

Abstract
CRISPR/Cas9-gene editing has emerged as a revolutionary technology to easily modify specific genomic loci by designing complementary sgRNA sequences and introducing these into cells along with Cas9. Self-Cloning CRISPR/Cas9 (scCRISPR) uses a self-cleaving palindromic sgRNA plasmid (sgPal) that recombines with short PCR-amplified site-specific sgRNA sequences within the target cell by homologous recombination to circumvent the process of sgRNA plasmid construction. Through this mechanism, scCRISPR enables gene editing within two hours once sgRNA oligos are available, with high efficiency equivalent to conventional sgRNA targeting: >90% gene knockout in both mouse and human embryonic stem cells and cancer cell lines. Furthermore, using PCR-based addition of short homology arms, we achieve efficient site-specific knock-in of transgenes such as GFP without traditional plasmid cloning or genome-integrated selection cassette (2–4% knock-in rate). The methods in this paper described the most rapid and efficient means of CRISPR gene editing.
Introduction
CRISPR/Cas9 gene editing technology has greatly advanced genetics and molecular biology research. By design of a site-specific single guide RNA (sgRNA) that interacts with the Cas9 endonuclease, DNA cleavage can be directed to almost any genomic site of interest1–3. It is the most efficient technology to mutate, delete, and insert genomic DNA sequences at specific genomic loci that has been developed to date, offering exciting opportunities to improve understanding of genome function4.
Site-specific genomic targeting generally requires molecular cloning of sgRNA plasmids for each novel targeted locus. This multistep process is both time-consuming and prone to missteps that delay an otherwise rapid process of genome editing. Furthermore, the cost associated with plasmid ligation, transformation, purification, and sequence verification of each newly generated sgRNA over the course of roughly 1 week are prohibitive to large-scale arrayed screening applications necessary for high-throughput genome editing platforms5.
We have overcome this barrier by the development of a self-cleaving palindromic sgRNA plasmid (sgPal) that targets and cuts its own sgRNA sequence once expressed in cells in the presence of Cas9 (Figure 1a)4. We harness the ability of embryonic stem cells (ESCs) and cancer cell lines to repair double-stranded breaks by homologous recombination to generate a new site-specific genome-targeting sgRNA from a short, PCR amplified sgRNA template within target cells. By self-cloning CRISPR (scCRISPR), we are able to target cells within two hours after sgRNA oligos are obtained, without compromising on mutational targeting efficiency while saving significant time, effort, and cost.
Figure 1. Self-Cloning CRISPR Gene Editing.
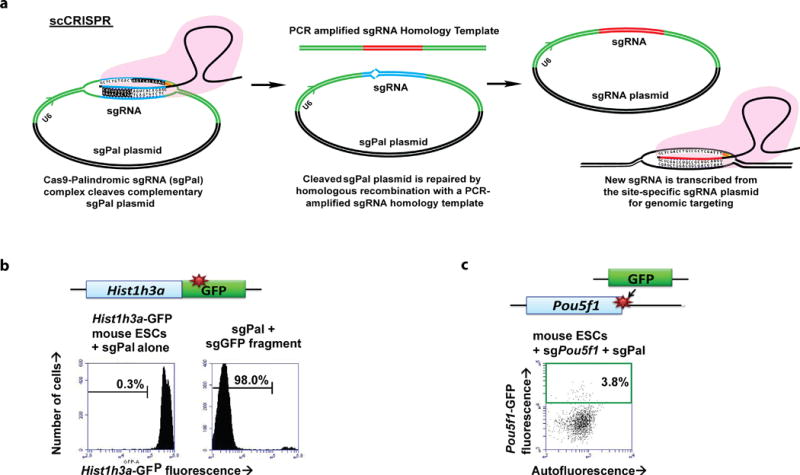
(A) Schematic of the scCRISPR process within target cells. The self-cleaving sgPal plasmid recombines with the short PCR-amplified sgRNA template to form a new site-specific sgRNA plasmid, to facilitate genome editing.
(B) Histogram of flow cytometric GFP fluorescence (x-axis) of Hist1h3a mouse ESCs after electroporation with sgPal plasmid alone (left), or together with sgGFP homology fragment (right).
(C) Flow cytometric plot of efficient generation of Pou5f1-GFP knock-in mouse ESCs (y-axis) using PCR-amplified GFP fragment.
For the generation of knock-in cell lines, additional constructs are traditionally made with long homology regions to facilitate insertion of the desired sequence at the locus of interest. Typically, flanking homology arms range from 600 – 6000bp long each, and must be cloned on either side of the insertion sequence to generate a targeting plasmid. Obtaining large genomic sequences from the genome by PCR amplification frequently requires multiple rounds of amplification, ligation and cloning, spanning weeks, and is often unsuccessful. With recent advances in oligonucleotide synthesis, these sequences can be bought, though at significant expense. Either method of homology-plasmid construction becomes a substantial investment to be made for each new insert and every knock-in site. As such, CRISPR gene editing is currently under-utilized for generating reporter cell models.
We demonstrate that short, ± 80bp flanking homology sequences are sufficient and highly effective at knock-in of ±2kb long sequences. The necessary homology arms are easily extended from the insert sequence by PCR amplification, in a rapid protocol spanning less than 2 hours. As such, knock-in cell targeting with scCRISPR is done within a day, and knock-in lines can be generated and verified in 2–3 weeks, depending on the knock-in sequence and genomic locus.
