

abstract
Epithelial tissues use adherens junctions to maintain tight interactions and coordinate cellular activities. Adherens junctions are remodeled during epithelial morphogenesis, including instances of epithelial-mesenchymal transition, or EMT, wherein individual cells detach from the tissue and migrate as individual cells. EMT has been recapitulated by growth factor induction of epithelial scattering in cell culture. In culture systems, cells undergo a highly reproducible series of cell morphology changes, most notably cell spreading followed by cellular compaction and cell migration. These morphology changes are accompanied by striking actin rearrangements. The current evidence suggests that global changes in actomyosin-based cellular contractility, first a loss of contractility during spreading and its activation during cell compaction, are the main drivers of epithelial scattering. In this review, we focus on how spreading and contractility might be controlled during epithelial scattering. While we propose a central role for RhoA, which is well known to control cellular contractility in multiple systems and whose role in epithelial scattering is well accepted, we suggest potential roles for additional cellular systems whose role in epithelial cell biology has been less well documented. In particular, we propose critical roles for vesicle recycling, calcium channels, and calcium-dependent kinases.
KEYWORDS: actin, calcium, calmodulin, epithelial-mesenchymal transition, myosin, rhoA, TRP channels
Introduction
Epithelial barriers require that individual epithelial cells are tightly integrated into the tissue. Integration of individual cells occurs through cell-cell adhesions systems, most notably the cadherins.1 The physical connection of cells provides the foundation for coordinating physiological processes of individual cells and a fully functional tissue is the result. However, the high degree of coordination required by epithelial barriers must be balanced to allow to tissue plasticity. A host of epithelial morphogenetic programs that remodel epithelial tissue architectures are observed throughout development. Among the most striking are epithelial- mesenchymal transitions (EMTs), which occur when individual cells detach from the tissue and become solitary, migratory cells.2 EMT events are thought to be highly relevant to cancer progression in solid tumors of epithelial origin, particularly cancer metastasis. Cancer metastasis is thought to result from inappropriate activation of developmental EMT pathways, resulting in detachment of solitary, migratory, and invasive tumor cells that then colonize distant tissues.
EMT is a highly complex cellular process and much can be learned from cell model systems. In tissue culture, epithelial cells can be induced to undergo epithelial scattering, a process akin to EMT. Epithelial scattering was first observed in cultured epithelial MDCK cells treated with so-called scatter factor,3 a growth factor that later turned out to be hepatocyte growth factor (HGF).4-7 HGF stimulation of MDCK cells results in a dramatic change in cell morphology and cell behavior that ultimately results in cells wrenching apart their cell-cell adhesions in dramatic fashion and ‘scattering’ about the culture surface.3,8,9 Cellular imaging, summarized in Figure 1, has shown that scattering is accompanied by a transient cell spreading phase, with cells increasing spreading on the culture surface by roughly double. The spreading phase is immediately followed by compaction of cells as spreading is reduced, an increase in cell migration, and disruption of cell-cell adhesions.8,9
Figure 1.
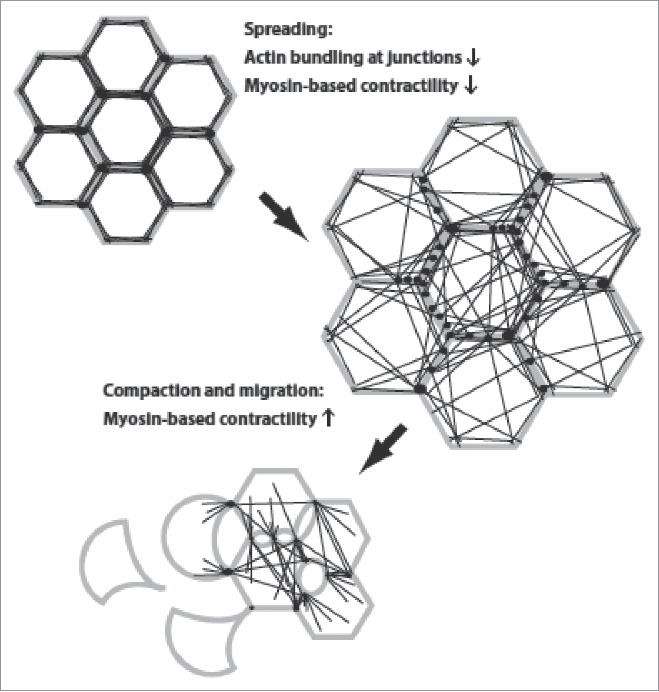
Changes in cell morphology and actin organization during epithelial scattering. Cells, with plasma membrane in gray, points of adhesion-actin connection as black spots, and actin cables as black lines, undergo a cell spreading phase that is accompanied by rearrangement of the actin cytoskeleton from a circumferential, highly bundled actin organization into transcellular networks. Spread colonies of cells then undergo subsequent actomyosin-based contractility that ruptures cell-cell adhesions and allows detachment of individual cells.
The underlying actin dynamics during epithelial scattering are equally striking. Cortical actin bundles that run parallel and immediately adjacent to cell-cell junctions resolve into smaller cables whose ends remain affixed to points of cell-cell adhesion and which now radiate across the cell.9 The reorganization of actin at cadherin-mediated junctions is preceded by redistribution of the actin bundling protein α–actinin from cell-cell junctions, perhaps suggesting that actin bundling proteins maintain a tight association of actin with cell-cell junctions and that this activity must be released to initiate cell scattering.9 The resulting ‘transcellular networks’ are reminiscent of structures seen during epithelial morphogenesis in developmental systems.10 Also similar is evidence of cellular contractility in transcellular networks. Actin becomes highly decorated with non-muscle myosin II during the reorganization of actin into transcellular networks and obvious deformations are visible at points where these cables abut cell-cell junctions.9 It is also at this point where cell spreading reverses and cells revert to having a smaller ‘footprint’ on the culture surface. Consistent with a role for myosin contractility in this process, changes in myosin phosphorylation are observed during epithelial scattering of MDCK cells; phosphorylation is reduced in early scattering, when spreading is observed, and increases in late scattering, when cell compaction, migration, and detachment occur.11
One dominant idea is that tension forces, coming from an increase in cell migration, are sufficient to break cell-cell junctions.12 An alternative view is that molecular changes in cell-cell adhesion systems is a key step in cell-cell detachment, that the molecular linkages of cell-cell adhesion are actively remodeled to allow cell-cell detachment. Though originally adherents to the latter view, our own observation suggest that cellular contractility is the main force behind cell-cell detachment; MDCK cells recovering from blebbistatin treatment, and thus experiencing a restoration in cellular contractility, wrench apart their cell-cell junctions in a manner that looks highly reminiscent of the same cells responding to HGF, but without an increase in migration.11 Interestingly, blebbistatin treatment drives cell spreading as myosin activity becomes suppressed in these experiments. Thus, restoration of myosin contractility occurs from a spread phase, suggesting that both spreading and compaction might be required for successful epithelial scattering. Consistent with this idea, dynamic changes in cell morphology accompanying HGF-induced epithelial scattering correlate with phosphomyosin levels; the cell spreading phase of epithelial scattering is accompanied by a drop in phosphomyosin levels, whereas times associated with cellular contractility display an increase in phosphomyosin levels.11 Thus, control of cellular contractility is likely a central mechanism for driving epithelial scattering. This review will focus on how molecular control of cellular mechanics, particularly spreading and compaction, during epithelial scattering.
