

ABSTRACT
The cell tightly controls histone protein levels in order to achieve proper packaging of the genome into chromatin, while avoiding the deleterious consequences of excess free histones. Our accompanying study has shown that a histone modification that loosens the intrinsic structure of the nucleosome, phosphorylation of histone H3 on threonine 118 (H3 T118ph), exists on centromeres and chromosome arms during mitosis. Here, we show that H3 T118ph localizes to centrosomes in humans, flies, and worms during all stages of mitosis. H3 abundance at the centrosome increased upon proteasome inhibition, suggesting that excess free histone H3 localizes to centrosomes for degradation during mitosis. In agreement, we find ubiquitinated H3 specifically during mitosis and within purified centrosomes. These results suggest that targeting of histone H3 to the centrosome for proteasome-mediated degradation is a novel pathway for controlling histone supply, specifically during mitosis.
KEYWORDS: centrosome, degradation, histone phosphorylation, proteasome
Introduction
The eukaryotic genome resides within the nucleoprotein structure termed chromatin, which comprises an equal mass of histone proteins and DNA. The presence of histones limits access to the DNA sequences. Therefore, the histones have to be dynamically disassembled and reassembled to permit genomic processes that require intimate access to the DNA to occur, including transcription, DNA repair and DNA replication. Chromatin disassembly and assembly is mediated by histone chaperones together with ATP-dependent chromatin remodelers.1 Chromatin disassembly and assembly are also modulated by specific histone post-translational modifications that either loosen the intrinsic nucleosome structure or alter the affinity of the histones for histone chaperones.2 Canonical histones are also removed to allow incorporation of histone variants into the chromatin, which have specific functions. For example, histone H3 is removed from centromeres to enable the incorporation of the centromere specific variant CENP-A after mitosis.3
Maintenance of an appropriate level of histone proteins in the cell is critically important. Experimental reduction of histone protein levels by 50 percent in budding yeast causes arrest at the subsequent metaphase-anaphase transition due to activation of the spindle assembly checkpoint. Intriguingly, histone levels go down naturally during mitotic aging, and their experimental restoration extends lifespan in both yeast and human cells.4,5 Conversely, experimental elevation of histones H3-H4 levels in yeast leads to enhanced DNA damage sensitivity6 and altered gene expression.7 Meanwhile, experimental elevation of H3-H4 in mammalian cells leads to S phase arrest.8
Histone protein levels are not only controlled at the transcription and mRNA stability level,9 but also by mechanisms that remove excess histone proteins that the cell no longer requires. In budding yeast, excess non-chromatin bound histones are degraded after DNA replication in a Rad53 kinase dependent manner whereby histone H3 is phosphorylated at tyrosine 99 via Rad53.6,7 This H3 phosphorylation is followed by polyubiquitination and proteasome-mediated degradation in yeast.7 In mammals, the histone chaperone NASP controls the levels of histones through an autophagy mediated degradation pathway.8 In response to DNA damage in mammalian cells, H3 and H4 are ubiquitinated by the Cul4-DDB-ROC1 ubiquitin ligase and are degraded.10 It is unknown whether mechanisms exist to degrade excess histone proteins during other cell cycle stages or during other processes that entail chromatin disassembly.
Recently, our lab has discerned that two histone H3 modifications, phosphorylation of H3 threonine 118 (H3 T118ph) 11 and phosphorylation of H3 threonine 80 (H3 T80ph),12 exist at the centrosome during mitosis. The centrosome is the microtubule-organizing center important for nucleating and orchestrating microtubule spindle dynamics to achieve equal segregation of genomic DNA into daughter cells during mitosis.13 In addition, functional components of the proteasome have been identified in purified centrosome fractions,14 suggesting that protein degradation may occur at the centrosome.15 In the current work, we show that overexpression of histones leads to detectable H3 at the centrosome upon proteasome inhibition. We demonstrate that histone H3 is ubiquitinated in mitosis and that ubiquitinated H3 exists in purified centrosome fractions. Endogenous histones with H3 T118ph were found at the centrosomes during mitosis in human cells, fly and worm, where H3 T118ph promotes, but is not essential for, centrosomal localization. We propose that non chromatin-bound histones localize to centrosomes during mitosis for their proteasome-mediated degradation.
Results & discussion
Histone H3 T118ph is at centrosomes during mitosis in metazoans
We set out to follow up on our recent observation that H3 T118ph localizes to the centrosome, as determined using an H3 T118ph specific antibody.11 In order to focus on the centrosomal localization of H3 T118ph, we used immunofluorescence analysis of HeLa cells with methanol fixation, which prevents the H3 T118ph antibody from accessing its epitope within chromosomes. We found that H3 T118ph colocalized with the centrosomal protein γ-tubulin during prophase through anaphase of mitosis (Fig. 1A). To obtain a higher resolution view of H3 T118ph at the centrosome, we used electron microscopy in conjunction with immunogold labeling of the H3 T118ph antibody. This assay showed that H3 T118ph localizes near the centrioles in an irregular distribution in the pericentriolar material (Fig. 1B). This localization pattern of H3 T118ph was not unique to HeLa cells, nor cancer cell lines, because MCF10A, MCF12A, HMEC, and WI-38 cell lines displayed a similar centrosomal staining of H3 T118ph (Fig. 1C). In these analyses in Fig. 1C, we used formaldehyde fixation, which also reveals the chromosomal localization of H3 T118ph as we described recently.11
Figure 1.