The cloning-free methods of CRISPR/Cas9 gene editing presented in this article considerably lower the bar on genetic engineering and transgenesis of in vitro cell models, as far as cost, time, and effort, without compromising targeting efficiency. Thus, we present scCRISPR as an ideal technique for inducing individual gene modifications, and large-scale and high-throughput gene editing applications alike.
NOTE: Cells for scCRISPR targeting are cultured and maintained according to standard practices in a Class II biological hazard flow hood or a laminar-flow hood.
BASIC PROTOCOL 1 scCRISPR Site Specific Targeting for Non-Homologous End-Joining
We design site-specific sgRNA oligos with short flanking regions homologous to the sgPal plasmid, and then further extend the homology regions by consecutive PCR amplification steps for optimal efficiency of recombination with the sgPal plasmid. Only one oligo must be designed and ordered for each desired target site, and the homology arm primers are all stock reagents. This locus-specific sgRNA PCR product is introduced into target cells along with plasmids encoding Cas9 and the self-cleaving sgPal, and cells that have received these plasmids are enriched by transient antibiotic selection. The locus-specific sgRNA recombines with sgPal inside the host cell, routinely yielding mutation in >90% of cells.
Materials
-
-
sgRNA oligos
-
-
PCR polymerase (OneTaq)
-
-
Agarose
-
-
Gel electrophoresis unit
-
-
Minelute DNA purification kit (or standard DNA purification followed by vacuum concentration)
-
-
sgPal7-HygR plasmid maxiprep DNA (Addgene #71484)
-
-
CBH-Cas9-BlastR plasmid maxiprep DNA (Addgene (#71489)
-
-
Sterile 1.5ml microcentrifuge tubes
-
-
Cell electroporator and cuvettes
-
-
Cells and appropriate cell culture media and reagents
-
-
Y-27632 ROCK-inhibitor
-
-
Hygromycin
-
-
Blasticidin
Step 1 – scCRISPR sgRNA preparation
Design sgRNAs for target locus by identifying the genomic Protospacer Adjacnt Motif (PAM)-sequence (…NGG) of interest. Note that spCas9 cleavage occurs between bases 3–4 upstream of the PAM: for mutation or knockout, this should be chosen to disrupt the function of interest. For gene knock-in, guidelines are given in Basic Protocol 2. The “NGG” can be on either strand, so the PAM-sequence “CCN” is also an acceptable target.
- Once you have identified the appropriate PAM, you will design an oligonucleotide from the genomic sequence upstream of the PAM, to be ordered as the protospacer, following the guidelines outlined below. The protospacer sequence should always be 19–21 bp long. Because the U6 promoter which will be used to transcribe the gRNA is most efficient when the first base of the protospacer is a ‘G’, we use the following scheme to design the oligonucleotide:
- Protospacers should have 19–21 bp of homology to the genome immediately preceding the NGG “PAM” sequence:
- If the genome sequence is GNNNNNNNNNNNNNNNNNNN NGG (GN19NGG), the protospacer sequence should be GNNNNNNNNNNNNNNNNNNN (GN19)
- If “i” is not satisfied but GNNNNNNNNNNNNNNNNNN NGG (GN18NGG) is satisfied, the protospacer sequence should be GNNNNNNNNNNNNNNNNNN (GN18)
- If “i” and “ii” are not satisfied, the protospacer sequence should be GNNNNNNNNNNNNNNNNNNNN (GN20) immediately upstream of the NGG, regardless of whether the “G” at position 1 is in the genome or not.
- If the genomic sequence targeted is CCN(N20), then reverse complement the entire stretch and apply these rules to the reverse complemented sequence.
- After applying these rules, you should have a protospacer sequence that is 19–21 bp long. The sgRNA oligonucleotide to order for scCRISPR order contains this 19–21 bp sequence, flanked by sgPal homology sequences that will be used to PCR amplify the protospacer into a functional homology template for the scCRISPR system. The oligonucleotide should obey the following format:
60bp scCRISPR oligo design
For 20 bp sgRNA TGGAAAGGACGAAACACCGN19GTTTAAGAGCTATGCTGGAAAC
For 21 bp sgRNA GGAAAGGACGAAACACCGN20GTTTAAGAGCTATGCTGGAAAC
For 19 bp sgRNA TGGAAAGGACGAAACACCGN18GTTTAAGAGCTATGCTGGAAACA
- Order standard scCRISPR homology directed repair (HDR) extension primers for 3 consecutive PCR steps:
PCR step Primer Sequence
Step 1 sgRNA_HDRstep1_fw TGTTTTAAAATGGACTATCATA
TGCTTACCGTAACTTGAAAGTATTTCGATTTCT
TGGCTTTATATATCTTGTGGAAAGGACGAAACACC
Step 1 sgRNA_HDRstep1_rv GTTGATAACGGACTAG
CCTTATTTAAACTTGCTATGCTGTTTCCAGCATAGCTC
TTAAAC
Step 1 sgRNA_HDRstep2_fw GTACAAAATACGTGACGTAGAAA
GTAATAATTTCTTGGGTAGTTTGCAGTTTT
AAAATTATGTTTTAAAATGGACTATCATATGCTTACC
Step 1 sgRNA_HDRstep2_rv ATTTTAACTTGCTATTTCTAGCTCT
AAAACAAAAAAGCACCGACTCGGTGCCAC
TTTTTCAAGTTGATAACGGACTAGCCTTATTTAAAC
Step 2 sgRNA_HDRstep3_fw CGATACAAGGCTGTTAGAGAGATA
ATTAGAATTAATTTGACTGTAAACACAAA
GATATTAGTACAAAATACGTGACGTAGAAAGTAATAA
Step 2 sgRNA_HDRstep3_rv TCAATGTATCTTATCATGTCTGC
TCGATTTTAACTTGCTATTTCTAGCTCTAAAA
CAAAA
Step 3 sgRNA_HDRstep4_fw GGCCTATTTCCCATGATTCCTT
CATATTTGCATATACGATACAAGGCTGTTAGA
GAGATA
Step 3 sgRNA_HDRstep4_rv TCAATGTATCTTATCATGTCTGCTCGA
Step 2 – sgRNA PCR amplification
The protocols below are for amplification with OneTaq polymerase as this is the most cost-effective, but other polymerases also work for scCRISPR. We PCR amplify sgRNA oligos in three successive PCR steps for maximum recombination efficiency, and PCR purification at intermediate steps is not required. PCR steps 1 and 2 are run with 10 amplification cycles to save time, and the final PCR step 3 is run with 35 cycles to generate a large number of amplicons. Note that the initial scCRISPR protocol employed the first two PCR steps only and worked at 70–90% efficiency, but we have since found that adding a third PCR step further increases efficiency to >90%. If targeting with the product of PCR step 2 is desired, make sure to perform 35 cycles of the final PCR step to produce sufficient amplicons for sgRNA recombination. For maximal efficiency we recommend the protocol as described below.
-
4PCR step 1 on 60 bp sgRNA oligo template with HDRstep1 and HDRstep2 forward and reverse primers using Onetaq polymerase (Ta=60°C), and run for 10 amplification cycles. Amplification product is 293 bp. PCR mix below is for typical 20ul reaction volume, but can be scaled if needed.
Volume Reagent % Reaction Volume
10ul OneTaq 2× Master Mix with standard buffer 50%
0.5ul 20uM stock of 60bp sgRNA oligo 2.5%
1ul 10uM HDRstep1 Fw + Rv primer mix 5%
1ul 10uM HDRstep2 Fw + Rv primer mix 5%
7.5ul mQ water 37.5%
-
5PCR step 2 on 293bp amplification product of PCR step 1, with HDRstep3 forward and reverse primers using Onetaq polymerase (Ta=60°C), and run for 10 amplification cycles. Amplification product of PCR step 1 is used directly without purification, and it is unnecessary to determine DNA concentration. If desired, amplicon size can be validated by running a small aliquot of PCR step 1 on a 2% agarose by gel electrophoresis as described below in step of this protocol, though this is not necessary at this stage. Amplification product is 379 bp. PCR mix below is for typical 20ul reaction volume, but can be scaled if needed.
Volume Reagent % Reaction Volume
10ul OneTaq 2× Master Mix with standard buffer 50%
1ul Unpurified product of PCR 1 5%
1ul 10uM HDRstep3 Fw + Rv primer mix 5%
8ul mQ water 40%
-
6PCR step 3 on 379bp amplification product of PCR step 2, with HDRstep3 forward and reverse primers using Onetaq polymerase (Ta=60°C). Amplification product of PCR step 2 is used directly as described above. Amplification is done for 35 cycles, as this is the last PCR step to generate templates for scCRISPR sgPal homologous recombination. Amplification product is 415 bp. PCR mix below is for typical 100ul reaction volume, but can be scaled if needed
Volume Reagent % Reaction Volume
50ul OneTaq 2× Master Mix with standard buffer 50%
5ul Unpurified product of PCR 1 5%
5ul 10uM HDRstep4 Fw + Rv primer mix 5%
40ul mQ water 40%
-
7
Final amplicon is validated by gel electrophoresis, running a 2ul aliquot on a 2% agarose gel. Product is as follows:
sgRNA oligo PCRstep1 amplicon PCRstep2 PCRstep3 GGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCTGTTAGAGAGATAATTAGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAATACGTGACGTAGAAAGTAATAATTTCTTGGGTAGTTTGCAGTTTTAAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGAGGCGTCTGGGTGGCTCTTGGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTGTTTTAGAGCTAGAAATAGCAAGTTAAAATCGAGCAGACATGATAAGATACATTGA
-
8
Minelute PCR purify the remaining 98ul of PCR step 3 in 10ul.
Step 3 – scCRISPR cell targeting for NHEJ
Cells can be targeted by electroporation, transfection, or nucleofection. We achieve the highest efficiency cutting by electroporation, and lowest efficiency by transfection, for which the protocols follow below:
- For electroporation
-
9For electroporation into 12-well, combine the 10ul eluate of PCRstep3 from above with 4ug CBH-Cas9-BlastR plasmid and 4ug sgPal7-HygR plasmid. If total volume exceeds 20ul, concentrate by vacuum centrifugation until it is reduced below this threshold: be vigilant so that the DNA mix does not get contaminated in this process and the DNA does not dry out.
-
10Prepare single cell suspension by enzymatic passaging and concentrate cells by centrifugation for 5 minutes at 200g.
-
11Transfer appropriate number of cells to electroporation cuvette in 120 ul EmbryoMax Electroporation buffer and gently mix well with DNA. For mouse and human embryonic stem cells (ESCs), we use ±106 cells and 0.4cm electroporation cuvettes.
