

Abstract
Human brain is a high energy consuming organ that mainly relies on glucose as a fuel source. Glucose is catabolized by brain mitochondria via glycolysis, tri-carboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) pathways to produce cellular energy in the form of adenosine triphosphate (ATP). Impairment of mitochondrial ATP production causes mitochondrial disorders, which present clinically with prominent neurological and myopathic symptoms. Mitochondrial defects are also present in neurodevelopmental disorders (e.g. autism spectrum disorder) and neurodegenerative disorders (e.g. amyotrophic lateral sclerosis, Alzheimer's and Parkinson's diseases). Thus, there is an increased interest in the field for performing 3D analysis of mitochondrial morphology, structure and distribution under both healthy and disease states. The brain mitochondrial morphology is extremely diverse, with some mitochondria especially those in the synaptic region being in the range of <200 nm diameter, which is below the resolution limit of traditional light microscopy. Expressing a mitochondrially-targeted green fluorescent protein (GFP) in the brain significantly enhances the organellar detection by confocal microscopy. However, it does not overcome the constraints on the sensitivity of detection of relatively small sized mitochondria without oversaturating the images of large sized mitochondria. While serial transmission electron microscopy has been successfully used to characterize mitochondria at the neuronal synapse, this technique is extremely time-consuming especially when comparing multiple samples. The serial block-face scanning electron microscopy (SBFSEM) technique involves an automated process of sectioning, imaging blocks of tissue and data acquisition. Here, we provide a protocol to perform SBFSEM of a defined region from rodent brain to rapidly reconstruct and visualize mitochondrial morphology. This technique could also be used to provide accurate information on mitochondrial number, volume, size and distribution in a defined brain region. Since the obtained image resolution is high (typically under 10 nm) any gross mitochondrial morphological defects may also be detected.
Keywords: Neuroscience, Issue 113, SBFSEM, mitochondria, OXPHOS, brain, synapse, 3D reconstruction
Introduction
Mitochondria are dynamic organelles which change their shape and location depending on the cellular cues and needs, in tight interaction with cell cytoskeleton, and in response to cellular events such as calcium currents in neurons 1. Mitochondria also interact with other cellular organelles e.g. endoplasmic reticulum, which in turn regulates their dynamics and metabolism2. Mitochondrial morphology shows heterogeneity in different cell types i.e. the shape of the organelle varies from tubular to that consisting of sheets, sacks and ovals 3. It has been shown that mitochondrial fusion and fission cycle proteins can regulate the location, size, shape and distribution of mitochondria 4. Moreover, changes in mitochondrial shape are associated with neurodegeneration, neuronal plasticity, muscle atrophy, calcium signaling, reactive oxygen species generation as well as lifespan and cell death implicating that cell-specific mitochondrial morphology is critical for the maintenance of normal cellular function 5-11.
A major bioenergetic function of mitochondria is to generate adenosine triphosphate (ATP) by executing a series of metabolic reactions that involve complete breakdown of nutrients (i.e. glucose, fatty-acids or amino-acids) via the TCA cycle and OXPHOS pathways 12. The human brain constitutes only 2% of body weight however it consumes ~20% of total energy produced making it an extremely energy demanding organ 13. It is therefore not surprising that mitochondrial dysfunction in humans leads to a large number of neurological manifestations 14-17. Genetic mutations in OXPHOS components that impair ATP generation leads to mitochondrial disorders 17,18, which are clinically heterogeneous group of disorders with a prevalence of ~1 : 5,000 individuals, and one of the most common cause of metabolic disorders in children and adults. Deficit of mitochondria-derived ATP affects multiple organ systems with high energy demanding organs such as brain, heart and skeletal muscles being predominantly affected in these patients 14,17,18. In recent years, multiple studies have provided evidence for mitochondrial dysfunction in both neurodevelopmental and neurodegenerative disorders 15-17,19,20. Since mitochondria are essential and critical for brain development and function, it is imperative to develop protocols that can analyze changes in brain mitochondrial morphology, structure, size, number and distribution under both healthy and diseased states. Mouse models with mitochondrially-targeted green fluorescent protein (GFP) have been produced to visualize mitochondrial movements and localization in the brain 21,22. While this is an extremely useful tool to examine mitochondrial motility and general distribution, there are some drawbacks which include limited resolution and sensitivity of fluorescence microscopy. These attributes make it difficult to track the relatively small sized mitochondria. Similarly, serial transmission electron microscopy has been successfully used to view synaptic mitochondria 23, but this method is very time consuming. Mitochondrial morphology is known to be highly dynamic as they undergo continuous fission and fusion cycles, and in most cells mitochondria maintain a highly connected network 24-26. Neurons are highly polarized cells with multiple dendrites and extended axons, and mitochondria that form a connected reticular network in the cell body may have to separate as they make their way through these neurites (Figure 1). This makes brain mitochondria extremely varied in size and shape. For example, using serial block-face scanning electron microscopy (SBFSEM) technique, we previously observed that the difference in the volume or size of extrasynaptic mitochondria to mitochondria present in the nerve terminals may be as much as sixteen fold 27.
There are several approaches for performing volume analyses 28, which includes serial section TEM 29, automated tape collecting ultramicrotome SEM 30, focused ion beam SEM 31, and SBFSEM 32. The SBFSEM analysis has advantages in that it has the resolution to provide quantitative data on the morphological shape, size, distribution and number of organelles such as mitochondria in areas up to 1 mm of the brain. The technical operation is also the least demanding, with data acquisition and analysis within capabilities of many biological labs that lack previous EM experience. The advent of commercial instruments for generating serial section-like images has made 3D ultrastructural analysis of tissues a routine technique, which further permits an unbiased volumetric analysis in a rapid and repeatable manner28. The SBFSEM was first described and used in the field of neurobiology in 2004 32, based on an idea introduced by Leighton in 1981 33. Multiple studies since then have established this technique as a major tool in reconstruction analysis of neuronal circuitry 34. Furthermore, for many smaller scale projects, it provides reconstruction analysis to identify cellular organelles 27,35-39. Since, the acquired images are derived from low voltage back scatter electrons, new staining protocols which combine different known heavy metal staining techniques were developed to increase the resolution 40.