The central regulator of cellular contractility: RhoA
The small GTPase RhoA is well documented to play a central role in controlling cellular contractility and actin dynamics in many cellular processes.13-14 Epithelial scattering appears to be no exception; expression of a dominant-negative RhoA mutant prevents HGF-induced scattering, in particular preventing cell-cell detachment that is associated with increased cellular contractility.11 RhoA is best known as a master regulator of cellular contractility, activating myosin-based tension forces in cells. ROCK is a downstream effector of Rho most known for its role in regulating cellular contractility in smooth muscle contraction. RhoA induces increased cellular contractility through 2 parallel mechanisms: ROCK phosphorylates and inactivates myosin light chain (MLC) phosphatase,15 while also directly phosphorylating MLC,16-18 thus synergistically increasing MLC phosphorylation and myosin activation. Recent evidence shows that myosin light chain kinase (MLCK) may also be a direct substrate for ROCK, which would cause additional myosin phosphorylation on RhoA activation.19 It is likely that cellular contractility is the central RhoA function required during epithelial scattering and that is blocked to prevent cell-cell detachment when MDCK cells expressing a dominant-negative RhoA mutant are triggered to scatter with HGF.11
How RhoA alters actin dynamics has been well documented.20-22 RhoA can drive changes in actin dynamics through mDia and cofilin, as well as profilin II. Mammalian diaphanous (mDia) is a formin protein that is activated upon Rho binding, resulting in nucleation of new actin filaments and polymerization that generates unbranched filaments, which is important in the formation of stress fiber contractile units.23-25 Active RhoA recruits profilin, which binds G-actin to facilitate F-actin barbed end polymerization.26 Phosphorylation of cofilin by LIM kinase results in a loss of cofilin activity and stabilization of actin filaments, which is the end result of RhoA-mediated activation of ROCK and, subsequently, of LIM kinase, the so-called RhoA-ROCK-LIM kinase-cofilin pathway.27
While RhoA almost certainly drives epithelial scattering by modulating actomyosin-based contractility, how might RhoA activity levels be controlled? Suppression of RhoA activity during the spreading phase of epithelial scattering may come from early activation of Rac1, another small GTPase that often acts antagonistically to RhoA. Consistent with this, Rac1 controls spreading during epithelial scattering.28 Rac1 is transiently activated shortly after HGF stimulation of epithelial scattering, resulting in membrane protrusion and spreading, and expression of dominant negative Rac1 blocks spreading. A transient activation of Rac1 may also lower RhoA activity and, thus, myosin-based contractility. Meanwhile, obvious candidates for RhoA activation later in epithelial scattering are Rho GEFs and GAPs that directly affect the nucleotide binding state of RhoA, but how these are controlled is less clear. In the next sections, we propose a mechanism by which RhoA activity can be modulated by intracellular calcium levels.
The case for intracellular calcium
In 1992, a report showed HGF induces a rapid increase in cytosolic free calcium in hepatocytes.29 This increase in intracellular calcium is dependent on the presence of extracellular calcium, as well as tyrosine kinase activity.29 Another group confirmed that after HGF stimulation, intracellular calcium waves originate from a region near the cell membrane and propagated across the cell.30 Electrophysiological recording of MDCK cells treated with hepatocyte growth factor reveals 2 bursts of high frequency calcium influxes at the plasma membrane.31 In MDA-MB-468 cells, which undergo epithelial scattering in response to epidermal growth factor (EGF), cytosolic calcium levels quickly rise after EGF stimulation, then gradually level off in under 800 seconds.32 This transient pulse results in a prolonged increase in vimentin expression,32 indicating that transient calcium increases have long-lasting impacts on cell behavior. Treatment of cells with cytosolic calcium chelator BAPTA-AM prior to EGF stimulation prevents a rise in vimentin expression,32 showing the importance of intracellular calcium during EMT. EMT in embryonic development also seems to depend on a rise in cytosolic calcium levels. In Xenopus embryos, Wnt11-R expression in the neural crest and certain somite cell populations is essential for EMT, allowing the cells to migrate into the dorsal fin matrix. Loss of Wnt11-R results in dorsal fin defects and fewer fin core cells.33 Inducing calcium signaling with thapsigargin, which stimulates calcium influxes, alleviates these defects, and increases fin core cells to near-control levels,33 suggesting that calcium channels mediate Wnt-induced EMT in this developmental model.
Transient receptor potential (TRP) channels are permeable to cations, and some are highly Ca2+ selective.34 Various TRP channels are involved in cancer progression that recapitulates EMT. TRPC6 has been shown to have a role in HGF-induced cellular processes. HGF normally stimulates proliferation of adenocarcinoma cells, but knocking down TRPC6 abolishes this proliferation.35,36 TRPC6 plays the same role in prostate cancer cells, allowing an HGF-induced calcium influx.36 TRPC6 inhibitors prevent HGF-induced proliferation, migration, and morphogenesis of HK2 cells.37 Additionally, Langford et al.31 concluded that TRPC6 is specifically required for HGF-induced scattering in MDCK cells, though calcium fluxes through TRPV4 and other channels are all increased at the plasma membrane following HGF stimulation.
While these data point to TRPC6 in epithelial scattering, the identity of the specific TRP family member may not be critical. Expressing TRP channel family member polycystin-1 (PC1) in MDCK cells leads to cytoskeletal rearrangements, increased cell migration, and scattering.38 In HepG2 cells, HGF stimulation causes calcium influxes through TRPV1 at both plasma membrane and sarcoplasmic reticulum.39 In MDA-MB-468 cells, EGF induces a calcium influx resulting in an increase in the EMT marker, vimentin. However, knocking down TRPM7 in these cells decreases vimentin levels after EGF stimulation.32 In BTEC cells, activation of TRPV4 channels with its agonist, 4αPDD, promotes cell migration, while silencing TRPV4 abolishes arachidonic acid-induced migration in these same cells.40
Despite that the association between changes in intracellular calcium levels during EMT has been documented in a number of studies, a role for calcium influxes has seldom been incorporated into broader molecular models of EMT. This is partially due to an unclear connection between cellular signaling that initiates epithelial scattering and increased calcium fluxes through calcium channels.
Molecular control of calcium influxes upon induction of epithelial scattering
A key finding of Langford et al.31 is that HGF-induced calcium fluxes recorded at the plasma membrane require an intact microtubule cytoskeleton; pretreatment of cells with microtubule destabilizing agents prevents bursts of high frequency calcium influxes that normally follow HGF stimulation.31 One obvious interpretation of this result is that microtubule-dependent delivery of vesicles to the plasma membrane is required for the occurrence of high frequency calcium influxes seen after HGF stimulation. This interpretation is further supported by the observation that calcium influxes increase simultaneously for all the 3 distinct channel types detected at the plasma membrane by electrophysiological recording.31 This is most easily accounted for by different channel types being cargoes in the same vesicle populations, rather than by different channels sharing the same mechanism of post-translational control at the plasma membrane. Further supporting this argument, EGF has been shown to induce translocation of TRPC5 channels to the cell surface, an event that is mediated by PI-3 kinase.41 EGF also induces Src family tyrosine kinase-mediated phosphorylation of TRPC4, leading to increased translocation and surface expression, as well as calcium influx.42 And changes in surface levels of TRP channels in response to cellular cues are not limited to growth factors that bind receptor tyrosine kinase systems; arachidonic acid stimulation causes an increase of TRPV4 at the cell surface.40 In our opinion, control of calcium channel surface expression through altered vesicle trafficking is likely the basis of how cellular signaling networks increase intracellular calcium during EMT.
Rab GTPases, which aid in cargo sorting, uncoating, motility, and docking of vesicles,43 are almost certain to play a central role in TRP channel trafficking. However, connections between TRP channels and specific rab GTPases have not been extensively studied, particularly in epithelial cells. One report shows that activation of Rab11in epithelial cells leads to an increase in TRPV5 and TRPV6 expression at the cell membrane.44 Rab11 interacts with TRPV5 and TRPV6 while in its GDP-bound form, yet the GTP-bound form is necessary to recycle these channels to the cell membrane.44 In a separate series of studies, Rab11, Rab8, and Rab6 have been shown to specifically immunoprecipitate with the polycystin-1 TRP channel.45 Importantly, silencing Rab8 prevents trafficking of polycystin-1 to the cell surface.45 Rab6 controls the budding and motility of exocytic vesicles,46 and mediates the recruitment of Rab8, which controls docking and fusion.47 Thus Rab6 and 8 may act in concert to deliver newly synthesized TRP channels to the plasma membrane from the Golgi. However, given how quickly calcium fluxes increase at the plasma membrane following HGF stimulation,31 channel recycling better accounts for the timing of high frequency plasma membrane calcium fluxes than delivery of newly synthesized TRP channels. Two routes could carry internalized TRP channels back to the plasma membrane: ‘fast’ endocytic recycling pathway, wherein internalized TRP channels would return immediately to the cell surface in vesicles decorated with Rab4, or ‘slow’ endocytic recycling pathway, wherein TRP channels are retained as an internal pool in the endocytic recycling compartment (ERC). Consistent with the latter, Rab11 is a marker for the endocytic recycling compartment.48,49
In recycling from the ERC, Rab8 transports vesicles and tubules from the ERC to the cell membrane.48 Rab11 stimulates Rab8-mediated trafficking through the Rab8 guanine-nucleotide exchange factor, Rabin8. Rabin8 has been shown to release GDP from Rab8 at a much higher rate in the presence of Rab11.50 In lumenal cells, Rab11, Rabin8, and Rab8 colocalize when Rab11 is active.51 How might cells responding to cellular cues that trigger epithelial scattering trigger a sudden change in vesicle trafficking of TRP channels? A recent study shows Rab8 activation following EGF stimulation of cells is mediated by ERK-dependent phosphorylation of Rabin8; ERK-mediated phosphorylation releases Rabin8 from an autoinhibitory conformation, allowing it to dissociate from Rab11 and activate Rab8.52 ERK1/2 is a downstream effector of receptor tyrosine kinase systems,53 and might serve to trigger large and sudden changes in vesicle trafficking associated with early epithelial scattering following HGF stimulation.