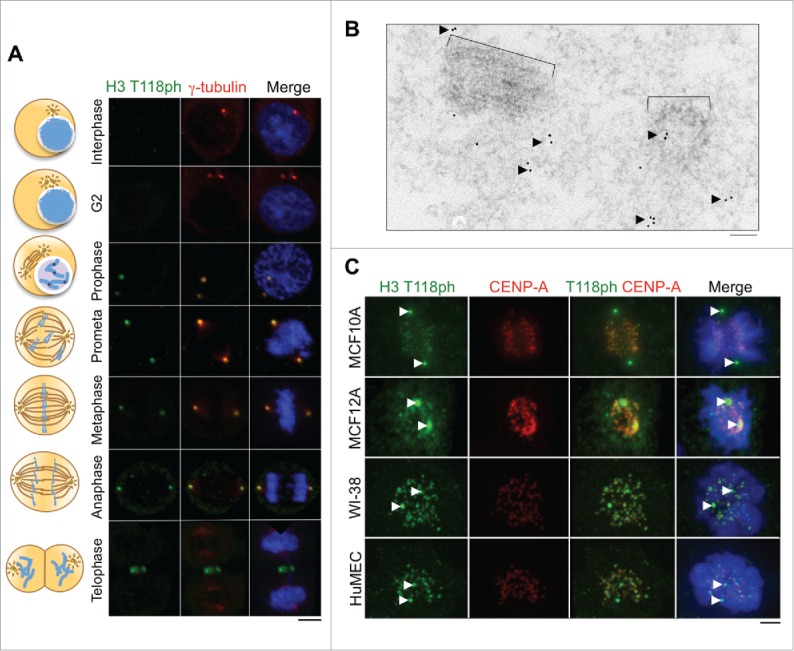
Localization of H3 T118ph to the centrosome during mitosis in human cells. (A) Immunofluorescence analysis of H3 T118ph in HeLa cells. Scale bar = 10 μm. (B) EM images of a mitotic centriole pair with immunogold labeling of H3 T118ph. Black arrows indicate groups of gold beads in the pericentriolar material that surrounds the centriole pair. Brackets indicate centrioles. Scale bar = 100 nm. (C) Immunofluorescence analysis of prometaphase cells from the indicated cell lines. CENP-A marks the centromeres. Arrows mark centrosomes. Scale bar = 10 μm.
To investigate whether the centrosomal localization of H3 T118ph was conserved among metazoans, we examined its localization in flies and worms. In Drosophila embryos at the syncytial blastoderm stage, H3 T118ph marks the centrosomes nucleating mitotic spindle microtubules as shown by theα–tubulin (Fig. 2A). Similarly, in C. elegans, H3 T118ph also demonstrated the centrosomal localization pattern during mitosis (Fig. 2B). Taken together, these data indicate that the centrosomal localization of H3 T118ph is evolutionarily conserved in metazoans.
Figure 2.

H3 T118ph localization to the centrosome is dependent on PLK-1, and occurs in worms and flies. (A) Drosophila embryos at the syncytial blastoderm stage in mitosis. White box indicates magnified area. Arrows mark centrosomes. (B) Control RNAi and PLK-1 (RNAi)-depleted C. elegans 4-cell embryos. Scale bars = 5 μm. White box indicates magnified prometaphase cell. (C) Immunofluorescence analysis of H3 T118ph in untreated and BI-2536 treated prometaphase HeLa cells. Scale bar = 5 μm.
The localization of H3 T118ph to centrosomes is dependent on PLK-1 kinase activity
The unexpected presence of histone H3 T118ph at the centrosome during mitosis (Fig. 1, 2) led us to investigate the mechanism by which H3 T118ph localizes to the centrosome. During our search for the H3 T118ph kinase (which was determined to be Aurora-A 11) we had observed that RNAi knockdown of polo like kinase 1 (PLK-1) in C. elegans caused delocalization of H3 T118ph from the centrosomes (Fig. 2B). This was also the case in mammalian cells, where inhibition of PLK-1 led to H3 T118ph exclusion from centrosomes (Fig. 2C). These results indicate that the centrosomal localization of histone H3 depends on PLK-1. Given the role of PLK-1 in centrosome maturation and assembly of the pericentriolar material,18 it is possible that the role of PLK-1 in localization of H3 T118ph to centrosomes is indirect. Regardless, these data suggest that the histones with H3 T118ph are transported to the centrosomes rather than being phosphorylated at the centrosome.
Histone H3 T118ph localization to centrosomes becomes more diffuse and abundant upon proteasome inhibition
Given that the centrosomes are known sites of proteasome-mediated protein degradation,14 we examined whether H3 T118ph localizes to the centrosome during mitosis to undergo proteasome-mediated degradation. In agreement with this idea, centrosomal accumulation of H3 T118ph became more abundant and diffuse after proteasome inhibition with MG132 (Fig. 3A). In addition to inhibiting the proteasome, MG132 has been reported to inhibit lysosomal proteases and calpains,19 which could also potentially cause increased H3 T118ph signal at the centrosome. Therefore, we utilized an additional proteasome inhibitor, Lactacystin20 and found that it also led to accumulation of H3 T118ph at the centrosome (Fig. 3B). These data are consistent with histones carrying H3 T118ph localizing to the centrosome during mitosis for proteasome-mediated degradation.
Figure 3.