In this paper, we provide a protocol for utilizing 3D electron microscopy imaging and volumetric analysis of brain mitochondria based on methods that have previously been used by us and others 38,39,41. The tissue post-processing methods used were as previously described by Deerinck et al40.
Protocol
Ethics Statement: Procedures involving animal subjects have been approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Tech.
Caution: Extreme precautions must be taken when handling and disposing several components used in this protocol. Before use, the local institutional guidelines and health and safety practices must be established and followed, particularly for osmium tetroxide, which is volatile and extremely poisonous, uranyl acetate, which is both a heavy metal and source of radioactivity, and lead nitrate, which is a heavy metal poison. Thiocarbohydrazide (TCH) can decompose to produce explosive and poisonous gases, if incorrectly handled. Many institutions will have an EM core facility in which these reagents are routinely utilized and can provide assistance.
1. Preparation of Brain Tissue and SBFSEM Imaging
Anesthetize a young (~2 - 4 months old) C57 black mouse (C57BL/6J strain) using 4 - 5% isoflurane following the institutional guidelines. Confirm anesthetization by monitoring the loss of muscle tone, lack of voluntary movements and responses to aversive stimuli like a tail pinch.
Pin the mouse on a dissection tray and make an incision on the skin along ventral midline. Make further incisions in the skin to expose the rib cage of the mouse. Incise the diaphragm, and carefully dissect out the chest cavity along the periphery to expose the beating heart, and then make an incision on the right atrium.
Using a butterfly cannula, cannulate the left ventricle. Perfuse the mouse transcardially using 10 - 20 ml of phosphate buffer saline (PBS) pH 7.2, until exsanguination is confirmed by a change in the color of the liver. Note: A change in the color of liver is used as a guide to determine the extent of exsanguination. Confirm that the liver color changes from reddish brown to pale pink.
Perfuse the mouse transcardially using 10 - 20 ml of 2% glutaraldehyde and 4% paraformaldehyde made in 0.10 M cacodylate buffer (pH 7.2), to fix the brain tissue rapidly from within. Fixation is monitored by observing the tail stiffening. Note: Prepare 0.10 M cacodylate buffer (pH 7.2) by dissolving 2.14 g of sodium cacodylate in 80 ml water, add hydrochloric acid to adjust the pH, and make the volume to 100 ml with water.
Following perfusion, decapitate the mouse, dissect the brain, followed by fixation in 0.10 M sodium cacodylate buffer (pH 7.2) containing 2% glutaraldehyde and 4% paraformaldehyde for 48 hr at 4 oC.
After 48 hr make 400 µm coronal sections using a vibratome and then carefully dissect the region of interest in the brain (for example, the hippocampus) under the dissection microscope 42. Carefully trim the tissue and take a picture for preserving orientation. Note: Post processing methods of tissue staining in SBFSEM combines a variety of heavy metal staining methods in order to improve resolution and are based on the method developed previously by Deerinck et al40.
Wash the glutaraldehyde-fixed tissues 3 times, 5 min each in 0.1 M cacodylate buffer (pH 7.2).
Prepare 0.1% tannic acid solution by dissolving tannic acid in 0.1 M sodium cacodylate buffer (pH 7.2). Swirl until dissolved and filter through 0.45 µm filter, if necessary.
Postfix the tissues with cacodylate buffered 0.1% tannic acid by incubating in 1 ml reagent for 30 - 60 min at RT. Note: The incubation time is dependent on tissue size, but 30 min works best for most tissues.
Wash tissues 3 times, 5 min each in cacodylate buffer (pH 7.2).
Dissolve 0.3 g potassium ferrocyanide and 0.86 g sodium cacodylate in 10 ml distilled H2O (dH2O). Keep the potassium ferrocyanide solution on ice. Just before use, add 10 ml of 4% osmium tetroxide (OsO4).
Stain the tissues with 2% osmium-ferrocyanide solution for 90 min, on ice. Wash 3 times, 5 min each in dH2O.
Prepare 1% thiocarbohydrazide (TCH) solution by dissolving 0.1 g TCH in 10 ml dH2O. Dissolve at 60 °C by swirling every 10 min until fully dissolved. Observe safety precautions while handling TCH, particularly take care to avoid use of metals, heating to high temperature, or allowing solution to dry out.
Treat the samples with freshly prepared 1% TCH for 20 min at RT. Wash 3 times, 5 min each in dH2O.
Dilute 4% OsO4 to 2% with dH2O, stain tissues with 2% aqueous osmium tetroxide by incubating for 1 hr. Wash tissues 3 times, 5 min each with dH2O.
Incubate the samples O/N in 1% uranyl acetate in dH2O at 4 °C.
- Prepare Walton's lead aspartate solution (as described by Deerinck et al40).
- Dissolve 0.998 g L-Aspartate in 250 ml dH2O and then add 10 N potassium hydroxide (KOH) in a dropwise fashion until the pH reaches 5.5. After pH adjustment, add 0.066 g of lead nitrate in 10 ml aspartic acid stock and heat to 60 °C for 30 min.
Rinse tissues in dH2O and incubate with Walton's lead aspartate stain for 30 min in a 60 °C oven. Wash 3 times, 5 min each in dH2O.
Dehydrate samples through a graded series of alcohol using chilled solutions of 20%, 50%, 75%, 85% and 95% ethanol for 5 min each, followed by 100% ethanol 3 times, 10 min each. Note: Use 100% ethanol from freshly opened bottle, as opened ethanol absorbs water from the air and cause embedding to fail. Longer incubations will be necessary with larger tissue samples.