Rab11 and Rab8 have both been implicated in EMT and cancer metastasis. In gastric cancer cells, overexpression of Rab11-FIP2, an effector of Rab11, promotes EMT, while inhibition of Rab11-FIP2 decreased hypoxia-induced migration in these same cell lines.54 Rab8 is known to regulate cell shape.46 Dominant-negative Rab8 mutants and silencing of Rab8 results in a loss of protrusions and increased cell-cell adhesions in HT1080 fibrosarcoma cells.55 Meanwhile, overexpression of active Rab8 in BHK cells causes an increase in actin-based cell protrusions, including lammelipodia and filopodia.56 Furthermore, Rab8 drives cellular invasion by trafficking the matrix metalloproteinase MT1-MMP to the plasma membrane.57 Most recently, Rab8 activation has been shown to induce Rac1 activity, causing cortical actin formation, focal adhesion disassembly, and stress fiber disassembly.58 This effect was dependent on Calpain, MT1-MMP, and Rho GTPases.58 Furthermore, Rab8 is necessary for persistent cell motility, as well as EGF-induced polarity and chemotaxis.58 These data all point to a direct link between the slow endocytic recycling pathway and initiation of cellular motility required for EMT. Additionally, it shows Rab8 affects RhoA activity.
As an alternative to control of surface levels of TRP and other calcium channels by vesicle trafficking, it is possible that signals inducing epithelial scattering affect the activity of individual channels. Importantly, no change in how long channels remain open or channel conductivity follows HGF stimulation.31 High frequency calcium fluxes at the membrane result from increased channel opening events alone. TRP channel opening frequency is reportedly controlled by PIP259-62 and inhibited by calmodulin.59,63-65 Studying these proteins in context of growth factor-stimulated calcium influx and EMT may be of interest. However, regulation by PIP2 or calmodulin does not explain why microtubule targeting agents inhibit high frequency calcium influxes following HGF stimulation.31
While highly speculative, we propose that activation of signaling networks that drive epithelial scattering and EMT mobilize calcium channels to the plasma membrane from an intracellular compartment, thereby increasing calcium influxes and raising intracellular calcium concentrations. Intracellular calcium affects the activity and function of a wide array of cellular proteins. We will now turn our attention to those that might participate in cellular mechanics of epithelial scattering.
Molecular regulation of epithelial scattering by calcium/calmodulin dependent protein kinase II
One principal downstream effector of calcium is Ca2+/calmodulin-dependent protein kinase II, or CaMKII. Calmodulin (CaM) directly binds calcium using its 4 EF-hand motifs to cause a conformational change,66 which results in binding and activation of CaMKII. CaMKII holoenzymes contain 8-14 subunits, each with a catalytic domain, a regulatory domain, and an association domain, all organized so that catalytic domains are placed in pairs.67-70 Ca2+/CaM binding to the regulatory domain causes a conformational change that removes the autoinhibitory pseudo-substrate segment from the active site, which allows for ATP binding and also exposes T286 of the autoinhibitory pseudo-substrate segment for phosphorylation.69,71-75 This phosphorylation sterically prevents the inhibitory segment from returning to the active site once Ca2+/CaM has dissociated, resulting in continued activation independent of calcium.75,76 CaMKII subunits undergo cis-phosphorylation; pairing of subunit's catalytic domains allows for phosphorylation in trans, but this only occurs if both subunits are activated through Ca2+/CaM binding.74,77-79
CaMKII has been implicated in EMT in development.80 In xenopus development, CaMKII activation is seen in EMT and migration of fin core cells.33 Early work in xenopus shows that different types of Wnts act on cell adhesion differently, with Xwnt-5A decreasing cadherin based cell-cell adhesion, and Xwnt-1 increasing cell-cell adhesion.81 Later it was elucidated that these opposing effects result from Xwnt-5a antagonizing downstream effects of Xwnt-8 (which acts similarly to Xwnt-1) through a non-canonical calcium pathway that activates CaMKII, allowing remodeling of both cadherin-based cell-cell adhesions and alterations in cell morphology.82 Interestingly, the connection of Wnts to CaMKII in development highlights the role of calcium influxes. Wnt11-R also elicits intracellular calcium fluctuations that result in activation of CaMKII, which cause the cell to undergo EMT. The CaMKII inhibitor KN-93 prevents Wnt11-R positive cells in xenopus somites from undergoing EMT, showing that this process works through CaMKII activation. Calcium influx due to thapsigargin can rescue EMT in Wnt11-R knockdown frog embryos.33
CaMKII has also been connected to cancer metastasis both in vitro and in patient samples. α-CaMKII is highly expressed and phosphorylation is increased in osteosarcoma samples taken from patients. Overexpression of α-CaMKII in several osteosarcoma cell lines results in an increase in migration, proliferation, and a dramatic increase in invasion, all hallmarks of EMT, while knockdown results in a significant reduction in these behaviors.83 CaMKID, a homolog of CaMKII, is overexpressed in invasive carcinomas and leads to EMT, increased cell proliferation, loss of cell-cell adhesions, and increases migration and invasion.84 In prostate cancer cells, the non-canonical Wnt5a calcium pathway was shown to be important in wound closure and cell migration; inhibition of CaMKII with AIP prevents wound closure in scratched confluent sheets and decreases cell motility in the same cell lines.85
CaMKII could be linked to cellular morphology changes and actin dynamics associated with epithelial scattering in several ways (Fig. 2). First, CaMKII can alter cellular contractility through myosin phosphorylation. It has been shown that CaMKII phosphorylates myosin light chain kinase (MLCK) near the CaM binding domain, thus decreasing MLCK's affinity for CaM and reducing its ability to phosphorylate and activate myosin in vitro and in vivo.86,87 Consistent with these findings, a CaMKII inhibitor peptide (CaMKIIN) increases MLCK activity.88 Thus, CaMKII activation could have a role in the spreading phase of EMT, when myosin phosphorylation is decreased, though this idea is untested.
Figure 2.

Regulation of actin dynamics by CaMKII. In its inactive state, CaMKII directly mediates actin bundling. Activation of CamKII, by calmodulin binding and subsequent phosphorylation reduces actin bundling by preventing CamKII-actin associations and by CaMKII-mediated phosphorylation of spinophilin and/or filamin. In addition, active CamKII impacts cellular contractility, either negatively (by phosphorylation and inhibition of myosin light chain kinase) or positively (by activating RhoA). CaMKII activation may thus act as a switch that alters global actin dynamics.
CaMKII also directly regulates actin dynamics by binding and bundling actin filaments in a stoichiometric manner. Since CaMKII is an oligomer, multiple subunits can bind individual F-actin filaments in vitro and form actin bundles that are visible by electron microscopy.20,89,90 In the presence of calcium, CaMKIIβ releases actin filaments allowing for actin rearrangements. Calcium-mediated regulation of CaMKII-mediated actin bundling explains how calcium influxes can rapidly alter dendritic spine structure after calcium influxes.20,89,90 Actin bundling activity has also been reported with δ, and γ isoforms, the latter of which creates a novel layered bundle structure.91 Whether CaMKII's actin bundling activity occurs in epithelial cells and whether negative regulation of this activity by calcium might facilitate epithelial scattering, perhaps by releasing actin bundling prior to rearrangement of actin at cell-cell junctions into transcellular networks, remains unclear.
CaMKII can also regulate actin bundling without directly binding actin filaments. CaMKII phosphorylates filamin, decreasing its affinity for F-actin. Filamin loosely crosslinks F-actin, causing the cytosol to become gel-like, an activity that must be antagonized to change cell morphology and increase motility.92 Increasing cytosolic calcium in head and neck squamous cell carcinoma cells increases filamin phosphorylation and decreases filamin-actin associations, while adding the CaMKII inhibitor KN-93 returns both phosphorylation and actin binding to normal.92 Spinophilin, which binds to and bundles F-actin, is also phosphorylated by CaMKII, reducing its affinity for F-actin.93 In MDCK cells undergoing HGF-induced epithelial scattering, the resolution of larger actin cables into a transcellular network does appear to involve a reduction in bundling, as demonstrated by the loss of actin bundling protein α-actinin shortly after HGF stimulation and prior to actin cable dissolution into smaller filaments.9 As with direct actin bundling binding by CaMKII itself, whether filamin and spinophilin participate in these actin rearrangements is unknown.