-
12Electroporate cells with appropriate settings for the cell type used. For mouse and human ESCs we electroporate at 230 V, 0.500 mF, and maximum resistance.
-
13Plate in 12 well and incubate at 37°C overnight. Mouse and human ESCs in complete ESC media can be plated with added 7.5uM Y-27632 if desired to help recovery and reduce cell death following electroporation. Continue with step 15.
-
9
- For transfection
-
14For transfection into 12-well, combine the 10ul eluate of PCRstep3 from above with 0.5ug CBH-Cas9-BlastR plasmid and 0.5ug sgPal7-HygR plasmid and transfect according to the standard protocol for transfection reagent and cell type used. Incubate cells overnight at 37°C.
-
15Select with Blasticidin and Hygromycin from hours 24–72 after targeting. For mouse ESCs we select with 10 ug/mL Blasticidin + 100 ug/mL Hygromycin. For human ESCs we select with 2 ug/mL Blasticidin + 66 ug/mL Hygromycin. Expect >90% cutting efficiency for electroporation and ~50% for transfection. Representative data for knock-out efficiency is shown in Figure 1b, by knock-out of Hist1h3a-GFP fluorescence.
-
14
BASIC PROTOCOL 2 scCRISPR Gene Insertion by Homologous Recombination
We design an scCRISPR sgRNA oligo as above for cutting at the genomic insertion site. In addition, we design ±150bp long sequences homologous to the genomic locus to flank the sequence that is to be inserted, such as GFP for the generation of a fluorescent reporter. We have found that introducing GFP with 75–80 bp homology arms is sufficient to induce knock-in at appreciable frequency. Because of cost and PCR efficiency, we add these homology arms using two rounds of PCR, each of which adds 35–40 bp of homology arm to GFP. Due to cost considerations for oligonucleotide production and PCR efficiency considerations, each primer will be 60 bp at maximum.
Significant overlap between the sgRNA and genomic homology arm sequence must be avoided to prevent cleavage of the insertion cassette. Thus, to avert further disruption of the genomic locus, it is important that the sgRNA targeting occurs as near to the insertion site as possible. Below details the strategy to create a C-terminal in-frame GFP knock-in, however this protocol can be adapted to knock in any insert at any locus following this sgRNA and homology-sequence design. Efficiencies reported here are representative for ±1–2kb sequences. Larger insertions are feasible if they can be PCR amplified, and they may have less efficient knock-in. Homologous recombination (HR) replacement of short regions such as SNP repair is possible using this protocol and facilitates knock-in in 20–50% of cells.
Materials
-
-
sgRNA oligos
-
-
Proofreading PCR polymerase (NEBNext)
-
-
Agarose
-
-
Gel electrophoresis unit
-
-
Minelute DNA purification kit (or standard DNA purification followed by vacuum concentration)
-
-
sgPal7-HygR plasmid maxiprep DNA (Addgene #71484)
-
-
CBH-Cas9-BlastR plasmid maxiprep DNA (Addgene (#71489)
-
-
Knock-in template
-
-
Sterile eppendorff 1.5ml microcentrifuge tubes
-
-
Cell electroporator and cuvettes
-
-
Cells and appropriate cell culture media and reagents
-
-
Y-27632 ROCK-inhibitor
-
-
Hygromycin
-
-
Blasticidin
-
-
Purelink Genomic DNA Mini Kit
-
-
gDNA Lysis Buffer (10 mM TrisHCl (pH7.5 or pH 8.0), 10 mM EDTA, 10 mM NaCl, 0.5% SDS) with 1mg/ml Proteinase K added fresh
Step 1 – scCRISPR sgRNA and insertion homology-arm design
-
Use UCSC genome browser (http://genome.ucsc.edu/) to identify the genomic sequence surrounding the gene of interest. If you are interested in making a C-terminal GFP fusion construct, identify the stop codon of the transcript you would like to tag (typically the primary RefSeq transcript), and copy ~500 bp of genomic DNA sequence centered on the stop codon. Make sure to copy the sequence in the orientation of the coding sequence which may require reverse complementing the entire sequence. Annotate the STOP-codon within the sequence, below is an example for the mouse Pou5f1 gene where the STOP-codon is in bold and underlined:
Pou5f1
CGAGTATGGTTCTGTAACCGGCGCCAGAAGGGCAAAAGATCAAGTATTGAGTATTCCCAACGAGAAGAGTATGAGGCTACAGGGACACCTTTCCCAGGGGGGGCTGTATCCTTTCCTCTGCCCCCAGGTCCCCACTTTGGCACCCCAGGCTATGGAAGCCCCCACTTCACCACACTCTACTCAGTCCCTTTTCCTGAGGGCGAGGCCTTTCCCTCTGTTCCCGTCACTGCTCTGGGCTCTCCCATGCATTCAAACTGAGGCACCAGCCCTCCCTGGGGATGCTGTGAGCCAAGGCAAGGGAGGTAGACAAGAGAACCTGGAGCTTTGGGGTTAAATTCTTTTACTGAGGAGGGATTAAAAGCACAACAGGGGTGGGGGGTGGGATGGGGAAAGAAGCTCAGTGATGCTGTTGATCAGGAGCCTGGCCTGTCTGTCACTCATCATTTTGTTCTTAAATAAAGACTGGGACACACAGTAGATAGCTGAATTTTGTTTTCCTTCAG
-
In order to make a C-terminal fusion protein, find the NGG on either strand that is closest to the stop codon, as homologous recombination works best when the homology arms are as close as possible to the double-strand break. Note that the sgRNA will continue to cleave any sequence with significant similarity to the target sequence, so it is pertinent to ensure the homology sequences used do not overlap with the protospacer sequence. It is best to avoid overlap between the homology sequence and sgRNA of more than 5bp, which means that a portion of the sgRNA sequence will be removed from the genome. To avoid loss of protein coding sequence, NGG sequences prior to or containing the stop codon should not be used, but NGG sequences after the stop codon and CCN sequences immediately prior to, including, or after the stop codon can be used.
Keeping this in mind, we design the targeting sgRNA as described above, as near to the insertion site as possible while avoiding the coding region. The sgRNA we designed is in the reverse complement, depicted above italicized. Note that the GGG and CCC PAM sequences prior to the stop codon and the AGG abutting the stop codon were not used because too much of their sgRNA sequence must be retained in the homology construct, so the sgRNA would cut in the homology construct and prevent its integration. The PAM sequence used was thus the closest acceptable PAM sequence to the stop codon, denoted above in italics.
Note: If no appropriate sgRNA sequence can be found outside of the coding region, or if it is desired to knock-in the insert within a coding region, the sgRNA recognition sequence will necessarily overlap with the homology-arms. In this case, to maintain the protein-coding sequence it is recommended to design the area of the homology-arms that overlaps with the sgRNA to have silent mutations, such that these codons remain unchanged, while the DNA sequences are significantly modified so that they are no longer recognized by the targeting sgRNA. The PAM sequence is the most important to Cas9 cutting, so ablating the PAM sequence from the desired homologous recombination-repaired genotype is the best way to avoid cutting.
Pou5f1_gRNA GGAAAGGACGAAACACCGACAGCATCCCCAGGGAGGGCGTTTAAGAGCTATGCTGGAAAC
Homology arms are designed to encode GFP in-frame immediately upstream of the stop codon of the gene and to include a stop codon after the GFP open reading frame (ORF). Homology arms were designed so as not to overlap with the gRNA sequence by more than 5 bp on either side. For the forward arm, the homology arm should always include the entire last codon before the stop codon to allow in-frame C-terminal GFP fusion. For the reverse arm, the rule of not overlapping with the gRNA sequence by more than 5 bp means that a portion of the 3′ UTR will be removed. We have not found this to be a problem for gene expression, but we limit the extent of this removal by designing gRNAs as close to the stop codon as possible (see above section) and designing homology arm primers with the maximum allowable 5 bp of overlap with the gRNA. Left and right homology sequences are depicted above, underlined.
-
Design primers to PCR amplify the insertion cassette with homology arms for the insertion locus. To insert GFP, we use the bolded primer sequences below. This GFP sequence can be ordered as a gBlock to use as PCR template for the knock-in cassette. Note the GFP STOP-codon at the 3′-end (bolded and double-underlined), and take care to remove this sequence when necessary. Genomic homology sequences are attached upstream of these primers to create the flanking homology arms in two consecutive PCR steps using 60bp oligos. The first primer pair is of the format in the table below, adding the 40bp of genomic homology sequence nearest to the insertion site, to each primer. The second homology-arm primer pair overlaps with the first homology arms by 20–30 bp. We choose the minimal overlap such that the overlapping region is estimated to have a Tm of >65 degrees Celsius using the NEB Tm calculator (http://tmcalculator.neb.com/#!/). We never use less than 20 bp of overlap to ensure efficient PCR. Then, we extend them by 30–40 bp on each end up to 60 bp maximum. Examples of homology-arm primers for Pou5f1 knock-in of GFP, based on the homology sequence denoted above, can be found in the table below:
GFP
GTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAAGCGGCCGCAATTCACTCCTCA
Design Primer Sequence
(Pre-STOP 40bp) – GFP_Fw GFPstep1_Fw TGTTCCCGTCACTGCTCTGGGCTCTCCCATGCATTCAAAC
GTGAGCAAGGGCGAGGAGCT
(Post-STOP 40bp) – GFP_Rv GFPstep1_Rv CCAGGTTCTCTTGTCTACCTCCCTTGCCTTGGCTCACAGC
TGAGGAGTGAATTGCGGCCG
(35–40bp ext) – GFP_step1_Fw GFPstep2_Fw TCTACTCAGTCCCTTTTCCTGAGGGCGAGGCCTTTCCCTC
TGTTCCCGTCACTGCTCTGG
(35–40bp ext) – GFP_step1_Rv GFPstep2_Rv TAATCCCTCCTCAGTAAAAGAATTTAACCCCAAAGCT
CCAGGTTCTCTTGTCTACCTCCC
- Finally, we design genomic DNA PCR primers to verify whether site specific homologous recombination was successful, using Primer3 (http://bioinfo.ut.ee/primer3/) with standard settings. Primers must be outside of the amplified homology arms to avoid background of unintegrated homology arm construct. To do this, paste the 500 bp genomic sequence into Primer 3 placing “[” and “]” at the end of the homology arms. Primer3 will give you one forward primer before the homology arm and one reverse primer after the homology arm that can typically be paired with the GFP primers below to look for locus-specific GFP integration. Examples for Pou5f1 flanking primers are also listed below:
GFPearly_Rv GTCCAGCTCGACCAGGATG
GFPlate_Fw GGATCACTCTCGGCATGGAC
Pou5f1insert_Fw TGTATCCTTTCCTCTGCCCC
Pou5f1insert_Rv GAGCTTCTTTCCCCATCCCA
Step 2 – Homology insertion cassette PCR amplification
The protocol below is for high-fidelity amplification with 2× NEBNext Mastermix, however other proofreading polymerases may also work for faithful gene knock-in. We PCR-amplify the insertion cassette in two successive PCR steps to generate a total of ± 150 bp of flanking homology sequence to the insertion site. PCR step 1 is run with 15 amplification cycles to save time, while PCR step 2 is run with 35 cycles to generate a large number of amplicons. The sgRNA-HDR oligo used for targeting is PCR amplified and prepared as explained in the protocol above with one minor modification: that the final PCR step 3 is scaled to a 200ul volume.
-
6PCR step 1 on GFP template with GFPstep1 forward and reverse primers using NEBNext polymerase (Ta=60–72°C, typically 72°C), and run for 15 amplification cycles. Amplification product is 819 bp. PCR mix below is for typical 20ul reaction volume, but can be scaled if needed.
Volume Reagent % Reaction Volume
10ul NEBNext 2× Master Mix 50%
0.1ul 100ng/ul stock of GFP template 0.5%
0.5ul 20uM GFPstep1 Fw primer 2.5%
0.5ul 20uM GFPstep1 Rv primer 2.5%
0.6ul DMSO 3%
8.3ul mQ water 41.5%
-
7PCR step 2 on 819bp amplification product of PCR step 1, with HDRstep3 forward and reverse primers using NEBNext polymerase (Ta=60–72°C, typically 72°C), and run for 30 amplification cycles. Amplification product of PCR step 1 is used directly without purification, and it is unnecessary to determine DNA concentration. If desired, amplicon size can be validated by running a small aliquot of PCR step 1 on a 2% agarose by gel electrophoresis as described below in step of this protocol, though this is not necessary at this stage. Amplification product is ±900 bp. PCR mix below is for typical 200ul reaction volume, but can be scaled if needed.
Volume Reagent % Reaction Volume
100ul NEBNext 2× Master Mix 50%
10ul Unpurified product of PCR 1 5%
5ul 20uM GFPstep2 Fw primer 2.5%
5ul 20uM GFPstep2 Rv primer 2.5%
6ul DMSO 3%
74ul mQ water 37%
-
8
Final amplicon is validated by gel electrophoresis, running a 2ul aliquot on a 2% agarose gel. Product is as follows:
GFP primers PCRstep1 homology primers PCRstep1 homology primers TCTACTCAGTCCCTTTTCCTGAGGGCGAGGCCTTTCCCTCTGTTCCCGTCACTGCTCTGGGCTCTCCCATGCATTCAAACGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAAGCGGCCGCAATTCACTCCTCAGCTGTGAGCCAAGGCAAGGGAGGTAGACAAGAGAACCTGGAGCTTTGGGGTTAAATTCTTTTACTGAGGAGGGATTA
-
9
There is no need to gel purify unless there is an abundance of smaller band. Minelute PCR purify the remaining 198ul of PCR step 2 in 10ul.
Step 3 – scCRISPR cell targeting for HR
Efficient cell targeting is achieved by electroporation as described below, or slightly lower efficiency by nucleofection. Transfection yields low knock-in efficiency and is not recommended.
- For electroporation
-
10For electroporation into 6-well, combine the 10ul eluate of the 200ul Minelute sgRNA PCRstep3, and the 10ul eluate of the 200ul Minelute insertion cassette PCRstep2 from above with 5ug CBH-Cas9-BlastR plasmid and 5ug sgPal7-HygR plasmid. If total volume exceeds 20ul, concentrate by vacuum centrifugation until it is reduced below this threshold: be vigilant so that the DNA mix does not get contaminated in this process and the DNA does not dry out.
-
11Prepare single cell suspension by enzymatic passaging and concentrate cells by centrifugation for 5 minutes at 200g.
-
12Transfer appropriate number of cells to electroporation cuvette in 120 ul EmbryoMax Electroporation buffer and gently mix well with DNA. For mouse and human embryonic stem cells (ESCs), we use ±106 cells and 0.4cm electroporation cuvettes.
-
13Electroporate cells with appropriate settings for the cell type used. For mouse and human ESCs we electroporate at 230 V, 0.500 mF, and maximum resistance.
-
14Plate in 6-well and incubate at 37°C overnight. Mouse and human ESCs in complete ESC media can be plated with added 7.5uM Y-27632 if desired, to help recovery and reduce cell death following electroporation.
-
15Select with Blasticidin and Hygromycin from hours 24–72 after targeting. For mouse ESCs we select with 10 ug/mL Blasticidin + 100 ug/mL Hygromycin. For human ESCs we select with 2 ug/mL Blasticidin + 66 ug/mL Hygromycin.
-
16Replace selection media with regular media and allow cells to recover and expand for ±7 days after electroporation. When passaging, take sample to analyze targeting efficiency as described below. Expect 2–4% insertion efficiency by electroporation in ESCs and >10% for HEK293T cells. Representative data for mESC knock-in efficiency is shown in Figure 1c, by C-terminal knock-in of GFP in the Pou5f1 gene.
-
10
Step 4 – Analyzing HR knock-in efficiency
Validating knock-in can be done in various ways, depending on the knock-in cassette. Insertion of a fluorescent, or antibiotic-resistance reporter of a gene that is expressed in the target cell type, may allow for swift selection of target clones by flow-cytometric sorting, or antibiotic selection of successfully targeted clones. Still, subsequent sequence verification of clones may still be recommended in these cases, or when phenotypic selection is not possible.
Below we describe a method to determine knock-in efficiency by PCR of bulk, and clonal targeted cells. For fastest identification of positive clones, we simultaneously plate cells for 96-well clonal analysis, while performing bulk population PCR validation, in the protocols below.
Bulk population genomic DNA analysis
-
17
When splitting confluent 6-well of targeted cells, collect genomic DNA on ~1/2 of cells using the Purelink Genomic DNA Mini Kit.
-
18Use the isolated bulk genomic DNA to check for integration using the primers ordered for this purpose during homology construct generation. There are three possible primer pairs to use that will confirm the left-end, right-end, and complete insertion size. All three can, and should be used to confirm insertion, but keep in mind that Pair 3 to check for insertion size will mainly show the wildtype locus in a bulk population in which knock-in genotypes are the minority, as this amplicon is smaller and will dominate the PCR. For the example of GFP insertion into the Pou5f1 C-terminus, we can use the following primer combinations:
Pair Forward Primer Reverse Primer
1 Pou5f1insert_Fw GFPearly_Rv
2 GFPlate_Fw Pou5f1insert_Rv
3 Pou5f1insert_Fw Pou5f1insert_Rv
PCR using the above pairs on 200ng of bulk isolated genomic DNA (gDNA) using Onetaq polymerase (Typically Ta=60°C), and run for 35 amplification cycles. PCR mix below is for typical 20ul reaction volume.
Volume Reagent % Reaction Volume
10ul OneTaq 2× Master Mix with standard buffer 50%
x.x ul 200ng Bulk gDNA
0.5ul 20uM Validation primer Fw 2.5%
0.5ul 20uM Validation primer Rv 2.5%
x.x ul mQ water Up to 100%
Check amplicon by gel electrophoresis, by running sample on a 2% agarose gel. If the bands are absent, weak, or have competing bands at incorrect sizes, optimize the Ta of this PCR and try PCR conditions both with, and without 3% DMSO. This step is important not only to ensure that GFP has integrated in the population but also to optimize primer combinations, as the 96-well genomic DNA PCRs are messier so require robust primers.
-
19
Optionally, qPCR can be used, after successful PCR conditions have been found, to estimate integration frequency. To do so, primer pairs that only give a product after correct integration should be used along with genomic DNA control primers that occur twice in every cell. By comparing amplification cycle number, approximate integration frequency can be established.
96-well genomic DNA analysis of feeder-free ESCs
Note that at this stage, individual colonies can be picked, or limiting dilution can be performed to reduce the number of colonies that eventually have to be picked. Given the typical 2–4% knock-in efficiency for a 1–2 kb insert, picking 96 colonies is a recommended minimum. The protocol below is for limiting dilution followed by single colony picking or limiting dilution from positive wells.
-
20
Count the cells and plate 96 wells of a 96-well plate at ~7 cells/well.
-
21
Once 96-wells are at least 1/3 confluent, prepare to split. To do so, prepare appropriate surface-coating for ESC type, and add 100 uL of media into wells.
-
22
Dissociate cells enzymatically using 25ul of Trypsin/EDTA solution, breaking up clumps with a multi-channel pipette.
-
23
Transfer half of the cell solution into the new 96well plate containing media to continue culture, and use the other half for 96-well gDNA isolation.
-
24
Add 50 uL genomic DNA lysis buffer with added Proteinase K.
-
25
Seal plate using parafilm and place plate in humidified staining chamber to avoid evaporation, then place chamber at 60 degrees. This step is ideally done overnight but can be shortened to >3 hrs.
-
26
Carefully add 100 uL/well of pre-chilled 100% EtOH + 75 mM NaCl (add NaCl fresh as it doesn’t really go into solution—150 uL 5 M NaCl per 10 mL EtOH) to each well and let sit on benchtop at room temp for 30 min.
-
27
Carefully invert plate to remove liquid then add 150 uL/well 70% EtOH. Invert and repeat for 2 total 70% EtOH washes.
-
28
After second wash, invert and then shake plate vigorously to remove all EtOH and blot upside down on paper towels to remove all EtOH. Shake vigorously and blot every few minutes, and let plate dry for 10 min at room temp
-
29
Add 30 uL/well TE Buffer/Elution Buffer and allow to dissolve. Optionally place back at 60 degrees for several minutes to facilitate DNA dissolving.
Prepare PCR mixture for Onetaq PCR reactions of each colony, according to optimized PCR conditions determined by bulk population gDNA analysis. Typically, we use a 15ul reaction volume and run for 35 amplification cycles.
-
30
Note which wells give positive PCR reactions. As these reactions were isolated from 7 clones per well originally, these wells must be further subcloned by limiting dilution or by colony picking, and new clones should again be verified by PCR. Clones should always be verified through complete sequencing using locus forward and reverse primers, as it is possible to get partial insertions or insertions with mutations.
COMMENTARY
Background Information
Genomic engineering is an invaluable tool in discerning the role of DNA and gene products in cell function1–3. Since the initial reports describing how CRISPR/Cas9 could be modified to target specific genomic loci, this technology has greatly impacted the course of genetic and cellular research. Design of site-specific targeting sgRNAs is much improved from the laborious construction of recombinant endonucleases, such as TALENs and ZFNs6,7, which makes CRISPR a far more amenable genome editing system.
Still, the requirement to clone novel sgRNA plasmids for each locus is disproportionately time consuming compared to the ease with which sgRNAs are designed and CRISPR cell targeting can be accomplished. Currently, sgRNA plasmid cloning can take up the same amount of time as targeting and validating correctly targeted cells. We bypass the lengthy, and relatively costly step of sgRNA plasmid construction by allowing the sgRNA plasmid recombination, normally achieved by molecular cloning, to occur within cells at the same time as genome targeting, without needing to compromise on targeting efficiency.
Similarly, the challenging steps involved in homology plasmid construction hold back CRISPR-mediated transgenesis from becoming a routine application. Presently, genomic insertion of gene sequences, such as to create reporters of gene expression, are reserved for only the most essential experiments due to the effort involved. Using the minimal parameters explained in this protocol, to add short ± 80bp long flanking homology regions onto the insertion sequence, the time, difficulty and cost of homology-mediated gene insertion are overcome.
Cloning-free approaches further improve on CRISPR engineering technology by avoiding the few demanding steps involved in the locus-specific gene editing process, to facilitate genomic modification and transgenesis of in vitro models and help to accelerate the progress of molecular research.
Critical Parameters
Good tissue culture practice and sterile technique are important to maintain healthy cell cultures and avoid contamination. When preparing single cell solutions for electroporation, care should be taken to treat cells gently so as to avoid unnecessary additional stress, as harsh treatment may lead to an increase in cell death and lower targeting efficiency. Limiting the volume of the DNA mix used for electroporation is also important to maintain the correct balance of the components in the electroporation solution. As described above, we electroporate in 120ul of electroporation buffer and restrict the DNA mix volume to 20ul or less.
Depending on the cell line used, the antibiotic concentrations may need to be adjusted to ensure optimal selection. If excessive cell death is observed, even after optimization of antibiotic concentrations, selection could be limited to only one antibiotic instead of two. If so, we find single selection by blasticidin to be more effective than hygromycin incubation.
Use of other sgRNA and Cas9 expressing plasmids than the ones used in this work may yield a different targeting efficiency. Note, that dual-expression plasmids that encode both the sgRNA and Cas9 will lead to lower targeting efficiency than when these components are expressed from two separate plasmids. We recommend the palindromic sgRNA sgPal7 (#71484) and CBH-Cas9 (#71489) plasmids available through Addgene that have been extensively validated for this protocol.
Anticipated results
Using the scCRISPR techniques described above, we routinely achieve mutational targeting efficiencies of > 90% in ESCs and HEK293T cells. Knock-in efficiency is somewhat variable per locus, and depending on the size of the gene insert. From the ± 20 GFP reporter cell lines we have generated, we predict 2–4% of cells will correctly insert the knock-in cassette in mouse and human ESCs. Efficiency of > 10% knock-in is routinely observed in HEK293T cells.
Time considerations
With scCRISPR, cell targeting for NHEJ as well as gene insertion by HR can be done in under 2 hours after receiving the designed oligonucleotides for PCR amplification, by following the successive PCR steps described in the protocols above. This is a massive advance over the standard required cloning time of roughly a week. After targeting, transient antibiotic selection from 24–72 hours after electroporation improves the percentage of targeted cells in the bulk population. We will usually allow cells to recover and expand for another one or two passages before utilizing these cells for further experimentation. In all, an NHEJ-targeted cell line can be generated in approximately 1 week. Knock-in lines can usually be validated and expanded for subsequent use in roughly 2–3 weeks from initial cell targeting.
Significance Statement.
CRISPR/Cas9 gene editing has been transformative in allowing genomic modification to be performed with unprecedented simplicity. Still, preparation of sgRNAs for gene knockout and homology constructs for gene knock-in are time-consuming. The self-cloning CRISPR (scCRISPR) techniques presented here circumvent time-consuming and costly steps of molecular cloning involved in sgRNA-plasmid construction and generation of homology-constructs for site-specific gene knockout and insertion in mouse and human ESCs and cancer cells. By scCRISPR, preparation for cell targeting takes 2 hours instead of 1 week after targeting oligos are obtained, which significantly improves the throughput of CRISPR-mediated gene editing without compromising on gene editing efficiency. Over 90% mutation and 2–4% gene knock-in rates are routinely attained.
Acknowledgments
The authors acknowledge technical assistance from May Sabry and funding from NIH 5UL1DE019581, RL1DE019021, 1K01DK101684-01, 1U01HG007037, and 5P01NS055923; the Harvard Stem Cell Institute’s Sternlicht Director’s Fund award; and Human Frontier Science Program grant to R.I.S.
References
- 1.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M, et al. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbab M, Srinivasan S, Hashimoto T, Geijsen N, Sherwood RI. Cloning-free CRISPR. Stem Cell Reports. 2015;5:1–10. doi: 10.1016/j.stemcr.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajagopal N, et al. High-throughput mapping of regulatory DNA. Nat Biotechnol. 2016 doi: 10.1038/nbt.3468. advance on. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–46. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]