Wash samples 2 times, 15 min each in propylene oxide.
Make plastic embedding resin using 25 ml resin, 10.5 ml DDSA (dodecenyl succinic anhydride), 15.5 ml NMA (nadic methyl anhydride) and 1 ml DMP-30 (2,4,6-Tris dimethylaminomethyl phenol). Mix the resin by shaking. Spin and allow the resin to stand until bubbles resolve. Note: This is the standard medium hardness recipe. Other types of electron microscopy (EM) plastic resin may be used, but should be tested with a non-essential control sample in advance as not all resins work for SBFSEM.
Incubate the tissues O/N in a 50 : 50 mix of embedding resin and propylene oxide, in a vial that is capped initially, then uncapped after 2 hr so that the propylene oxide evaporates over the period of about 8 - 10 hr.
Transfer the tissues to 100% fresh embedding resin in clean vials for 2 hr.
Embed samples in fresh embedding resin, in flat molds containing printed paper labels, and cure them in an oven at 60 °C for 48 hr. After about 1 hr, check the tissue placement and alignment again, and adjust if necessary.
Trim samples to the area of interest and mount on an aluminum pin using gelling cyanoacrylate superglue or a conductive epoxy resin, then coat with colloidal silver paste around the sides of the block to provide a conductive path to the aluminum pin.
Examine tissue specimens using a scanning electron microscope system equipped with an in-chamber ultramicrotome stage and low kV backscattered electron detector 32. Note: Obtain instrument- and site-training in scanning electron microscopy (SEM) use and become an authorized user. Alternatively, collaboration with a researcher or core facility for small projects may be possible. Radiation training may also be required as SEMs generate x-rays.
To image the samples, use the following settings: 2.25 kV, at 5 - 10 nm/pixel resolution, with field sizes between 80 - 250 µm in x,y (other field sizes possible), and slice thickness of 50 - 100 nm, with a total of 250 - 600 slices in a 16 - 20 hr time period. Note: Settings vary substantially between different microscopes, individual samples, and desired resolution. These settings should produce images that are readily interpretable for many samples.
2. Analyzing the Imaging Dataset
Note: The Image J/Fiji software is used to analyze the dataset and relies upon the TrakEM2 plugin. Preprocessing steps may be performed using a variety of software, and may be extensive or minor depending on experience level and the stacks obtained. The main transformations using the open-source software (ImageJ ver 1.50b, FIJI download Oct 1, 2015) are described here.
- Convert images to 8 bit tiff format from the original proprietary 16-bit images by opening in the software and selecting menu items Image→Type→8 bit.
- If automatic contrast/brightness conversion during this step is not ideal for images, reopen the 16-bit images, and press Image→Adjust→Brightness/Contrast. Select a range that works for all images, and press Apply. Then perform conversion. Note: On some SEMs, this step may require microscope manufacturer software.
- If required, due to unacceptable image movement between slices (e.g. drift due to charging), register/align the image stacks (menu items Plugins→Registration→Linear StackAlignmentWithSIFT). In most registration software, set for "translation-only" mode rather than "rigid body". Note: Many approaches and software may work: the SIFT registration plug-in works for many applications and there is a virtual stack version.
- If required, enlarge the canvas size prior to registration (Image→Adjust→CanvasSize) or reduce it to an area of interest (Image→Crop). Note: Some drift or splaying may occur, and trying other plugins or software may produce better results. Manual options are also available (e.g. ImageJ/FIJI, Plugins→Registration→ManualLandmarkSelection).
- If desired, scale images to smaller, more manageable size (e.g. 25%) using ImageJ (Image→Scale).
Launch the software, select File→import→image sequence and then select the tiff files.
Launch the plugin by selecting File→new→TrakEM2 (Blank). Two windows will open; one manages the project and area_lists and the other manages tracing, and is referred to as the 'canvas'.
Load the image stack into the plugin by making a right-click on the canvas import. Select import and click on import stack. In the pop-up options, check the box for virtual stacks. Note: Although the image stacks are already opened in the software, they must also be opened in the plugin. The computer may run more smoothly if the virtual stacks box is checked.
After the mipmaps are created and the stack is loaded, right click to name the project under the plugin window. Right click on 'new project' in the template column, and select 'add new child' for that project. Right click again to select 'area_list'.
Set the Z-axis scale of the project to agree with the original electron micrograph settings by right clicking on the canvas, then click display and select calibration. Also, set the Z-scale by selecting all layers in the plugin window, right click to select scale Z and thickness.
Click and drag the 'project' and all 'children' into the project objects section to create the 'area lists' under the 'Z space' tab in the canvas window.
Select the 'area list' and right click to select and set a color. Now, use the paint brush tool to trace objects such as mitochondria, in the electron micrograph images. Use 'Shift + click' to fill in an enclosed circle, 'Ctrl + scroll' to zoom in and out, and 'Alt + click' to turn paint brush cursor into an eraser.
Select two areas of ~10 - 15 µm by 10 - 15 µm at the top left corner and bottom right corner of the image and identify all mitochondria within these areas to allow unbiased sampling of mitochondria in each dataset.
On the 'canvas', observe and trace mitochondria throughout the sections. Note: Mitochondria are quite dark/dense appearing organelles, of similar size in diameter and loosely cylindrical. The unique cristae formed by the inner membrane are easily distinguishable inside this organelle.
- When finished with tracing, right click on the area_list under the Z space tab, select 'Show in 3D'. This will launch the 3D viewer plugin to view a 3D reconstruction of the traced image.
- To perform mitochondrial volume measurements, select object in 3D viewer, click on 'Edit' tab and select 'Object Properties'. The mitochondrial volume estimate is listed along with several other measurements.