While activities of CaMKII described above point to a role for CaMKII in the early spreading phase of epithelial scattering, is there a potential connection of calcium and CaMKII to RhoA? Rho-mediated activation of cofilin occurs downstream of calcium fluxes in dendritic spines is calcium-dependent.94 Interestingly, it is calcium-mediated activation of CaMKII that in turn drives longer term RhoA activation in this sytem; a CaMKII inhibitor blocks RhoA-ROCK pathway activation.94 Though this data suggests CaMKII could be the key mediator between calcium influxes and RhoA-mediated cellular contractility during epithelial scattering, the molecular mechanism by which CaMKII stimulates RhoA activation remains unidentified.
Molecular basis of calcium-mediated changes in actin dynamics and cellular mechanics
While a case for CaMKII in directing actin rearrangements in response to calcium influxes during EMT still remains to be verified, calcium can undoubtedly alter actin dynamics independently of CaMKII. RhoA has also been shown to be directly activated by calcium through the calcium-sensitive tyrosine kinase pyk2. Calcium is necessary and sufficient to stimulate pyk2 activity,95,96 leading to activation Rho specific GEFs that lead to RhoA-mediated cellular constriction.97,98 AM-BAPTA, a calcium chelator, abolishes Angiotensin II-induced RhoA activation by preventing pyk2 activation.97 Pyk2 is also important for cytoskeletal reorganization in monocyte cell motility in vitro, where it localizes to the leading edge.99 PDZ-RhoGEF, p190RhoGEF, and leukemia-associated Rho GEF (LARG) are examples of Rho-specific GEFs that could link pyk2 and RhoA.98,100-102
Calcium may also act on myosin independently of RhoA. Myosin light chain kinase (MLCK) is a Ca2+/Calmodulin dependent kinase that phosphorylates the regulatory light chains of myosin II isoforms.103 It may be important in the development of sarcomeres, hinting at a possible role in the development of other actin-myosin contractile units.104 Death-associated protein kinase (DAP kinase) is a calcium/calmodulin-dependent serine/threonine kinase that phosphorylates the regulatory light chain of myosin II (MLC) both in vitro and in vivo. In quiescent fibroblasts, DAP kinase stabilizes stress fibers through phosphorylation of MLC, and is necessary and sufficient for formation of stress fibers.105
S100A4 is a member of the S100 family of 25 calcium binding proteins that are widely expressed in vertebrates.106-109 All S100 proteins have 2 calcium binding EF-hands that cause a conformational change in the protein upon calcium binding, exposing protein binding surfaces and allowing S100 proteins to act as calcium sensors that modulate a range of downstream behaviors through several binding partners.107,108,110 S100A4 overexpression is associated with metastasis and decreased survival in many cancers, and it is considered an EMT promoter and marker.109,111,112 Overexpression in non-metastatic tumor cells leads to metastasis in xenograft models,111,113,114 whereas knockdown of S100A4 in metastatic tumor cells prevents metastases in animal models.113,115 This is likely because S100A4 interacts with F-actin, non-muscle myosin heavy chain (NMMHC) IIA, tubulin, and non-muscle tropomyosin to increase cell migration through force generation as well as forming and stabilizing lamellipodia.106,108,111-113,116
Removal of S100A4 from cells results in increased assembly of non-muscle myosin IIA complexes, while overexpression results in large lamellipodia with a loss of focal adhesion maturation and filopodia.112 S100A4 also interacts with Rhotekin, a Rho binding and regulating protein that participates in actin-based contractility during migration and invasion.112 The indirect interaction with Rho, along with direct binding to myosin and the prevention of maturation of focal adhesions allow S100A4 a role in the contractility and basement membrane detachment phases of EMT.
During EMT, S100A4 is expressed in cells stimulated with EGF and tumor growth factor β1.113,117 S100A4 is upregulated by canonical Wnt signaling, and inhibition of β-catenin decreases tumor cell migration and invasion through downregulation of S100A4.106,109 NFAT is expressed in metastatic breast cancer and also transcriptionally activates S100A4,118 Interestingly, NFAT activation is driven by calcium influxes during epithelial scattering of MDCK cells,31 indicating that there might be multiple connections between S100A4 and increased intracellular calcium concentration in EMT.
In short, a number of potential actin regulatory systems might be controlled by calcium influxes during EMT.
Discussion
Two hypotheses for how epithelial cells regulate cellular processes to drive epithelial scattering have been proposed. In the first, active disassembly of cell-cell junctions allows for rupture of adhesions between cells. This is supported by the observation that cadherin expression changes accompany EMT119 and that alterations in actin and actin-regulatory protein function at cell-cell adhesions can prevent or accelerate cell-cell detachment.9 In the second hypothesis, contractile forces centered on cell-cell junctions results in physical detachment.12 That transient treatment of MDCK cells with blebbistatin leads to cell dissociations identical to those observed in MDCK cells treated with HGF11 suggests that the latter theory, where cellular tension forces are primarily responsible for rupturing of cell-cell adhesion during epithelial scattering, holds. The reality is that molecular changes to the architecture of cell-cell junctions likely accompany force driven detachment of cell-cell adhesion. In fact, it may be impossible to distinguish these events, as linkage of cadherin-based adhesions to actin appears to be tension dependent120,121 and changes in cellular contractility would alter the molecular architecture at sites of cell-cell adhesion. In fact, a model whereby cell spreading precedes contractility and cell-cell detachment is attractive in this light, as the relaxation of myosin-based contractility occurring in cell spreading may in fact prime cell-cell junctions by releasing actin anchoring of cadherin complex just before cellular contractility is increased to rupture now weakened cell-cell junctions.
Whether cell-cell junctions are remodeled at the molecular level or not prior to cell-cell detachment, broader questions remain as to how cells control changes in cellular mechanics and actin organization. A growing number of studies highlight a critical role for calcium channels in the TRP family for playing a critical role in epithelial scattering and, more recently, in control of actin dynamics during this process. While TRP channels can be controlled through multiple mechanisms, control of surface expression by regulated trafficking of channels from intracellular stores is an attractive mechanism for controlling calcium influxes during epithelial scattering. Despite a lack of integration of the role of TRP channels into current models of EMT, we expect further studies linking calcium channels and their vesicle trafficking to changes in cell morphology and actin dynamics.
How does calcium exert its effects on cell morphology during epithelial scattering? A central role for RhoA, a master regulator of actin dynamics generally and of cellular contractility specifically, has, unsurprisingly, emerged in epithelial scattering. Expression of a dominant-negative RhoA mutant in MDCK cells prevents cellular contractility and resulting cell-cell detachment in response to HGF stimulation.11 The specific link between increased concentration of intracellular calcium ions and RhoA activation in scattering epithelial cells is likely multifaceted. Just as CaMKII links calcium fluxes to RhoA activation in the dendritic spine,94 CaMKII might link calcium fluxes to RhoA activation and then global myosin-based contractility in scattering epithelial cells.
Stronger reported connections of CaMKII with cellular activities involved in the earlier spreading phase of epithelial scattering suggest calcium may also act outside of regulating contractility. Here calcium may negatively regulate direct actin bundling by CaMKII itself, as well as stimulate phosphorylation of several target actin binding proteins, reducing their activity to trigger changes in actin organization and reduced myosin-based contractility through the resulting actin cytoskeleton.
If one views spreading as a result, at least in part, of reduced actomyosin contractility, it suggests calcium, and possibly CaMKII, may play opposing roles in early and late epithelial scattering, stimulating a reduction in myosin activity early and increasing it in late stages. How might a single signal, calcium influxes, generate opposing effects through the same machinery? We note that calcium sensors are often highly sensitive to the pattern of calcium concentration changes in the cytosol; rapid calcium oscillations are seen to activate NF-κB, NFAT and Oct/OAP, while only NFAT responds to slower oscillations.122,123 Could calcium sensors that affect actin dynamics and cellular contractility be similarly sensitive to patterns of intracellular calcium concentration changes? Clearly dissecting the role of calcium influxes in epithelial scattering will prove complex if such is the case.
We thus propose an overall, and speculative, model as outlined in Figure 3, with initiation of epithelial scattering triggering a redistribution of internal TRP channels to the plasma membrane. The resulting calcium ion influx activates changes in actin dynamics and cellular mechanics through downstream effectors. We argue, speculatively, for a central role played by CaMKII, which facilitates spreading in early scattering and drives contractility through RhoA in late scattering.
Figure 3.
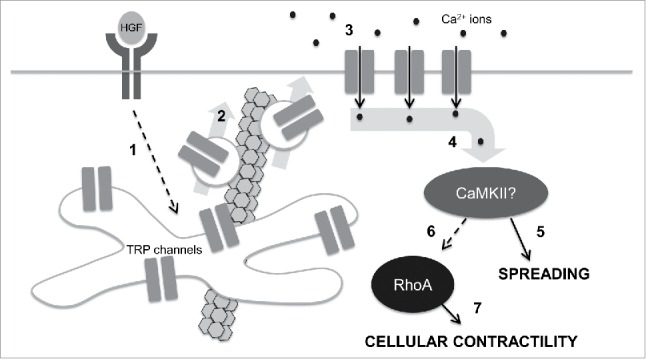
A speculative model for control of cell mechanics during epithelial scattering. In our model, 1.) HGF binding and activation of c-met receptor tyrosine kinases stimulates activates vesicle trafficking machinery in endosomal recycling compartments through an unidentified mechanism; 2.) Internal TRP channels are mobilized into vesicles that are destined for the plasma membrane in a microtubule-dependent vesicle trafficking step. 3.) TRP channels at the plasma membrane mediate a calcium influx that raises intracellular calcium concentrations. 4.) Intracellular calcium binds camodulin and in turn activates CaMKII. 5.) Early in epithelial scattering, CaMKII drives spreading, a result of reduced actin bundling at cell-cell junctions and myosin-based contractility. 6.) Late in epithelial scattering and through an as yet unidentified mechanism, CaMKII can stimulate an increase in RhoA activation. Seven. RhoA, through ROCK, increases myosin light chain phosphorylation, driving global cellular contractility and cell-cell detachment.
We note in closing that signal transduction downstream of receptor tyrosine kinases, such as c-met, has been extensively studied. Many signaling modules have been carefully reconstructed and are widely utilized by different receptor tyrosine kinases. Flow of ‘information’ through these modules is very rapid, allowing rapid responses to receptor activation. But the events of epithelial scattering play out on a time scale that takes hours, a process much slower than is provided by well-characterized signaling modules. In our view, controlling events of epithelial scattering in response to c-met and other cues, whether through receptor tyrosine kinases or other systems, will require testing hypotheses with novel mechanisms and slower information flow, such as slower vesicle trafficking steps.
Abbreviations
- CaM
Calmodulin
- CamKII
Calcium-dependent kinase II
- EGF
Epidermal growth factor
- EMT
Epithelial-mesenchymal transition
- ERC
Endosomal recycling compartment
- HGF
Hepatocyte growth factor
- MDCK
Madin-Darby canine kidney
- MLC
Myosin light chain
- MLCK
Myosin light chain kinase
- TRP
Transient receptor potential
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Ivanov AI, Naydenov NG. Chapter Two - Dynamics and Regulation of Epithelial Adherens Junctions: Recent Discoveries and Controversies In: Kwang WJ, ed. International Review of Cell and Molecular Biology: Academic Press, 2013:27-99 [DOI] [PubMed] [Google Scholar]
- [2].Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420-8; PMID:19487818; http://dx.doi.org/ 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stoker M, Perryman M. An epithelial scatter factor released by embryo fibroblasts. J Cell Sci 1985; 77:209-23; PMID:3841349 [DOI] [PubMed] [Google Scholar]
- [4].Weidner KM, Arakaki N, Hartmann G, Vandekerckhove J, Weingart S, Rieder H, Fonatsch C, Tsubouchi H, Hishida T, Daikuhara Y. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci U S A 1991; 88:7001-5; PMID:1831266; http://dx.doi.org/ 10.1073/pnas.88.16.7001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Furlong RA, Takehara T, Taylor WG, Nakamura T, Rubin JS. Comparison of biological and immunochemical properties indicates that scatter factor and hepatocyte growth factor are indistinguishable. J Cell Sci 1991; 100:173-7; PMID:1839027 [DOI] [PubMed] [Google Scholar]
- [6].Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell 1991; 67:901-8; PMID:1835669; http://dx.doi.org/ 10.1016/0092-8674(91)90363-4 [DOI] [PubMed] [Google Scholar]
- [7].Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, Narsimhan RP, Hartmann G, Zarnegar R, Michalopoulos GK. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J 1991; 10:2867-78; PMID:1655405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].LI Y, Joseph A, Bhargava MM, Rosen EM, Nakamura T, Goldberg I. Effect of scatter factor and hepatocyte growth factor on motility and morphology of mdck cells. In Vitro Cell Dev Biol 1992; 28:364-8; http://dx.doi.org/ 10.1007/BF02877060 [DOI] [PubMed] [Google Scholar]
- [9].Sperry RB, Bishop NH, Bramwell JJ, Brodeur MN, Carter MJ, Fowler BT, Lewis ZB, Maxfield SD, Staley DM, Vellinga RM, et al.. Zyxin controls migration in epithelial-mesenchymal transition by mediating actin-membrane linkages at cell-cell junctions. J Cell Physiol 2010; 222:612-24; PMID:19927303 [DOI] [PubMed] [Google Scholar]
- [10].Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature 2009; 457:495-9; PMID:19029882; http://dx.doi.org/ 10.1038/nature07522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hoj JP, Davis JA, Fullmer KE, Morrell DJ, Saguibo NE, Schuler JT, Tuttle KJ, Hansen MD. Cellular contractility changes are sufficient to drive epithelial scattering. Exp Cell Res 2014; 326:187-200; PMID:24780819; http://dx.doi.org/ 10.1016/j.yexcr.2014.04.011 [DOI] [PubMed] [Google Scholar]
- [12].de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol 2005; 171:153-64; PMID:16216928; http://dx.doi.org/ 10.1083/jcb.200506152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases 2014; 5:1-15; PMID:25483301; http://dx.doi.org/ 10.4161/21541248.2014.973760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Citalán-Madrid AF, García-Ponce A, Vargas-Robles H, Betanzos A, Schnoor M. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers 2013; 1:e26938; http://dx.doi.org/ 10.4161/tisb.26938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al.. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science 1996; 273:245-8; PMID:8662509; http://dx.doi.org/ 10.1126/science.273.5272.245 [DOI] [PubMed] [Google Scholar]
- [16].Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and Activation of Myosin by Rho-associated Kinase (Rho-kinase). J Biol Chem 1996; 271:20246-9; PMID:8702756; http://dx.doi.org/ 10.1074/jbc.271.34.20246 [DOI] [PubMed] [Google Scholar]
- [17].Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and Activation of Myosin by Rho-associated Kinase (Rho-kinase). J Biol Chem 1996; 271:20246-9; PMID:8702756; http://dx.doi.org/ 10.1074/jbc.271.34.20246 [DOI] [PubMed] [Google Scholar]
- [18].Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 2003; 4:446-56; PMID:12778124; http://dx.doi.org/ 10.1038/nrm1128 [DOI] [PubMed] [Google Scholar]
- [19].Jiang L, Wen J, Luo W. Rho associated kinase inhibitor, Y27632, inhibits the invasion and proliferation of T24 and 5367 bladder cancer cells. Mol Med Rep 2015; 12:7526-30; PMID:26459851 [DOI] [PubMed] [Google Scholar]
- [20].Okamoto K, Bosch M, Hayashi Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag? Physiology (Bethesda) 2009; 24:357-66; PMID:19996366; http://dx.doi.org/ 10.1152/physiol.00029.2009 [DOI] [PubMed] [Google Scholar]
- [21].Fonseca R. Activity-dependent actin dynamics are required for the maintenance of long-term plasticity and for synaptic capture. Eur J Neurosci 2012; 35:195-206; PMID:22250814; http://dx.doi.org/ 10.1111/j.1460-9568.2011.07955.x [DOI] [PubMed] [Google Scholar]
- [22].Schubert V, Da Silva JS, Dotti CG. Localized recruitment and activation of RhoA underlies dendritic spine morphology in a glutamate receptor-dependent manner. J Cell Biol 2006; 172:453-67; PMID:16449195; http://dx.doi.org/ 10.1083/jcb.200506136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lammers M, Meyer S, Kuhlmann D, Wittinghofer A. Specificity of interactions between mDia isoforms and Rho proteins. J Biol Chem 2008; 283:35236-46; PMID:18829452; http://dx.doi.org/ 10.1074/jbc.M805634200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev 2009; 28:65-76; PMID:19160018; http://dx.doi.org/ 10.1007/s10555-008-9170-7 [DOI] [PubMed] [Google Scholar]