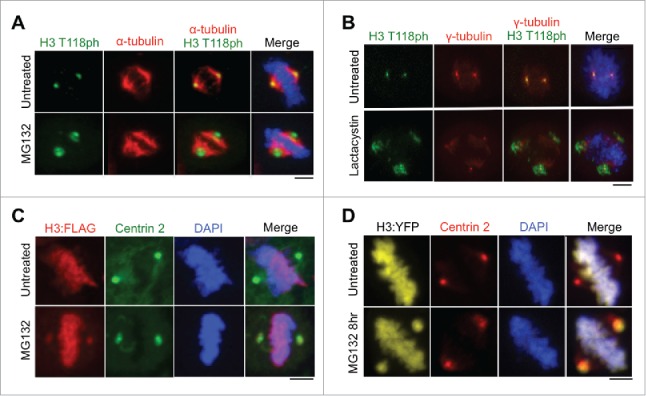
Abundance of histone H3 at the centromere increases upon proteasome inhibition. (A-B) Metaphase HeLa cells untreated or treated with MG132 (A) or lactacystin (B). Scale bars = 10 μm. (C-D) Metaphase 293TR cells stably expressing H3:FLAG (C) or transiently transfected with H3:YFP (D) upon treatment with or without MG132. Scale bars = 10 μm.
A previous report suggested that the histone variant macroH2A.1 localizes to centrosomes,21 but it was subsequently shown that this localization was an artifact of non-specific antibody staining.22 Therefore, it was important for us to validate that histone H3 localizes to centrosomes using a method that was independent of H3 T118ph antibodies. Using a stable cell line expressing H3-FLAG generated using 293TR Flp-in technology (Invitrogen),11 FLAG-tagged H3 was observed at the centrosomes specifically during mitosis, but only upon inhibition of the proteasome by MG132 treatment (Fig. 3C). Additionally, upon proteasome inhibition, histone H3 was detectable at the centrosome during mitosis using transient transfection of a plasmid expressing H3:YFP (Fig. 3D). These data indicate that detection of histone H3 at the centrosomes is not an artifact of non-specific antibody binding.
We were not able to detect endogenous H3 at the centrosomes by immunofluorescence, only upon its overexpression. This is likely due to the much higher abundance of H3 on the chromatin and the efficient degradation of free H3, precluding its detection at centrosomes. However, the fact that overexpression of H3-FLAG and H3-YFP (to about 5% the level of endogenous histones11) allows detection of H3 at the centrosomes upon proteasome inhibition (Fig. 3C, D), indicates that excess free H3 localizes to the centrosomes during mitosis for degradation. Also, because we observed H3 at the centrosomes by immunofluorescence using antibodies to H3 T118ph, H3 T80ph, and FLAG and via intrinsic fluorescence of an YFP tag fused to H3, the centrosomal localization of H3 is not an antibody artifact. These data indicate that excess non DNA-bound histone H3 localizes to the centrosomes during mitosis, to undergo a proteasome-mediated degradation pathway unique to mitosis.
T118ph promotes, but is not essential for, centrosomal localization of H3
Given that we readily detected endogenous histones carrying T118ph at centrosomes, it suggested to us that H3 T118ph may function to promote histone removal from the chromatin during mitosis. In agreement with this idea, H3 T118ph substantially destabilizes mononucleosomes in vitro.23 To determine whether phosphorylation of H3 T118 was required for localization of histones to centrosomes during mitosis, we examined the effect of mutating T118. To do this, we created a panel of stable cell lines using the HEK 293TR Flp-in system (Invitrogen) expressing histone H3:FLAG, H3 T118E:FLAG (to mimic constitutive phosphorylation), H3 T118I:FLAG (to mimic the yeast SIN mutant 24), as well as T118A-FLAG (unmodifiable) from the same locus.11 Structurally, H3 T118I more closely resembles the effect of H3 T118 phosphorylation than H3 T118E because the isoleucine side chain would mimic the rigidity of phosphate group compared to the flexible side chain of glutamic acid. Functionally, H3 T118I caused stronger phenotypes than H3 T118E and that were identical to overexpression of the H3 T118 kinase, Aurora-A.11 The H3:FLAG proteins were expressed at equivalent levels, regardless of the mutation (Fig. 4A).
Figure 4.
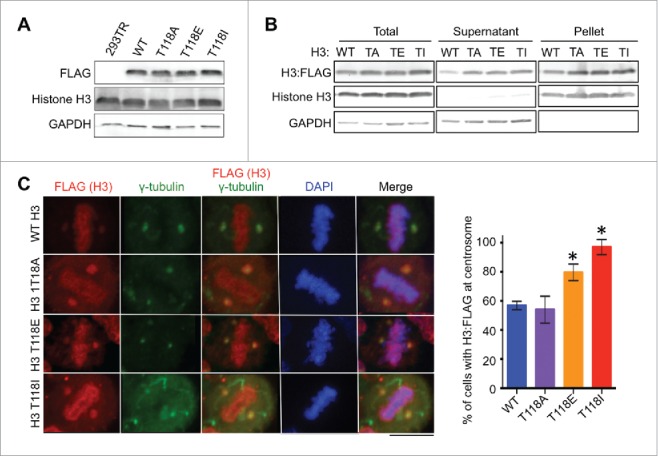
Mutations mimicking phosphorylation of H3 T118 lead to more accumulation of histone H3 at centrosomes. (A) Equal expression of H3:FLAG in stable 293TR cell lines expressing H3:FLAG that was wild type (WT) or with the T118A (TA), T118E (TE) or T118I (TI) substitution. (B) Fractionation showing that the H3 T118 mutants can assemble into chromatin, where “pellet” contains the chromatinized histones and “supernatant” contains the free histones. (C) Metaphase 293TR H3:FLAG cells treated with MG132. Quantitation of images, showing the percentage of cells with H3:FLAG detectable at the centrosomes (n = 50 cells per cell line). Error bars represent SD of the mean (SDM). * significant difference from the wild type, p < 0.05 calculated with Student t-test.
We found that all of the FLAG-tagged histones were incorporated into chromatin regardless of the mutation of H3 T118, indicating that the mutations do not prevent incorporation into nucleosomes (Fig. 4B). Upon proteasome inhibition, FLAG-tagged H3 with T118A, T118E, T118I substitutions were all detectable at the centrosomes (Fig. 4C), indicating that T118ph is not a pre-requisite for the centrosomal localization of histone H3 in mitosis. However, quantitation of the number of cells with FLAG-tagged H3 at centrosomes demonstrated that virtually all mitotic cells expressing H3 T118I have centrosomal histone staining (Fig. 4C), as compared to about half of mitotic cells for wild type histones and for H3 T118A mutants. The histones with the T118E mutation were also significantly more likely to be detected at centrosomes than wild type histones (Fig. 4C). Because these experiments were performed under conditions of proteasome inhibition (with MG132), the significant increase in the centrosomal detection of T118I, T118E and T118ph in comparison to wild type H3 and T118A, is unlikely to be due to potential differences in protein half-life, differences in ubiquitination or differences in rates of degradation. Furthermore, the T118A result indicates that the H3 T118 residue is not essential for localizing histone H3 to the centrosome for degradation. However, these results suggest that H3 T118ph may be more detectable at centrosomes than bulk H3 because this modification enables histones to be more readily removed from chromatin.23 In agreement, we had observed previously that H3 T118I is easier to extract from chromatin than wild type histone H3.11 Indeed, H3 T118I in vitro and in yeast, respectively, has been shown to loosen chromatin25 and to overcome the need for the ATP-dependent chromatin remodeler SWI/SNF.24 Given that H3 T118ph disappears from mitotic chromosomes during metaphase,11 we propose that histones carrying H3 T118ph are removed from the DNA upon formation of appropriate spindle attachments to the centromeres. Functionally, we have shown that the timely loss of H3 T118ph from chromatin in mitosis enables the timely removal of cohesion and condensin I to enable accurate chromosome segregation.11 It will be interesting in the future to determine if the disassembly of H3 T118ph at the centromere also serves to provide DNA binding sites for subsequent CENP-A incorporation after mitosis, given that newly-synthesized CENP-A is incorporated at the start of G1 phase.26 The fate of the H3 T118ph histones removed from chromatin in mitosis and the fate of overexpressed histones that may never have been assembled into chromatin appears to be the same: i.e. proteasomal degradation at the centrosomes during mitosis.
Ubiquitinated H3 is found in centrosomes purified from mitotic cells
Histone H3 can be ubiquitinated and degraded in response to UV damage in metazoans and during S-phase in yeast.7,10 To investigate if degradation of H3 at the centrosomes in mitosis is accompanied by its ubiquitination, we asked if histone H3 is ubiquitinated in mitotic HeLa cells. Western blotting of whole cell extracts revealed bands at positions that corresponded to mono and di-ubiquitinated histone H3 only in the cells that were mitotically arrested and treated with the proteasome inhibitor (Fig. 5A). To unequivocally determine whether the size shift of H3 in mitosis upon addition of MG132 was due to addition of ubiquitin moieties, we used antibodies to ubiquitin. When we performed anti-ubiquitin immunoprecipitation (using the FK2 antibody 27) followed by western blotting for H3, a band the size of mono ubiquitinated H3 was apparent (Fig. 5B). Conversely, when we performed anti-H3 immunoprecipitation, followed by protein gel blotting for ubiquitin (using the FK2 antibody), bands with the size of mono and di ubiquitinated H3 were apparent (Fig. 5C). Additional ubiquitinated proteins, some even more abundant than H3, presumably represent ubiquitinated proteins that co-immunoprecipitate with histone H3 (Fig. 5C). Taken together, these results demonstrate that inhibition of the proteasome leads to detectable levels of ubiquitinated H3 during mitosis.
Figure 5.
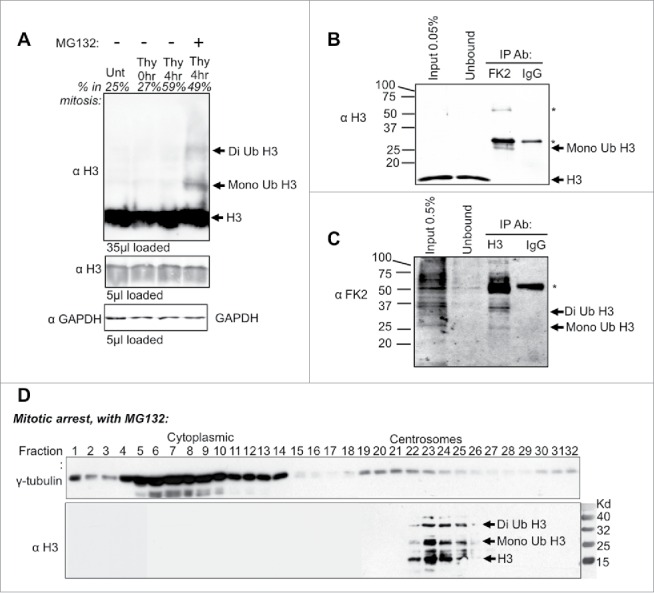
Histone H3 ubiquitin shift detected during mitosis and in purified centrosomes. (A) HeLa cell extracts were harvested at the indicated time points following release from a double thymidine block with or without MG132. Percent in mitosis is indicated above each lane, as determined by flow cytometry analysis. (B) Western analysis of anti-ubiquitin (FK2 antibody) immunoprecipitation from HeLa cells that were double synchronized with Thymidine and released into MG132 for 8 hrs. Immunoblot was probed with histone H3 antibody. Heavy and light chain is marked by “∗.” (C) Western analysis of anti-histone H3 Immunoprecipitation from HeLa cells that were treated as in B. Immunoblots were probed with anti-ubiquitin (FK2 antibody). The heavy chain is marked by “∗.” (D) Velocity sedimentation of HeLa cell extracts upon double thymidine arrested cells released into MG132. Extracts were separated on a 15–60% sucrose gradient.
To gain biochemical evidence for the centrosomal localization of H3 and potentially also ubiquitinated H3 at centrosomes, we fractionated mitotic centrosomes. To do this, we performed velocity sedimentation of MG132-treated HeLa extracts from mitotic cells.28 The H3-positive fractions co-sedimented with γ-tubulin-positive centrosomal fractions. Interestingly, above the H3 protein band, there are larger sized H3-positive bands spaced approximately 9 kD apart (Fig. 5C). This data is consistent with ubiquitinated histone H3 being present at the centrosomes. Taken together, these data indicate that non-chromatin bound histone H3 localizes to centrosomes for ubiquitination and degradation during mitosis, representing a novel mechanism for the removal of excess histone proteins from mammalian cells.
In summary, we have uncovered multiple lines of evidence to demonstrate that excess core histones localize to the centrosome, specifically during mitosis, to be degraded (Fig. 6). Furthermore, we provide the first solid evidence for histone localization to the centrosomes and for histone ubiquitination during mitosis. In agreement, histones have been discovered at the centrosome by mass spectrometry previously, although this was along with hundreds of other proteins.29 Additionally, our data indicates that free histone H3 at the centrosome is degraded in a proteasome-mediated pathway specifically during mitosis. By analogy, in yeast excess free histones that remain after DNA replication are degraded by a phosphorylation, ubiquitination and a proteasome-dependent pathway.7 However, in mammalian cells excess H3-H4 has been reported to be degraded by chaperone-mediated autophagy.8 Given that the proteasome inhibitor MG132 is known to increase autophagy,30 the pathway of degradation of histones that we have uncovered is distinct from autophagy-mediated degradation. It is noteworthy that we did not observe polyubiquitination of histone H3 during mitosis, given that a special form of proteasome containing the proteasome activator PA200 was shown to degrade histones in a polyubiquitination-independent manner during spermatogenesis and during DNA repair.31 It will be interesting in the future to determine if PA200 is also involved in the mitotic-specific degradation of histone at the centrosomes. Clearly, mammalian cells have evolved distinct pathways to regulate proper histone levels during different phases of the cell cycle and in response to different stresses, including a novel proteasome-dependent degradation pathway at centrosomes during mitosis.
Figure 6.
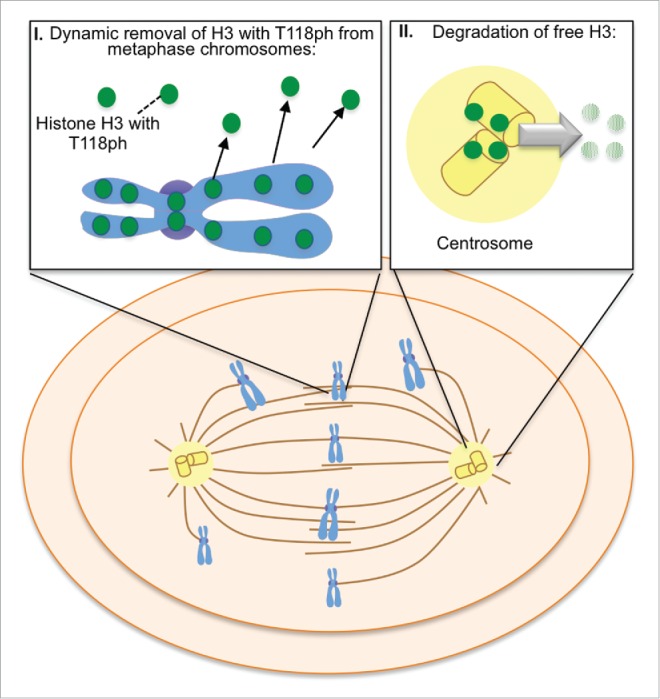
Model of proteasome-mediated histone H3 degradation of histones carrying H3 T118 phosphorylation at centrosomes during mitosis, as explained in the text.
Material and methods
Cell culture and plasmids
All cell lines were maintained and were cultured as described previously.11 Information describing plasmids and the site-directed mutagenesis has been previously published.11
Drug treatments
For proteasome inhibitors, cells were synchronized with 4 mM thymidine (Sigma-Aldrich, T1895, PBS) for 18 hour and then released into fresh medium for 8–10 hours, followed by incubation in 4 mM thymidine enriched medium for 18 hours. Cell were then released into fresh MG132 (Calbiochem, Billerica, MA USA, 474790-1MG, in ETOH) for 6 hour, or into Lactacystin (10μM, Enzo Life sciences, Farmingdale, NY USA, ALX-350-245-MC01 in DMSO) for 6 hours. PLK-1 was inhibited in HeLa cells with BI-2536 (100 nM, Selleck chemicals) for 3 hours.
C. elegans and RNAi mediated interference
Wild type N2 Bristol C. elegans were grown and maintained at 20°C as described.33 The feeding method of RNAi delivery was used to deplete PLK-1, as previously described by Timmons and Fire.34 RNAi plasmids for PLK-1 were obtained from the Geneservice Ltd. C. elegans feeding library.35 E. coli HT115 (DE3) bacteria was transformed with the control or PLK-1 RNAi plasmids. Plating of RNAi and feeding RNAi was described previously.36 The L4440 RNAi vector was used as an RNAi control.
Antibodies
The following primary antibodies were used: polyclonal H3 T118ph (Abcam Cambridge, MA USA ab33310), CENP-A (Abcam, ab8245), α-tubulin (Sigma-Aldrich, T9026), γ-tubulin (Abcam, ab27074), C-terminal H3 (Abcam, ab1791), Centrin 2 (Cell Signaling, Danvers, MA, USA, #2091), M2-FLAG (Sigma St. Louis, MO USA, F3165), GAPDH (Abcam, ab8245). The secondary antibodies used were Alexa Fluor® 488 goat anti-rabbit (Life Technologies, A11034) and Alexa Fluor® 488 goat anti-mouse (A11029).
C. elegans immunostaining
Embryos from adult hermaphrodites were performed as previously described.37 Rabbit anti-H3 T118ph antibody was used at 1:500 and mouse anti-α Tubulin was used at 1:2000.
Immunofluorescence in Drosophila embryos
Embryo stains were performed as previously described on yw embryos.38 Rabbit anti-H3 T118ph antibody was used at 1:500 and rat anti-α Tubulin was used at 1:100 (AbD Serotec #MCA78G).
Tissue culture immunofluorescence
Cells were grown on Poly-D-lysine-coated coverslips for at least 16 h and subsequently washed, fixed with (4% PFA or 100% ice cold methanol), permeabilized with (0.1% Triton-X-100/1xPBS), blocked (3% BSA/1xPBS) and incubated with primary and secondary antibodies.
Immunogold label scanning electron microscopy
HeLa cells were grown on a 12 well plate (Life sciences 3513) to confluency and treated 3 hours with Colcemid (0.01 µg/mL, Roche 10295892001) prior to collection. The immunofluorescence protocol was followed above except secondary was replaced with 10 nm gold anti-rabbit IgG (Ted Pella Inc., Redding, CA USA, 15726-1). Once labeled cells were fixed in 2% glutaraldehyde. Fixed samples were washed in 0.1 M sodium cacodylate buffer, post-fixed with 1% buffered osmium tetroxide for 30 min, washed in distilled water and stained en bloc with 1% Millipore-filtered uranyl acetate for 30 mins. After washing in distilled water, the samples were dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in LX-112 medium. The samples were polymerized in a 60 C oven for 2 d. Ultrathin sections were cut in a Leica Ultracut microtome (Leica, Deerfield, IL), stained with uranyl acetate and lead citrate in a Leica EM Stainer, and examined in a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA) at an accelerating voltage of 80 kV. Digital images were obtained using AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA).
Acquisition of images
The images were acquired on a 3i Marianas Spinning Disk Confocal equipped with a coolSNAP HQ2 CCD Camera. Slidebook 5.5 software was used with a 63 × 1.49 NA Plan Apo oil immersion objective and Z sections were acquired at 0.2 um steps.
Isolation of pellet and supernatant fractions
Two D150 plates, at 80% confluency, were collected by mitotic shake off. Cells were pelleted and washed in TB buffer (20 mM Hepes, pH 7.3, 110 nM K-acetate, 5 mM Na-acetate, 2 mM Mg-acetate, 1 mM EGTA, 2 mM DTT and a protease inhibitor cocktail (Roche, Complete-mini, cat#1187350001). All steps were done at 4°C. NP40. Extraction of detergent soluble proteins was performed by treatment with 0.1% NP40 for 5 mins, followed by centrifugation at 3000 rpm for 3 mins to separate the non-chromatin supernatant and chromatin pellet fractions. The pellet fractions were subsequently digested with 20 µg/ml DNaseI (Worthington Biochemical Corporation, Lakewood, NJ USA, LS006342) for 10 mins at 37°C. Total, supernatant (non-chromatin), and pellet (chromatin) fractions were resolved by SDS-PAGE and analyzed by western blotting.
Whole cell extracts
Generation and analysis of whole cell extracts were performed as previously described.11
Western blot analysis
Samples were resolved by 15% SDS-PAGE and transferred to nitrocellulose according to standard procedures. The Centrosome isolated samples were ran on a 4–20% gradient gel (Biorad Hercules, CA USA, Cat#567-1094) according to standard procedures For LICOR Odyssey detection the transfer blots were blocked in Sea Block buffer (Thermo Scientific, Cat#37527) in 1 x PBS for 1 hr. Blots were incubated with primary and secondary antibodies as described above. An Odyssey imager was used to analyze and quantitate bands. Specifically for the immunoprecipitation protein gel blots, the secondary antibodies used for detection for rabbit primary was Protein A-HRP (Biorad, 1706522) and for mouse primary antibodies was Protein G-HRP to minimize recognition of the antibody heavy and light chains (Biorad, 1706425). ECL detection used Amersham ECL Western Blotting Detection Reagents (GE, Pittsburgh, PA USA, RPN2106). Alpha viewer was used to analyze the gels (Proteinsimple, Santa Clara, CA, USA).
Immunoprecipitation
Cells were treated as described above in the “drug treatments” section. Cells were collected and resupended in denaturing buffer (20 mM tris pH 7.5, 50 mM NaCl, 0.5% NP-40, 0.5% NA-Deoxycholate, 0.5% SDS, 1mMEDTA) containing protease inhibitors. Samples were briefly sonicated to reduce the viscosity. Samples were centrifuged at high speed for 10mins. The supernatant was incubated with beads pre-blocked protein-A–Dynabeads (Thermo-Fischer, 10001D) overnight at 4°C. The antibody was added and the samples were incubated for 4hrs. Bead-bound protein complexes were extensively washed in RIPA buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8, 10 mM NaF, 0.4 mM EDTA, 10% glycerol and protease inhibitors) and were analyzed by western blotting.
Centrosome fractionation
HeLa cells were synchronized at the G1/S phase boundary by double thymidine block followed by arrest in MG132 to increase the amount of histones at the centrosome. Cells were treated with 4 mM thymidine (Sigma-Aldrich, T1895, PBS) for 18 hr and then released into fresh medium for 8–10 hr, followed by incubation in 4 mM thymidine enriched medium for 18 h. Cell were then released into fresh MG132 (20 µM) for 6 hrs. Cells were collected by trypsinization and spun down. The centrosome isolation was then performed using previously published methods.44
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are extremely grateful to Allan Tackett and Stephanie Byrum for assistance with our search for T118ph-binding proteins. We thank Zhenkun Lou for plasmids. We thank Henry Adams for his assistance with microscopy and analysis. We thank Kenneth Dunner Jr (High Resolution Electron Microscopy Facility, UTMDACC, funded by Institutional Core Grant #CA16672) for help with the SEM. We thank Andrew Davis for technical assistance and Jason Ford, Jessica Orbeta, and Tokiko Furuta for help with C. elegans experiments. We appreciate the continued support of Bradley Cairns during the review process. This research was performed with the help of the MD Anderson Cancer Center Flow Cytometry & Cellular Imaging Facility.
Author contributions
JT guided the overall project. JG guided AW on the centrosome purification aspect of the work. JS guided the worm work. CLW devised the experiments and did the majority of the experiments, with some experimental assistance from HG and RH. The manuscript was written by CLW and JKT.
Funding
This work was supported by NIH grant RO1GM64475 and a CPRIT rising star award to JKT.
References
- [1].Gurard-Levin ZA, Quivy JP, Almouzni G. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem 2014; 83:487-517; PMID:24905786; http://dx.doi.org/ 10.1146/annurev-biochem-060713-035536 [DOI] [PubMed] [Google Scholar]
- [2].Dahlin JL, Chen X, Walters MA, Zhang Z. Histone-modifying enzymes, histone modifications and histone chaperones in nucleosome assembly: Lessons learned from Rtt109 histone acetyltransferases. Crit Rev Biochem Mol Biol 2015; 50:31-53; PMID:25365782; http://dx.doi.org/ 10.3109/10409238.2014.978975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dunleavy EM, Almouzni G, Karpen GH. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus 2011; 2:146-57; PMID:21738837; http://dx.doi.org/ 10.4161/nucl.2.2.15211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].O'Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 2010; 17:1218-25; PMID:20890289; http://dx.doi.org/ 10.1038/nsmb.1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. Elevated histone expression promotes life span extension. Mol Cell 2010; 39:724-35; PMID:20832724; http://dx.doi.org/ 10.1016/j.molcel.2010.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gunjan A, Verreault A. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 2003; 115:537-49; PMID:14651846; http://dx.doi.org/ 10.1016/S0092-8674(03)00896-1 [DOI] [PubMed] [Google Scholar]
- [7].Singh RK, Kabbaj MH, Paik J, Gunjan A. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 2009; 11:925-33; PMID:19578373; http://dx.doi.org/ 10.1038/ncb1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cook AJ, Gurard-Levin ZA, Vassias I, Almouzni G. A specific function for the histone chaperone NASP to fine-tune a reservoir of soluble H3-H4 in the histone supply chain. Mol Cell 2011; 44:918-27; PMID:22195965; http://dx.doi.org/ 10.1016/j.molcel.2011.11.021 [DOI] [PubMed] [Google Scholar]
- [9].Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. The human and mouse replication-dependent histone genes. Genomics 2002; 80:487-98; PMID:12408966; http://dx.doi.org/ 10.1006/geno.2002.6850 [DOI] [PubMed] [Google Scholar]
- [10].Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell 2006; 22:383-94; PMID:16678110; http://dx.doi.org/ 10.1016/j.molcel.2006.03.035 [DOI] [PubMed] [Google Scholar]
- [11].Wike CL, Graves HK, Hawkins R, Gibson MD, Ferdinand MB, Zhang T, Chen Z, Hudson DF, Ottesen JJ, Poirier MG, et al.. Aurora-A mediated histone H3 phosphorylation of threonine 118 controls condensin I and cohesin occupancy in mitosis. Elife 2016; 5:e11402; PMID:26878753; http://dx.doi.org/ 10.7554/eLife.11402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hammond SL, Byrum SD, Namjoshi S, Graves HK, Dennehey BK, Tackett AJ, Tyler JK. Mitotic phosphorylation of histone H3 threonine 80. Cell Cycle 2014; 13:440-52; PMID:24275038; http://dx.doi.org/ 10.4161/cc.27269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Doxsey SJ, Stein P, Evans L, Calarco PD, Kirschner M. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 1994; 76:639-50; PMID:8124707; http://dx.doi.org/ 10.1016/0092-8674(94)90504-5 [DOI] [PubMed] [Google Scholar]
- [14].Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol 1999; 145:481-90; PMID:10225950; http://dx.doi.org/ 10.1083/jcb.145.3.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schatten G, Palazzo RE, ScienceDirect (Online service) . The centrosome in cell replication and early development. San Diego; London: Academic Press, 2000. [Google Scholar]
- [16].Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 1996; 135:1701-13; PMID:8991084; http://dx.doi.org/ 10.1083/jcb.135.6.1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee DH, Goldberg AL. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol Cell Biol 1998; 18:30-8; PMID:9418850; http://dx.doi.org/ 10.1128/MCB.18.1.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995; 268:726-31; PMID:7732382; http://dx.doi.org/ 10.1126/science.7732382 [DOI] [PubMed] [Google Scholar]
- [19].Chadwick BP, Willard HF. Cell cycle-dependent localization of macroH2A in chromatin of the inactive X chromosome. J Cell Biol 2002; 157:1113-23; PMID:12082075; http://dx.doi.org/ 10.1083/jcb.200112074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Friedman N, Barzily-Rokni M, Isaac S, Eden A. The histone H2A variant macroH2A1 does not localize to the centrosome. PLoS One 2011; 6:e17262; PMID:21364955; http://dx.doi.org/ 10.1371/journal.pone.0017262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].North JA, Simon M, Ferdinand MB, Shoffner MA, Picking JW, Howard CJ, Mooney AM, van Noort J, Poirier MG, Ottesen JJ. Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure. Nucleic Acids Res 2014; 42:4922-33; PMID:24561803; http://dx.doi.org/ 10.1093/nar/gku150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kruger W, Peterson CL, Sil A, Coburn C, Arents G, Moudrianakis EN, Herskowitz I. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev 1995; 9:2770-9; PMID:7590252; http://dx.doi.org/ 10.1101/gad.9.22.2770 [DOI] [PubMed] [Google Scholar]
- [23].Muthurajan UM, Bao Y, Forsberg LJ, Edayathumangalam RS, Dyer PN, White CL, Luger K. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J 2004; 23:260-71; PMID:14739929; http://dx.doi.org/ 10.1038/sj.emboj.7600046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 2009; 137:485-97; PMID:19410545; http://dx.doi.org/ 10.1016/j.cell.2009.02.040 [DOI] [PubMed] [Google Scholar]
- [25].Fujimuro M, Yokosawa H. Production of antipolyubiquitin monoclonal antibodies and their use for characterization and isolation of polyubiquitinated proteins. Methods Enzymol 2005; 399:75-86; PMID:16338350; http://dx.doi.org/ 10.1016/S0076-6879(05)99006-X [DOI] [PubMed] [Google Scholar]
- [26].Meigs TE, Kaplan DD. Isolation of centrosomes from cultured Mammalian cells. CSH Protoc 2008; 2008:pdb prot5039; PMID:21356896 [DOI] [PubMed] [Google Scholar]
- [27].Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003; 426:570-4; PMID:14654843; http://dx.doi.org/ 10.1038/nature02166 [DOI] [PubMed] [Google Scholar]
- [28].Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513-24; PMID:17620365; http://dx.doi.org/ 10.2353/ajpath.2007.070188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Qian MX, Pang Y, Liu CH, Haratake K, Du BY, Ji DY, Wang GF, Zhu QQ, Song W, Yu Y, et al.. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013; 153:1012-24; PMID:23706739; http://dx.doi.org/ 10.1016/j.cell.2013.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974; 77:71-94; PMID:4366476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Timmons L, Fire A. Specific interference by ingested dsRNA. Nature 1998; 395:854; PMID:9804418; http://dx.doi.org/ 10.1038/27579 [DOI] [PubMed] [Google Scholar]
- [32].Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al.. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003; 421:231-7; PMID:12529635; http://dx.doi.org/ 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- [33].Arur S, Ohmachi M, Nayak S, Hayes M, Miranda A, Hay A, Golden A, Schedl T. Multiple ERK substrates execute single biological processes in Caenorhabditis elegans germ-line development. Proc Natl Acad Sci U S A 2009; 106:4776-81; PMID:19264959; http://dx.doi.org/ 10.1073/pnas.0812285106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Severson HH, Andrews JA, Lichtenstein E, Gordon JS, Barckley M, Akers L. A self-help cessation program for smokeless tobacco users: comparison of two interventions. Nicotine Tobacco Res 2000; 2:363-70; http://dx.doi.org/ 10.1080/713688152 [DOI] [PubMed] [Google Scholar]
- [35].Gunesdogan U, Jackle H, Herzig A. A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO Rep 2010; 11:772-6; PMID:20814422; http://dx.doi.org/ 10.1038/embor.2010.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gopalakrishnan J, Mennella V, Blachon S, Zhai B, Smith AH, Megraw TL, Nicastro D, Gygi SP, Agard DA, Avidor-Reiss T. Sas-4 provides a scaffold for cytoplasmic complexes and tethers them in a centrosome. Nat Communications 2011; 2:359; http://dx.doi.org/ 10.1038/ncomms1367 [DOI] [PMC free article] [PubMed] [Google Scholar]