Representative Results
We demonstrate that the brain mitochondrial morphology and size is heterogeneous in different neuronal sub-compartments. Confocal microscopy on low density neuronal cultures transduced with lentivirus expressing mitochondrially-targeted green fluorescent protein showed that mitochondria residing in neuronal soma form a reticular network, whereas those residing in distal neurites exhibit a discrete elongated morphology (Figure 1 A-B). Using the SBFSEM technique, the mitochondrial morphology, size, volume and distribution were analyzed in the mouse brain. Three dimensional (3D) images of mitochondria were reconstructed from both neurites and synapses in the mouse brain hippocampus. The presynaptic mitochondria were identified and the number of presynaptic boutons harboring resident mitochondria quantified. Heterogeneity in the size of mitochondria was observed in the dendritic versus axonal compartments (Figure 1 C-D). Moreover, many small presynaptic compartments in the mouse brain hippocampus were devoid of mitochondria (Figure 2 A-C). The volumetric measurements of both presynaptic and extrasynaptic mitochondria revealed that the volume or size of mitochondria residing within the neuronal presynapses was significantly smaller than those residing in the extrasynaptic region (Figure 2 D-F). Interestingly, the two dimensional (2D) size distribution of mitochondria measured from an unbiased sampling of 200 non-somatic mitochondria showed that ~11% of mitochondrial fraction lies below the resolution limit of light microscopy (Figure 2G). In addition, the 2D images acquired by SBFSEM analysis provide increased resolution to visualize the ultrastructural characteristics of individual mitochondria (Figure 3 A-B). Due to the clearly visible topography of the tissue, it is further possible to classify the cellular compartment of mitochondria by determining its proximity to readily identifiable structures like nucleus (somatic) and synaptic vesicles (presynaptic) (Figure 3 A-B). In order to identify mitochondria present in the neuronal processes, reconstruct the individual process as axon or dendrite based on the presence or absence of presynaptic boutons respectively 42. Based on these results, we propose that SBFSEM is an extremely valuable analytical technique to identify mitochondrial shape, size, number and distribution in brain tissue and that any gross defects in mitochondrial structure and distribution can also be determined using this method. Since the ultrastructural characteristics of different cell types in the brain are distinct, for e.g. presence of glycogen granules in astrocytes, it is also possible to analyze the cell-type-specific differences in brain mitochondria.
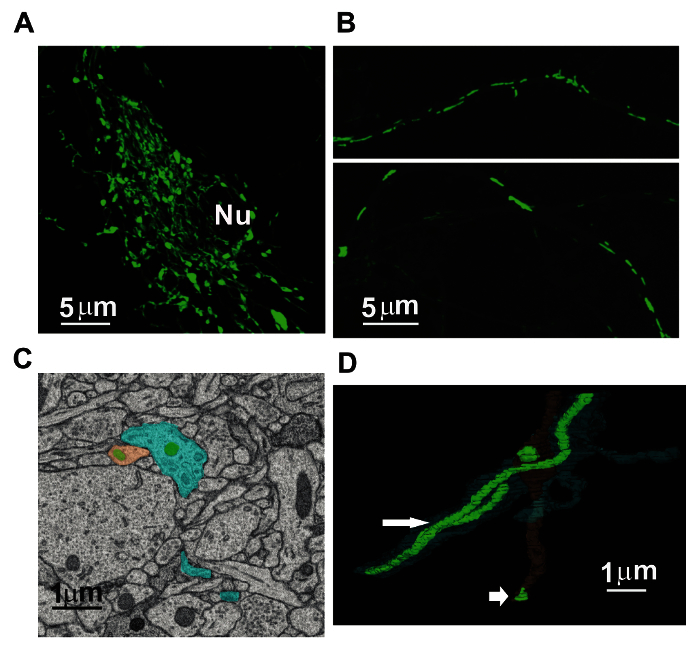 Figure 1:Heterogeneity in Neuronal Mitochondrial Morphology and Distribution. Cortical cultures from postnatal day 1 mouse pups were transduced with lentivirus expressing mitochondrially-targeted green fluorescent protein (mito-GFP). (A) Shows neuronal soma expressing mito-GFP, note the reticulate network of mitochondrial morphology; Nu = nucleus; scale bar = 5 µm. (B) Shows elongated mitochondrial morphology in distal neurites; scale bar = 5 µm. (C) Representative 2D image from the SBFSEM dataset generated from the mouse brain tissue, the mitochondria are stained in green, dendrites are stained in blue and axonal varicosities are stained in brown; scale bar = 1 µm. (D) 3D reconstructions of mitochondria in the neurites of mouse brain tissue, note the difference in size between dendritic and axonal mitochondria indicated by large and small arrowhead respectively; scale bar = 1 µm. Please click here to view a larger version of this figure.
Figure 1:Heterogeneity in Neuronal Mitochondrial Morphology and Distribution. Cortical cultures from postnatal day 1 mouse pups were transduced with lentivirus expressing mitochondrially-targeted green fluorescent protein (mito-GFP). (A) Shows neuronal soma expressing mito-GFP, note the reticulate network of mitochondrial morphology; Nu = nucleus; scale bar = 5 µm. (B) Shows elongated mitochondrial morphology in distal neurites; scale bar = 5 µm. (C) Representative 2D image from the SBFSEM dataset generated from the mouse brain tissue, the mitochondria are stained in green, dendrites are stained in blue and axonal varicosities are stained in brown; scale bar = 1 µm. (D) 3D reconstructions of mitochondria in the neurites of mouse brain tissue, note the difference in size between dendritic and axonal mitochondria indicated by large and small arrowhead respectively; scale bar = 1 µm. Please click here to view a larger version of this figure.
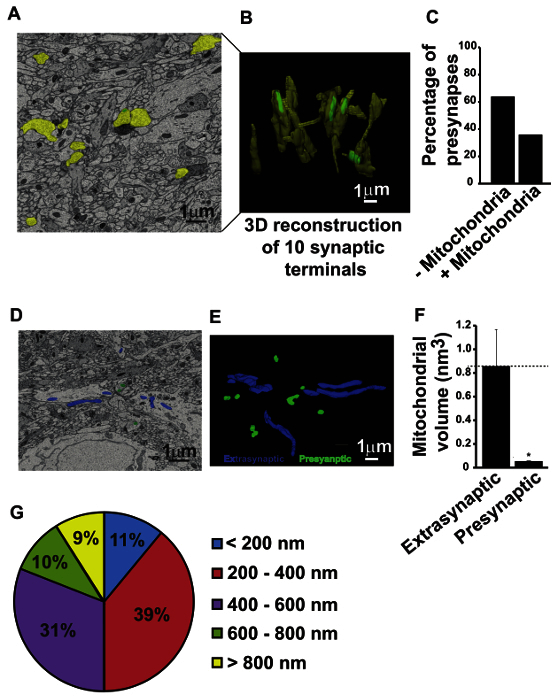 Figure 2:Serial Block-face Scanning Electron Microscopy (SBFSEM) Analysis Revealed Low Abundance of Mitochondria at the Presynaptic Terminals. (A) Representative 2D ultramicrograph from the SBFSEM dataset analysis obtained from the hippocampi of P15 wild-type mice; scale bar = 1 µm. (B) Displays 3D reconstruction of 10 presynaptic nerve terminals. Note that only 4 out of 10 presynaptic terminals showed discernible mitochondria; scale bar = 1 µm. (C) Bar graph showing the quantitation of 173 reconstructed presynaptic nerve terminals from the SBFSEM dataset analysis. (D) Representative 2D image from SBFSEM dataset showing the extrasynaptic mitochondria in blue and presynaptic mitochondria in green; scale bar = 1 µm. (E) 3D reconstructions of the presynaptic and extrasynaptic mitochondria from the SBFSEM dataset using the software; scale bar = 1 µm. (F) Bar graph showing the volume of extrasynaptic and presynaptic mitochondria. Data are plotted as mean ± SEM; n = 3 different datasets (includes 62 presynaptic mitochondria and 80 extrasynaptic mitochondria in total); * depicts p value = 0.0405. (G) A 15 by 15 µm area in all four corners of images were marked and all non-somatic mitochondria were identified to allow random sampling. The least dimension of ~200 mitochondria was measured. Pie graph shows that ~11 % of non-somatic mitochondria were <200 nm in dimension which is below the resolution limit of light microscopy. This figure has been adapted from Chavan et al27. Please click here to view a larger version of this figure.
Figure 2:Serial Block-face Scanning Electron Microscopy (SBFSEM) Analysis Revealed Low Abundance of Mitochondria at the Presynaptic Terminals. (A) Representative 2D ultramicrograph from the SBFSEM dataset analysis obtained from the hippocampi of P15 wild-type mice; scale bar = 1 µm. (B) Displays 3D reconstruction of 10 presynaptic nerve terminals. Note that only 4 out of 10 presynaptic terminals showed discernible mitochondria; scale bar = 1 µm. (C) Bar graph showing the quantitation of 173 reconstructed presynaptic nerve terminals from the SBFSEM dataset analysis. (D) Representative 2D image from SBFSEM dataset showing the extrasynaptic mitochondria in blue and presynaptic mitochondria in green; scale bar = 1 µm. (E) 3D reconstructions of the presynaptic and extrasynaptic mitochondria from the SBFSEM dataset using the software; scale bar = 1 µm. (F) Bar graph showing the volume of extrasynaptic and presynaptic mitochondria. Data are plotted as mean ± SEM; n = 3 different datasets (includes 62 presynaptic mitochondria and 80 extrasynaptic mitochondria in total); * depicts p value = 0.0405. (G) A 15 by 15 µm area in all four corners of images were marked and all non-somatic mitochondria were identified to allow random sampling. The least dimension of ~200 mitochondria was measured. Pie graph shows that ~11 % of non-somatic mitochondria were <200 nm in dimension which is below the resolution limit of light microscopy. This figure has been adapted from Chavan et al27. Please click here to view a larger version of this figure.
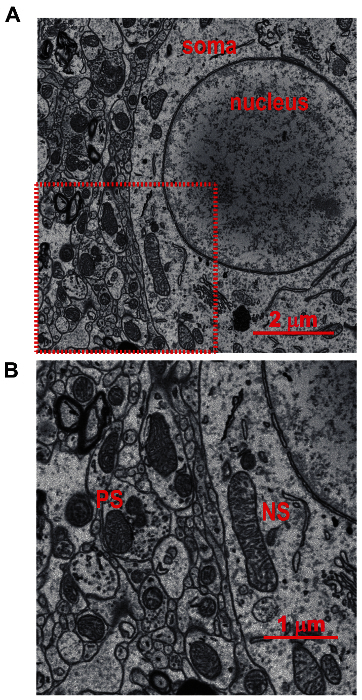 Figure 3:2D SBFSEM Images from the Lateral Geniculate Nuclei of Mouse Brain. (A) Two dimensional representative SBFSEM image from the lateral geniculate nuclei (8/8 µm) demonstrating a detailed topography of the region. Soma and nucleus are indicated; scale bar = 2 µm. (B) A magnification of the area indicated by red square in panel A. Note that the outer and inner membranes of mitochondria as well as cristae formation are clearly visible. The topography and relations to other organelles and cellular structure is also observed. PS indicates example of presynaptic mitochondria and NS indicates example of non-synaptic (NS) somatic mitochondria; scale bar = 1 µm. Please click here to view a larger version of this figure.
Figure 3:2D SBFSEM Images from the Lateral Geniculate Nuclei of Mouse Brain. (A) Two dimensional representative SBFSEM image from the lateral geniculate nuclei (8/8 µm) demonstrating a detailed topography of the region. Soma and nucleus are indicated; scale bar = 2 µm. (B) A magnification of the area indicated by red square in panel A. Note that the outer and inner membranes of mitochondria as well as cristae formation are clearly visible. The topography and relations to other organelles and cellular structure is also observed. PS indicates example of presynaptic mitochondria and NS indicates example of non-synaptic (NS) somatic mitochondria; scale bar = 1 µm. Please click here to view a larger version of this figure.
Discussion
The complexity of the nervous system poses a significant challenge in reconstructing large tissue volumes and analyzing the morphology and distribution of organelles such as mitochondria with adequate resolution. Multiple cells including neurons, oligodendrocytes and astrocytes with numerous processes extended in three dimensions interact within the brain tissue 43. Since mitochondria resides both in the soma of cells and distant processes, mitochondrial morphology is extremely pleomorphic in the nervous system (Figure 1). Adequate 3D structural information with sufficient resolution therefore cannot be acquired by conventional light microscopy techniques such as confocal or two-photon microscopy 44,45. Electron microscopy is the only currently available technique that allows reconstruction of large volumes of neural tissue with sufficiently high resolution. Traditionally, 3D reconstruction of neural tissue has been achieved by serial section transmission electron microscopy (SSTEM) of ultrathin sections 29. However, recent technological advancements have improved the quality of volume electron microscopy data acquisition and automation.
SBFSEM is an automated block-face imaging technique which combines serial sectioning inside the chamber of a scanning electron microscope, in order to reconstruct 3D tissue structure over hundreds of micrometers, with a resolution that is sufficient to follow the thinnest cellular processes and identify small organelles. This technique has opened up the prospect of routinely reconstructing both invertebrate and vertebrate nervous systems. It involves multiple steps including sample preparation, scanning electron microscope operation, data acquisition, image post-processing, and image analysis. These procedures require specialized hands-on training and certification for scanning electron microscope operation. Three factors are critical i.e. dissecting the correct brain anatomical region, obtaining high resolution images, and performing proper image analysis. After dissecting the anatomical region of interest from the vibratome slice of fixed brain tissue, it is crucial to take the picture for preserving proper orientation. It can otherwise be difficult to ascertain the correct site for imaging. Obtaining high resolution images depends on adequate fixation and proper post-fixing staining procedures. The brain tends to undergo rapid deterioration in its ultrastructure after death. Therefore, it is critical to fix the brain in a glutaraldehyde solution using proper transcardial perfusion method. This should be followed by further fixation of the brain in glutaraldehyde solution for at least 24 hr. Since the image resolution is completely dependent on the combination of a variety of heavy metal staining methods, it is critical to follow the post-fixing method as detailed in the protocol to acquire quantifiable images. Solutions should be made as described in the methods. Finally, for accurate image analysis the organelle/s to be analyzed should be identified unequivocally (e.g. by visualizing ultrastructural characteristics) before tracing, and the software guidelines must be strictly adhered to. The resolution of the acquired images should be known for conversion from pixels to nanometers for volumetric measurements.
Besides the software described in the protocol for performing image analysis, other available software's like Reconstruct and Knossos can also be used. Although the emphasis of the described protocol is on viewing mitochondria in the nervous system, it can be modified to generate high resolution 3D information on various other subcellular structures (e.g. endoplasmic reticulum, lysosomes etc.) in a variety of tissues.
While a detailed troubleshooting guide for scanning electron microscope operation is beyond the scope of this article (see instrument user manuals and training information), a few problems routinely occur during imaging experiments and understanding how to troubleshoot them may help new users or those obtaining images through collaboration/ commercial sources. A common problem is charging of the sample which occurs principally in resin regions that contain little or no stained material. In a "positive" image (i.e. cytosol appears white), the nuclei, blood vessels, and empty resin expanses may appear "blackened". In addition, charging also promotes local beam drift resulting in apparent image warping. Possible solutions vary between instruments and samples. Reducing kV settings reduces the "blackening" artifact but may promote more beam drift. Using a lower vacuum mode, and including nitrogen (N2) gas, water vapor, etc. in the chamber also reduces charging, but at a substantial cost of resolution and signal-to-noise ratio. A second common problem is knife skipping. Slower scanning may cause beam induced damage to the block surface, which cuts unevenly or not at all for some imaging/ cutting cycles (i.e. successive images appears the same). Several solutions exist including increasing the cutting depth/ slice thickness with reduced z-resolution, increasing scan speed and consequentially accepting noisier images, reducing pixel size and accepting lower x/y-resolution, and choosing samples or areas with more intense staining (as poorly-stained areas charge more, damage more easily, and require longer beam exposure to obtain images with acceptable signal-to -noise ratios). Debris redeposition on the block-face is an occasional source of artifact in images, and if this occurs frequently it may require pausing the acquisition, and carefully cleaning the knife (blowing air), or retrimming the sample to a smaller size. Block-face charging also promotes redeposition, and steps above may help. Correction of focus and stigmation may also be required as the microscope images samples continuously for many hours to weeks, during which time the vacuum depth increases in the chamber, requiring stigmation corrections. Each scanning electron microscope differs according to age and characteristics, and experience is the best guide. Sudden dramatic changes in stigmation or focus may occur when sectioned material adheres to the microscope imaging components. The chamber must be opened and material carefully dislodged through vacuum or compressed nitrogen. This absolutely requires specialized training.
There are a few disadvantages of SBFSEM technology with regards to its utility in analyzing brain mitochondria. A major disadvantage, common to all microscopy of fixed tissues, is that it provides static images of an extremely dynamic organelle. Mitochondria undergo continuous fission and fusion cycles 26, are mobile and trafficked along the neurite processes 21. Defects in mitochondrial trafficking and dynamics which are not purely numerical or structural can be easily missed by this approach, although, by looking at the number of mitochondria in an anatomically defined space such as presynapse one can interpret if the transport of mitochondria may be altered 39. A second disadvantage is that it is performed only in a small portion of the brain, therefore circuitry specific defects in mitochondrial distribution may be missed. Correlative approaches 28,46 afford the opportunity to combine the confocal and multiphoton imaging with SBFSEM technology to generate both the molecular and ultrastructural data. For mitochondrial analysis, therefore, it may be useful to perform the SBFSEM after light microscopic examination of brain sections either by using the mitochondrial antigen specific antibody or by using a transgene which expresses mitochondrially-targeted fluorescent protein. This combinational strategy may provide a robust methodology to identify mitochondrial defects in mouse models of neurological and mitochondrial disorders.
There are also technical limitations common to many forms of electron microscopy due to use of harsh fixatives, heavy metal staining and uneven penetration of chemicals. Other limitations may stem from the plastic embedding of tissue samples. Empty regions of resin and sparsely stained structures retain electrons during imaging, causing beam deflection and drift (image warping) as well as artifactual charging signals (e.g. dark nuclei and blood vessel lumen), which both impinge on resolution. Beam damage to the resin also promotes uneven cutting, and for this reason, imaging typically requires slices of 50 - 100 nm to be cut from the block-face, producing non-isotrophic voxels in the datasets. Other limitations for some applications include that the sections are destroyed and cannot be reexamined subsequently, which may force users to collect high resolution images of areas that might not contain useful data.
A major advantage of SBFSEM is that it automates the process of sectioning and imaging blocks of tissue by incorporating a custom microtome into a low vacuum SEM chamber 32,33. Since the images are obtained directly from the block-face prior to each cut, the problems of section wrinkling, compression and loss during handling are substantially avoided, although debris deposition and warping due to block-face charging do contribute to some image loss and distortion. Furthermore, the images obtained in raw datasets are already aligned and require only micrometer-scale registration to accommodate beam drift in order to be amenable to most analysis. Because of the automated sectioning process, once the system vacuum has stabilized, large volumes of tissue can be imaged without significant operator involvement. There are multiple advantages of using this technology in performing morphological and quantitative studies on organelles and intracellular structures. One advantage is getting the information on 3D structure of mitochondria within a reasonable amount of time. The availability of open source reconstruction software packages like Reconstruct 47, TrakEM 48 and Knossos 49-51 have made this technology a powerful analytical tool where detailed 3D ultrastructure of neuronal network from experimental animal models can be directly compared. The semiautomated and fully automated image analysis approaches 52 are likely to produce highly mechanistic data in animals where mitochondrial morphology, function and/or biogenesis are expected to be affected. Earlier SBFSEM analysis was greatly hindered due to lower resolution, however newer sample preparation techniques involving enhanced staining methods 40 have considerably improved the resolution to an extent that even a single synaptic vesicle can be easily resolved. At this resolution ultrastructural defects in mitochondria such as vacuolation, membrane disruption, and loss of cristae can be easily observed, and the distribution of defective mitochondria within cells can be determined 38,39. The high resolution of this technique provides a major advantage over light microscopy where the resolution is limited to ~200 nm in XY axis and ~500 nm in Z-axis. Mitochondria are therefore just at the resolution limit of light microscopy 53. Although mitochondria having dimensions below the resolution limit can still be visualized by light microscopy, their dimensions cannot be reliably measured. Importantly, mitochondria that are separated by a distance lower than the resolution limit of a light microscope cannot be resolved by light microscopy. Another advantage is that the mitochondrial structural analysis can be performed within the context of the tissue, without isolation of the organelle. This can allow for appropriate comparisons within the tissue based on mitochondrial localization, for e.g. axonal vs. somatic and/or dendritic mitochondria. An additional advantage is that it is usually possible to determine whether the mitochondria are neuronal or neuroglial in localization by following the processes to a defining organelle, such as glial filaments and glycogen for astrocytes or myelin for oligodendrocytes. The processes of neurons and neuroglial cells are often in close proximity, and with 2D approaches such as TEM it may be difficult to assign with confidence which processes are neuronal and which are neuroglial.
In conclusion, the SBFSEM technology provides stacks of serial images covering tissue areas 20 - 1,000 µm in size with ultrastructural resolution of 5 - 10 nm or higher, which allows entire central nervous system cells and organelles such as mitochondria to be imaged, measured and reconstructed. In this paper, we have provided some practical approaches to making use of this technology. Future applications include analyzing the mitochondrial structure and distribution in variety of animal models of neurological diseases such as neurodevelopmental and neurodegenerative disease models as well as analyzing the various subcellular ultrastructures such as endoplasmic reticulum, nucleus and/or lysosomes in whole cells or tissues under healthy and disease states.
Disclosures
The authors declare that they have no competing financial interests. Emily K Benson is a paid employee of Renovo Neural Inc., and Grahame J Kidd serves as Scientific Director for 3DEM at Renovo Neural Inc., which is a commercial provider of serial block-face SEM services and analysis.
Acknowledgments
We thank Sidney Walker for providing technical help. This work was supported in part by a grant from the National Institute of Health (1R01EY024712-01A1).
References
- Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Friedman JR, et al. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Voth M, Mai S, Jendrach M. Structural implications of mitochondrial dynamics. Biotechnol J. 2008;3:765–780. doi: 10.1002/biot.200800024. [DOI] [PubMed] [Google Scholar]
- Campello S, Scorrano L. Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep. 2010;11:678–684. doi: 10.1038/embor.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer PA, et al. Abnormal mitochondrial morphology in sporadic Parkinson's and Alzheimer's disease cybrid cell lines. Exp Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet. 2009;18:169–176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Romanello V, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, et al. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckhuber CQ, et al. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol. 2007;9:99–105. doi: 10.1038/ncb1524. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM, 3rd , Bohr VA. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015;25:158–170. doi: 10.1016/j.tcb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macke JH, et al. Contour-propagation algorithms for semi-automated reconstruction of neural processes. J Neurosci Methods. 2008;167:349–357. doi: 10.1016/j.jneumeth.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Parikh S. The neurologic manifestations of mitochondrial disease. Dev Disabil Res Rev. 2010;16:120–128. doi: 10.1002/ddrr.110. [DOI] [PubMed] [Google Scholar]
- Frye RE, Rossignol DA. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr Res. 2011;69:41–47. doi: 10.1203/PDR.0b013e318212f16b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anitha A, et al. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013;23:294–302. doi: 10.1111/bpa.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Takihara Y, et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A. 2015;112:10515–10520. doi: 10.1073/pnas.1509879112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3→CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, et al. Dynamic tubulation of mitochondria drives mitochondrial network formation. Cell Res. 2015;25(10):1108–1120. doi: 10.1038/cr.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617–620. doi: 10.1038/nature14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics in mammals. Curr Top Dev Biol. 2004;59:119–144. doi: 10.1016/S0070-2153(04)59005-1. [DOI] [PubMed] [Google Scholar]
- Chavan V, et al. Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS One. 2015;10:e0125185. doi: 10.1371/journal.pone.0125185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddie CJ, Collinson LM. Exploring the third dimension: volume electron microscopy comes of age. Micron. 2014;61:9–19. doi: 10.1016/j.micron.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Harris KM, et al. Uniform serial sectioning for transmission electron microscopy. J Neurosci. 2006;26:12101–12103. doi: 10.1523/JNEUROSCI.3994-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayworth KJ, et al. Imaging ATUM ultrathin section libraries with WaferMapper: a multi-scale approach to EM reconstruction of neural circuits. Front Neural Circuits. 2014;8:68. doi: 10.3389/fncir.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushby AJ, et al. Imaging three-dimensional tissue architectures by focused ion beam scanning electron microscopy. Nat Protoc. 2011;6:845–858. doi: 10.1038/nprot.2011.332. [DOI] [PubMed] [Google Scholar]
- Denk W, Horstmann H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol. 2004;2:e329. doi: 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton SB. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scan Electron Microsc. 1981. pp. 73–76. [PubMed]
- Briggman KL, Denk W. Towards neural circuit reconstruction with volume electron microscopy techniques. Curr Opin Neurobiol. 2006;16:562–570. doi: 10.1016/j.conb.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Shomorony A, et al. Combining quantitative 2D and 3D image analysis in the serial block face SEM: application to secretory organelles of pancreatic islet cells. J Microsc. 2015;259:155–164. doi: 10.1111/jmi.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinali C, Kitmitto A. Serial block face scanning electron microscopy for the study of cardiac muscle ultrastructure at nanoscale resolutions. J Mol Cell Cardiol. 2014;76:1–11. doi: 10.1016/j.yjmcc.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Miyazaki N, Esaki M, Ogura T, Murata K. Serial block-face scanning electron microscopy for three-dimensional analysis of morphological changes in mitochondria regulated by Cdc48p/p97 ATPase. J Struct Biol. 2014;187:187–193. doi: 10.1016/j.jsb.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Traka M, et al. WDR81 is necessary for purkinje and photoreceptor cell survival. J Neurosci. 2013;33:6834–6844. doi: 10.1523/JNEUROSCI.2394-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno N, et al. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc Natl Acad Sci U S A. 2014;111:9953–9958. doi: 10.1073/pnas.1401155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerinck TJ, Bushong EA, Thor A, Ellisman MH. NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. San Diego, CA: University of California San Diego; 2010. Available at http://ncmir.ucsd.edu/sbfsem-protocol.pdf. [Google Scholar]
- Ohno N, et al. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci. 2011;31:7249–7258. doi: 10.1523/JNEUROSCI.0095-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer S, Monavarfeshani A, Lemon T, Su J, Fox MA. Multiple Retinal Axons Converge onto Relay Cells in the Adult Mouse Thalamus. Cell Rep. 2015;12:1575–1583. doi: 10.1016/j.celrep.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasthuri N, et al. Saturated Reconstruction of a Volume of Neocortex. Cell. 2015;162:648–661. doi: 10.1016/j.cell.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Conchello JA, Lichtman JW. Optical sectioning microscopy. Nat Methods. 2005;2:920–931. doi: 10.1038/nmeth815. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Bohorquez D, Haque F, Medicetty S, Liddle RA. Correlative Confocal and 3D Electron Microscopy of a Specific Sensory Cell. J Vis Exp. 2015. p. e52918. [DOI] [PMC free article] [PubMed]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Cardona A, et al. TrakEM2 software for neural circuit reconstruction. PLoS One. 2012;7:e38011. doi: 10.1371/journal.pone.0038011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter M, Mitra PP. Computational methods and challenges for large-scale circuit mapping. Curr Opin Neurobiol. 2012;22:162–169. doi: 10.1016/j.conb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter M, Briggman KL, Denk W. High-accuracy neurite reconstruction for high-throughput neuroanatomy. Nat Neurosci. 2011;14:1081–1088. doi: 10.1038/nn.2868. [DOI] [PubMed] [Google Scholar]
- Saalfeld S, Cardona A, Hartenstein V, Tomancak P. CATMAID: collaborative annotation toolkit for massive amounts of image data. Bioinformatics. 2009;25:1984–1986. doi: 10.1093/bioinformatics/btp266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuly RJ, Martone ME, Ellisman MH. Method: automatic segmentation of mitochondria utilizing patch classification, contour pair classification, and automatically seeded level sets. BMC Bioinformatics. 2012;13:29. doi: 10.1186/1471-2105-13-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs S, Wurm CA. Super-resolution microscopy of mitochondria. Curr Opin Chem Biol. 2014;20:9–15. doi: 10.1016/j.cbpa.2014.03.019. [DOI] [PubMed] [Google Scholar]