- [25].Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol 1999; 1:136-43; PMID:10559899; http://dx.doi.org/ 10.1038/11056 [DOI] [PubMed] [Google Scholar]
- [26].Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci 2003; 6:1194-200; PMID:14555951; http://dx.doi.org/ 10.1038/nn1135 [DOI] [PubMed] [Google Scholar]
- [27].Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the Actin Cytoskeleton Through Protein Kinases ROCK and LIM-kinase. Science 1999; 285:895-8; PMID:10436159; http://dx.doi.org/ 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- [28].Royal I, Lamarche-Vane N, Lamorte L, Kaibuchi K, Park M. Activation of cdc42, rac, PAK, and rho-kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Mol Biol Cell 2000; 11:1709-25; PMID:10793146; http://dx.doi.org/ 10.1091/mbc.11.5.1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Baffy G, Yang L, Michalopoulos GK, Williamson JR. Hepatocyte growth factor induces calcium mobilization and inositol phosphate production in rat hepatocytes. J Cell Physiol 1992; 153:332-9; PMID:1429853; http://dx.doi.org/ 10.1002/jcp.1041530213 [DOI] [PubMed] [Google Scholar]
- [30].Kawanishi T, Kato T, Asoh H, Uneyama C, Toyoda K, Momose K, Takahashi M, Hayashi Y. Hepatocyte growth factor-induced calcium waves in hepatocytes as revealed with rapid scanning confocal microscopy. Cell Calcium 1995; 18:495-504; PMID:8746948; http://dx.doi.org/ 10.1016/0143-4160(95)90012-8 [DOI] [PubMed] [Google Scholar]
- [31].Langford PR, Keyes L, Hansen MD. Plasma membrane ion fluxes and NFAT-dependent gene transcription contribute to c-met-induced epithelial scattering. J Cell Sci 2012; 125:4001-13; PMID:22685327; http://dx.doi.org/ 10.1242/jcs.098269 [DOI] [PubMed] [Google Scholar]
- [32].Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW Jr., Goodhill GJ, Thompson EW, Roberts-Thomson SJ, Monteith GR. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014; 33:2307-16; PMID:23686305; http://dx.doi.org/ 10.1038/onc.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garriock RJ, Krieg PA. Wnt11-R signaling regulates a calcium sensitive EMT event essential for dorsal fin development of Xenopus. Dev Biol 2007; 304:127-40; PMID:17240368; http://dx.doi.org/ 10.1016/j.ydbio.2006.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol 2011; 12:218; PMID:21401968; http://dx.doi.org/ 10.1186/gb-2011-12-3-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Song J, Wang Y, Li X, Shen Y, Yin M, Guo Y, Diao L, Liu Y, Yue D. Critical role of TRPC6 channels in the development of human renal cell carcinoma. Mol Biol Rep 2013; 40:5115-22; PMID:23700295; http://dx.doi.org/ 10.1007/s11033-013-2613-4 [DOI] [PubMed] [Google Scholar]
- [36].Wang Y, Yue D, Li K, Liu Y-L, Ren C-S, Wang P. The role of TRPC6 in HGF-induced cell proliferation of human prostate cancer DU145 and PC3 cells. Asian J Androl 2010; 12:841-52; PMID:20835261; http://dx.doi.org/ 10.1038/aja.2010.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rampino T, Gregorini M, Guidetti C, Broggini M, Marchini S, Bonomi R, Maggio M, Roscini E, Soccio G, Tiboldo R, et al.. KCNA1 and TRPC6 ion channels and NHE1 exchanger operate the biological outcome of HGF/scatter factor in renal tubular cells. Growth Factors 2007; 25:382-91; PMID:18365869; http://dx.doi.org/ 10.1080/08977190801892184 [DOI] [PubMed] [Google Scholar]
- [38].Boca M, D'Amato L, Distefano G, Polishchuk RS, Germino GG, Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3β-dependent Cell–cell mechanical adhesion. Mol Biol Cell 2007; 18:4050-61; PMID:17671167; http://dx.doi.org/ 10.1091/mbc.E07-02-0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vriens J, Janssens A, Prenen J, Nilius B, Wondergem R. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium 2004; 36:19-28; PMID:15126053; http://dx.doi.org/ 10.1016/j.ceca.2003.11.006 [DOI] [PubMed] [Google Scholar]
- [40].Fiorio Pla A, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, Paria B, Munaron L, Ambudkar IS. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012; 31:200-12; PMID:21685934; http://dx.doi.org/ 10.1038/onc.2011.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 2004; 6:709-20; PMID:15258588; http://dx.doi.org/ 10.1038/ncb1150 [DOI] [PubMed] [Google Scholar]
- [42].Odell AF, Scott JL, Van Helden DF. Epidermal growth factor induces tyrosine phosphorylation, membrane insertion, and activation of transient receptor potential channel 4. J Biol Chem 2005; 280:37974-87; PMID:16144838; http://dx.doi.org/ 10.1074/jbc.M503646200 [DOI] [PubMed] [Google Scholar]
- [43].Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009; 10:513-25; PMID:19603039; http://dx.doi.org/ 10.1038/nrm2728 [DOI] [PubMed] [Google Scholar]
- [44].van de Graaf SFJ, Chang Q, Mensenkamp AR, Hoenderop JGJ, Bindels RJM. Direct interaction with Rab11a targets the epithelial Ca(2+) channels TRPV5 and TRPV6 to the plasma membrane. Mol Cell Biol 2006; 26:303-12; PMID:16354700; http://dx.doi.org/ 10.1128/MCB.26.1.303-312.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ward HH, Brown-Glaberman U, Wang J, Morita Y, Alper SL, Bedrick EJ, Gattone VH 2nd, Deretic D, Wandinger-Ness A. A conserved signal and GTPase complex are required for the ciliary transport of polycystin-1. Mol Biol Cell 2011; 22:3289-305; PMID:21775626; http://dx.doi.org/ 10.1091/mbc.E11-01-0082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Peranen J. Rab8 GTPase as a regulator of cell shape. Cytoskeleton 2011; 68:527-39; PMID:21850707; http://dx.doi.org/ 10.1002/cm.20529 [DOI] [PubMed] [Google Scholar]
- [47].Grigoriev I, Yu Ka L, Martinez-Sanchez E, Serra-Marques A, Smal I, Meijering E, Demmers J, Peränen J, Pasterkamp RJ, van der Sluijs P, et al.. Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr Biol 2011; 21:967-74; PMID:21596566; http://dx.doi.org/ 10.1016/j.cub.2011.04.030 [DOI] [PubMed] [Google Scholar]
- [48].Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Revi Mol Cell Biol 2009; 10:597-608; PMID:19696797; http://dx.doi.org/ 10.1038/nrm2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol 2000; 149:901-14; PMID:10811830; http://dx.doi.org/ 10.1083/jcb.149.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Knödler A, Feng S, Zhang J, Zhang X, Das A, Peränen J, Guo W. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A 2010; 107:6346-51; PMID:20308558; http://dx.doi.org/ 10.1073/pnas.1002401107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol 2010; 12:1035-45; PMID:20890297; http://dx.doi.org/ 10.1038/ncb2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang J, Ren J, Wu B, Feng S, Cai G, Tuluc F, Peränen J, Guo W. Activation of Rab8 guanine nucleotide exchange factor Rabin8 by ERK1/2 in response to EGF signaling. Proc Natl Acad Sci U S A 2015; 112:148-53; PMID:25535387; http://dx.doi.org/ 10.1073/pnas.1412089112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Frémin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol 2010; 3:8; http://dx.doi.org/ 10.1186/1756-8722-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dong W, Qin G, Shen R. Rab11-FIP2 promotes the metastasis of gastric cancer cells. Int J Cancer 2016; 138:1680-8; PMID:26502090; http://dx.doi.org/ 10.1002/ijc.29899 [DOI] [PubMed] [Google Scholar]
- [55].Hattula K, Furuhjelm J, Tikkanen J, Tanhuanpää K, Laakkonen P, Peränen J. Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J Cell Sci 2006; 119:4866-77; PMID:17105768; http://dx.doi.org/ 10.1242/jcs.03275 [DOI] [PubMed] [Google Scholar]
- [56].Peränen J, Auvinen P, Virta H, Wepf R, Simons K. Rab8 promotes polarized membrane transport through reorganization of actin and microtubules in fibroblasts. J Cell Biol 1996; 135:153-67; http://dx.doi.org/ 10.1083/jcb.135.1.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bravo‐Cordero JJ, Marrero‐Diaz R, Megías D, Genís L, García‐Grande A, García MA, Arroyo AG, Montoya MC. MT1‐MMP proinvasive activity is regulated by a novel Rab8‐dependent exocytic pathway. EMBO J 2007; 26:1499-510; PMID:17332756; http://dx.doi.org/ 10.1038/sj.emboj.7601606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bravo-Cordero JJ, Cordani M, Soriano SF, Díez B, Muñoz-Agudo C, Casanova-Acebes M, Boullosa C, Guadamillas MC, Ezkurdia I, González-Pisano D, et al.. A novel high content analysis tool reveals Rab8-driven actin and FA reorganization through Rho GTPases and calpain/MT1. J Cell Sci 2016; 129:1734-49; PMID:26940916 [DOI] [PubMed] [Google Scholar]
- [59].Grycova L, Holendova B, Lansky Z, Bumba L, Jirku M, Bousova K, Teisinger J. Ca2+ binding protein S100A1 competes with calmodulin and PtdIns(4,5)P2 for binding site on the C-terminus of the TPRV1 receptor. ACS Chem Neurosci 2015; 6:386-92; PMID:25543978; http://dx.doi.org/ 10.1021/cn500250r [DOI] [PubMed] [Google Scholar]
- [60].Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R. The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 2007; 54:905-18; PMID:17582331; http://dx.doi.org/ 10.1016/j.neuron.2007.05.027 [DOI] [PubMed] [Google Scholar]
- [61].Lukacs V, Thyagarajan B, Varnai P, Balla A, Balla T, Rohacs T. Dual regulation of TRPV1 by phosphoinositides. J Neurosci 2007; 27:7070-80; PMID:17596456; http://dx.doi.org/ 10.1523/JNEUROSCI.1866-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mercado J, Gordon-Shaag A, Zagotta WN, Gordon SE. Ca(2+)-dependent desensitization of TRPV2 channels is mediated by hydrolysis of phosphatidylinositol 4,5-bisphosphate. J Neurosci 2010; 30:13338-47; PMID:20926660; http://dx.doi.org/ 10.1523/JNEUROSCI.2108-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].de Groot T, Kovalevskaya NV, Verkaart S, Schilderink N, Felici M, van der Hagen EAE, Bindels RJM, Vuister GW, Hoenderop JG. Molecular mechanisms of calmodulin action on TRPV5 and modulation by parathyroid hormone. Mol Cell Biol 2011; 31:2845-53; PMID:21576356; http://dx.doi.org/ 10.1128/MCB.01319-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Holakovska B, Grycova L, Jirku M, Sulc M, Bumba L, Teisinger J. Calmodulin and S100A1 protein interact with N terminus of TRPM3 channel. J Biol Chem 2012; 287:16645-55; PMID:22451665; http://dx.doi.org/ 10.1074/jbc.M112.350686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhu M. Multiple roles of calmodulin and other Ca2+-binding proteins in the functional regulation of TRP channels. Pflügers Archiv 2005; 451:105-15; PMID:15924238; http://dx.doi.org/ 10.1007/s00424-005-1427-1 [DOI] [PubMed] [Google Scholar]
- [66].Black DJ, Leonard J, Persechini A. Biphasic Ca2+ -dependent switching in a calmodulin-IQ domain complex. Biochemistry 2006; 45:6987-95; PMID:16734434; http://dx.doi.org/ 10.1021/bi052533w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nguyen TA, Sarkar P, Veetil JV, Davis KA, Puhl HL 3rd, Vogel SS. Covert changes in CaMKII holoenzyme structure identified for activation and subsequent interactions. Biophys J 2015; 108:2158-70; PMID:25954874; http://dx.doi.org/ 10.1016/j.bpj.2015.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang Y-y, Zhao R, Zhe H. The emerging role of CaMKII in cancer. Oncotarget 2015; 6:11725-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hudmon A, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem 2002; 71:473-510; PMID:12045104; http://dx.doi.org/ 10.1146/annurev.biochem.71.110601.135410 [DOI] [PubMed] [Google Scholar]
- [70].Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell 2003; 11:1241-51; PMID:12769848; http://dx.doi.org/ 10.1016/S1097-2765(03)00171-0 [DOI] [PubMed] [Google Scholar]
- [71].Colbran RJ, Fong Y-L, Schworer CM, Soderling TR. Regulatory interactions of the calmodulin-binding, inhibitory, and autophosphorylation domains of Ca2+/calmodulin-dependent protein kinase 1. J Biol Chem 1988; 263:18145-51 [PubMed] [Google Scholar]
- [72].Rellos P, Pike AC, Niesen FH, Salah E, Lee WH, von Delft F, Knapp S. Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol 2010; 8:e1000426; PMID:20668654; http://dx.doi.org/ 10.1371/journal.pbio.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hoffman L, Stein RA, Colbran RJ, McHaourab HS. Conformational changes underlying calcium/calmodulin-dependent protein kinase II activation. EMBO J 2011; 30:1251-62; http://dx.doi.org/ 10.1038/emboj.2011.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Mukherji S, Soderling TR. Mutational analysis of Ca2+-independent autophosphorylation of calcium/calmodulin-dependent protein kinase II. J Biol Chem 1995; 270:14062-7; http://dx.doi.org/ 10.1074/jbc.270.23.14062 [DOI] [PubMed] [Google Scholar]
- [75].Yang E, Schulman H. Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J Biol Chem 1999; 274:26199-208 [DOI] [PubMed] [Google Scholar]
- [76].Hanson PtdIns, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem 1992; 61:559-61; http://dx.doi.org/ 10.1146/annurev.bi.61.070192.003015 [DOI] [PubMed] [Google Scholar]
- [77].Lucic V, Greif GJ, Kennedy MB. Detailed state model of CaMKII activation and autophosphorylation. Eur Biophys J 2008; 38:83-98; http://dx.doi.org/ 10.1007/s00249-008-0362-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mukherji S, Soderling TR. Regulation of Ca2+/Calmodulin-dependent Protein Kinase I1 by Inter- and Intrasubunit-catalyzed Autophosphorylations. J Biol Chem 1994; 269:13744-7 [PubMed] [Google Scholar]
- [79].Bradshaw JM, Hudmon A, Schulman H. Chemical quenched flow kinetic studies indicate an intraholoenzyme autophosphorylation mechanism for Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 2002; 277:20991-8; PMID:11925447; http://dx.doi.org/ 10.1074/jbc.M202154200 [DOI] [PubMed] [Google Scholar]
- [80].Sun X, Zhao D, Li YL, Sun Y, Lei XH, Zhang JN, Wu MM, Li RY, Zhao ZF, Zhang ZR, et al.. Regulation of ASIC1 by Ca2+/calmodulin-dependent protein kinase II in human glioblastoma multiforme. Oncol Rep 2013; 30:2852-8; PMID:24100685 [DOI] [PubMed] [Google Scholar]
- [81].Tortes MA, Yang-Snyder JA, Purcell SM, DeMarais AA, McGrew LL, Moon RT. Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherinin early xenopus development. J Cell Biol 1996; 133:1123-37; PMID:8655584; http://dx.doi.org/ 10.1083/jcb.133.5.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kuhl M, Geis K, Sheldahl LC, Pukrop T, Moon RT, Wedlich D. Antagonistic regulation of convergent extention movements in zenopus by Wnt/Beta-catenin and Wnt/Ca2+ signaling. Mech Dev 2001; 106:61-76; PMID:11472835; http://dx.doi.org/ 10.1016/S0925-4773(01)00416-6 [DOI] [PubMed] [Google Scholar]
- [83].Daft PG, Yuan K, Warram JM, Klein MJ, Siegal GP, Zayzafoon M. Alpha-CaMKII plays a critical role in determining the aggressive behavior of human osteosarcoma. Mol Cancer Res 2013; 11:349-59; PMID:23364534; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bergamaschi A, Kim YH, Kwei KA, La Choi Y, Bocanegra M, Langerod A, Han W, Noh DY, Huntsman DG, Jeffrey SS, et al.. CAMK1D amplification implicated in epithelial-mesenchymal transition in basal-like breast cancer. Mol Oncol 2008; 2:327-39; PMID:19383354; http://dx.doi.org/ 10.1016/j.molonc.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang Q, Symes AJ, Kane CA, Freeman A, Nariculam J, Munson P, Thrasivoulou C, Masters JR, Ahmed A. A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PloS One 2010; 5:e10456; PMID:20454608; http://dx.doi.org/ 10.1371/journal.pone.0010456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tansey MG, Word RA, Hidaka H, Singer HA, Schworer CM, Kamm KE, Stull JT. Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent protein kinase II in smooth muscle cells. J Biol Chem 1992; 267:12511-6; PMID:1319999 [PubMed] [Google Scholar]
- [87].Hashimoto Y, Soderling TR. Phosphorylation of smooth muscle myosin light chain kinase by Ca2+/calmodulin-dependent protein kinase II: Comparative study of the phosphorylation sites. Arch Biochem Biophys 1990; 278:41-5; PMID:2157362; http://dx.doi.org/ 10.1016/0003-9861(90)90228-Q [DOI] [PubMed] [Google Scholar]
- [88].Prasad AM, Nuno DW, Koval OM, Ketsawatsomkron P, Li W, Li H, Shen FY, Joiner M-lA, Kutschke W, Weiss RM, et al.. Differential control of calcium homeostasis and vascular reactivity by CaMKII. Hypertension 2013; 62:434-41; PMID:23753415; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.113.01508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A 2007; 104:6418-23; PMID:17404223; http://dx.doi.org/ 10.1073/pnas.0701656104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lin YC, Redmond L. CaMKIIbeta binding to stable F-actin in vivo regulates F-actin filament stability. Proc Natl Acad Sci U S A 2008; 105:15791-6; PMID:18840684; http://dx.doi.org/ 10.1073/pnas.0804399105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hoffman L, Farley MM, Waxham MN. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 2013; 52:1198-207; PMID:23343535; http://dx.doi.org/ 10.1021/bi3016586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bourguignon LY, Gilad E, Brightman A, Diedrich F, Singleton P. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation, phospholipase C epsilon-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. J Biol Chem 2006; 281:14026-40; PMID:16565089; http://dx.doi.org/ 10.1074/jbc.M507734200 [DOI] [PubMed] [Google Scholar]
- [93].Grossman SD, Futter M, Snyder GL, Allen PB, Nairn AC, Greengard P, Hsieh-Wilson LC. Spinophilin is phosphorylated by Ca2+/calmodulin-dependent protein kinase II resulting in regulation of its binding to F-actin. J Neurochem 2004; 90:317-24; PMID:15228588; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02491.x [DOI] [PubMed] [Google Scholar]
- [94].Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 2011; 472:100-4; PMID:21423166; http://dx.doi.org/ 10.1038/nature09823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, et al.. Activation of a novel calcium-dependent protein-tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated protein kinase activation. J Biol Chem 1996; 271:29993-8; PMID:8939945; http://dx.doi.org/ 10.1074/jbc.271.47.29993 [DOI] [PubMed] [Google Scholar]
- [96].Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature 1995; 376:737-45; PMID:7544443; http://dx.doi.org/ 10.1038/376737a0 [DOI] [PubMed] [Google Scholar]
- [97].Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol 2009; 29:1657-63; PMID:19759375; http://dx.doi.org/ 10.1161/ATVBAHA.109.190892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, Uryu SA, Canete-Soler R, Zhai J, Lin H, et al.. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol 2008; 180:187-203; PMID:18195107; http://dx.doi.org/ 10.1083/jcb.200708194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Watson JM, Harding TW, Golubovskaya V, Morris JS, Hunter D, Li X, Haskill JS, Earp HS. Inhibition of the calcium-dependent tyrosine kinase (CADTK) blocks monocyte spreading and motility. J Biol Chem 2001; 276:3536-42; PMID:11062241; http://dx.doi.org/ 10.1074/jbc.M006916200 [DOI] [PubMed] [Google Scholar]
- [100].Mariette H.E., Driessensa CO, Koh-ichi Nagatab, Masaki Inagakib, Collarda JG. B plexins activate Rho through PDZ-RhoGEF. FEBS Lett 2002; 529:168-72 [DOI] [PubMed] [Google Scholar]
- [101].Oinuma I, Katoh H, Harada A, Negishi M. Direct interaction of Rnd1 with Plexin-B1 regulates PDZ-RhoGEF-mediated Rho activation by Plexin-B1 and induces cell contraction in COS-7 cells. J Biol Chem 2003; 278:25671-7; PMID:12730235; http://dx.doi.org/ 10.1074/jbc.M303047200 [DOI] [PubMed] [Google Scholar]
- [102].Perrot V, Vazquez-Prado J, Gutkind JS. Plexin B regulates Rho through the guanine nucleotide exchange factors leukemia-associated Rho GEF (LARG) and PDZ-RhoGEF. J Biol Chem 2002; 277:43115-20; PMID:12183458; http://dx.doi.org/ 10.1074/jbc.M206005200 [DOI] [PubMed] [Google Scholar]
- [103].Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem 2001; 276:4527-30; PMID:11096123; http://dx.doi.org/ 10.1074/jbc.R000028200 [DOI] [PubMed] [Google Scholar]
- [104].Ferrari MB, Podugu S, Eskew JD. Assembling the Myofibril. Cell Biochem Biophys 2006; 45:317-37; PMID:16845177; http://dx.doi.org/ 10.1385/CBB:45:3:317 [DOI] [PubMed] [Google Scholar]
- [105].Kuo J-C, Lin J-R, Staddon JM, Hosoya H, Chen R-H. Uncoordinated regulation of stress fibers and focal adhesions by DAP kinase. J Cell Sci 2003; 116:4777-90; PMID:14600263; http://dx.doi.org/ 10.1242/jcs.00794 [DOI] [PubMed] [Google Scholar]
- [106].Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 Proteins. Curr Mol Med 2013; 13:24-57; PMID:22834835; http://dx.doi.org/ 10.2174/156652413804486214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Emberley ED, Murphy LC, Watson PH. S100 proteins and their influence on pro-survival pathways in cancer. Biochem Cell Biol 2004; 82:508-15; PMID:15284904; http://dx.doi.org/ 10.1139/o04-052 [DOI] [PubMed] [Google Scholar]
- [108].Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer 2015; 15:96-109; PMID:25614008; http://dx.doi.org/ 10.1038/nrc3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Chen H, Xu C, Jin Qe, Liu Z. S100 protein family in human cancer. Am J Cancer Res 2014; 4:89-115; PMID:24660101 [PMC free article] [PubMed] [Google Scholar]
- [110].Donato R. Functional roles of S100 proteins, calcium binding proteins of the EF-hand type. Biochim Biophys Acta 1999; 1450:190-231 [DOI] [PubMed] [Google Scholar]
- [111].Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol 2008; 34:357-64; PMID:17566693; http://dx.doi.org/ 10.1016/j.ejso.2007.04.009 [DOI] [PubMed] [Google Scholar]
- [112].Gross SR, Sin CG, Barraclough R, Rudland PS. Joining S100 proteins and migration: for better or for worse, in sickness and in health. Cell Mol Life Sci 2014; 71:1551-79; PMID:23811936; http://dx.doi.org/ 10.1007/s00018-013-1400-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lukanidin E, Sleeman JP. Building the niche: the role of the S100 proteins in metastatic growth. Semin Cancer Biol 2012; 22:216-25; PMID:22381352; http://dx.doi.org/ 10.1016/j.semcancer.2012.02.006 [DOI] [PubMed] [Google Scholar]
- [114].LLoyd BH, Platt-Higgins A, Rudland PS, Barraclough R. Human S100A4 (p9Ka) induces the metastatic phenotype upon benign tumor cells. Oncogene 1998; 17:465-73; PMID:9696040; http://dx.doi.org/ 10.1038/sj.onc.1201948 [DOI] [PubMed] [Google Scholar]
- [115].Maelandsmo GM, Hovig E, Skrede M, Engebraaten O, Florenes VA, Mykiebost O, Grigorian M, Lukanidin E, Scanlon KJ, Fodstad O. Reversal of the in vivo metastatic phenotype of human tumor cells by an anti-CAPL (mts1) ribozyme. Cancer Res 1996; 56:5490-8 [PubMed] [Google Scholar]
- [116].Ji YF, Huang H, Jiang F, Ni RZ, Xiao MB. S100 family signaling network and related proteins in pancreatic cancer (Review). Int J Mol Med 2014; 33:769-76; PMID:24481067 [DOI] [PubMed] [Google Scholar]
- [117].Strutz F, Zeisberg M, Kalluri R, Muller GA, Ziyadeh FN, Yang C-Q, Neilson EG. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 2002; 61:1714-28; PMID:11967021; http://dx.doi.org/ 10.1046/j.1523-1755.2002.00333.x [DOI] [PubMed] [Google Scholar]
- [118].Li J-T, Wang L-F, Zhao Y-L, Yang T, