovers CP. Q fever: still more queries than answers. Adv Exp Med Biol. 2011;719:133–143. doi: 10.1007/978-1-4614-0204-6_12. [DOI] [PubMed] [Google Scholar]
- Delsing CE, Kullberg BJ, Bleeker-Rovers CP. Q fever in the Netherlands from 2007 to. Neth J Med. 2010;68:382–387. [PubMed] [Google Scholar]
- van der Hoek W, et al. Epidemic Q fever in humans in the Netherlands. Adv Exp Med Biol. 2012;984:329–364. doi: 10.1007/978-94-007-4315-1_17. [DOI] [PubMed] [Google Scholar]
- Madariaga MG, Rezai K, Trenholme GM, Weinstein RA. Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3:709–721. doi: 10.1016/s1473-3099(03)00804-1. [DOI] [PubMed] [Google Scholar]
- Hoover TA, Culp DW, Vodkin MH, Williams JC, Thompson HA. Chromosomal DNA deletions explain phenotypic characteristics of two antigenic variants, phase II and RSA 514 (crazy), of the Coxiella burnetii nine mile strain. Infect Immun. 2002;70:6726–6733. doi: 10.1128/IAI.70.12.6726-6733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, et al. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol. 2011;77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio. 2011;2:e00175–e00111. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, et al. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A. 2010;107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, et al. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J Bacteriol. 2011;193:1493–1503. doi: 10.1128/JB.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Sandoz KM, Omsland A, Rockey DD, Heinzen RA. Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front Microbiol. 2011;2:97. doi: 10.3389/fmicb.2011.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Larson CL, Gilk SD, Heinzen RA. Two systems for targeted gene deletion in Coxiella burnetii. Appl Environ Microbiol. 2012;78:4580–4589. doi: 10.1128/AEM.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun. 2010;78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol. 2007;9:891–909. doi: 10.1111/j.1462-5822.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- Beare PA. Genetic manipulation of Coxiella burnetii. Adv Exp Med Biol. 2012;984:249–271. doi: 10.1007/978-94-007-4315-1_13. [DOI] [PubMed] [Google Scholar]
- Carey KL, Newton HJ, Luhrmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 2011;7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal G, Shuman HA. Possible origin of the Legionella pneumophila virulence genes and their relation to Coxiella burnetii. Mol Microbiol. 1999;33:669–670. doi: 10.1046/j.1365-2958.1999.01511.x. [DOI] [PubMed] [Google Scholar]
- Zamboni DS, McGrath S, Rabinovitch M, Roy CR. Coxiella burnetii express type IV secretion system proteins that function similarly to components of the Legionella pneumophila Dot/Icm system. Mol Microbiol. 2003;49:965–976. doi: 10.1046/j.1365-2958.2003.03626.x. [DOI] [PubMed] [Google Scholar]
- Zusman T, Yerushalmi G, Segal G. Functional similarities between the icm/dot pathogenesis systems of Coxiella'burnetii and Legionella pneumophila. Infect Immun. 2003;71:3714–3723. doi: 10.1128/IAI.71.7.3714-3723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton HJ, McDonough JA, Roy CR. Effector Protein Translocation by the Coxiella burnetii Dot/Icm Type IV Secretion System Requires Endocytic Maturation of the Pathogen-Occupied Vacuole. PLoS One. 2013;8:e54566. doi: 10.1371/journal.pone.0054566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz Z, et al. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc Natl Acad Sci U S A. 2013;110:E707–E715. doi: 10.1073/pnas.1215278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, et al. The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J Bacteriol. 2009;191:4232–4242. doi: 10.1128/JB.01656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, et al. Identification of C. burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol. 2013. [DOI] [PMC free article] [PubMed]
- Klingenbeck L, Eckart RA, Berens C, Luhrmann A. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol. 2013;15(4):675–678. doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- Larson CL, Beare PA, Howe D, Heinzen RA. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci U S A. 2013;110:E4770–E4779. doi: 10.1073/pnas.1309195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrmann A, Nogueira CV, Carey KL, Roy CR. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci U S A. 2010;107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton HJ, et al. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Luhrmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. 2008;320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier X, Oswald E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J Bacteriol. 2004;186:5486–5495. doi: 10.1128/JB.186.16.5486-5495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe KS, et al. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 2008;4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong MF, Sun YH, den Hartigh AB, van Dijl JM, Tsolis RM. Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol Microbiol. 2008;70:1378–1396. doi: 10.1111/j.1365-2958.2008.06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffatellu M, et al. Host restriction of Salmonella enterica serotype Typhi is not caused by functional alteration of SipA, SopB, or SopD. Infect Immun. 2005;73:7817–7826. doi: 10.1128/IAI.73.12.7817-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RE, Newton P, Latomanski EA, Newton HJ. Dot/Icm Effector Translocation by Legionella longbeachae Creates a Replicative Vacuole Similar to That of Legionella pneumophila despite Translocation of Distinct Effector Repertoires. Infect Immun. 2015;83:4081–4092. doi: 10.1128/IAI.00461-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller KE, Fields KA. Application of beta-lactamase reporter fusions as an indicator of effector protein secretion during infections with the obligate intracellular pathogen Chlamydia trachomatis. PLoS One. 2015;10 doi: 10.1371/journal.pone.0135295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N, et al. RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev. 2003;67:657–685. doi: 10.1128/MMBR.67.4.657-685.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Lam JK, Chow MY, Zhang Y, Leung SW. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids. 2015;4:e252. doi: 10.1038/mtna.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E, Cantet F, Fava L, Norville I, Bonazzi M. Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog. 2014;10:e1004013. doi: 10.1371/journal.ppat.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaton K, Peter O, Raoult D, Tissot JD, Greub G. Development of a high throughput PCR to detect Coxiella burnetii and its application in a diagnostic laboratory over a 7-year period. New Microbes New Infect. 2013;1:6–12. doi: 10.1002/2052-2975.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prashar A, Terebiznik MR. Legionella pneumophila: homeward bound away from the phagosome. Curr Opin Microbiol. 2014;23C:86–93. doi: 10.1016/j.mib.2014.11.008. [DOI] [PubMed] [Google Scholar]
- McDonough JA, et al. Host Pathways Important for Coxiella burnetii Infection Revealed by Genome-Wide RNA Interference Screening. MBio. 2013;4(1):e00606–e00612. doi: 10.1128/mBio.00606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfan MJ, Toro CS, Barry EM, Nataro JP. Shigella enterotoxin-2 is a type III effector that participates in Shigella-induced interleukin 8 secretion by epithelial cells. FEMS Immunol Med Microbiol. 2011;61:332–339. doi: 10.1111/j.1574-695X.2011.00778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodor S, Price CT, Habyarimana F, Kalia A, Abu Kwaik Y. A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Mol Microbiol. 2008;70:908–923. doi: 10.1111/j.1365-2958.2008.06453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes K, Worley M, Niemann G, Heffron F. Identification of new secreted effectors in Salmonella enterica serovar Typhimurium. Infect Immun. 2005;73:6260–6271. doi: 10.1128/IAI.73.10.6260-6271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sory MP, Boland A, Lambermont I, Cornelis GR. Identification of the YopE and YopH domains required for secretion and internalization into the cytosol of macrophages, using the cyaA gene fusion approach. Proc Natl Acad Sci U S A. 1995;92:11998–12002. doi: 10.1073/pnas.92.26.11998. [DOI] [PMC free article] [PubMed] [Google Scholar]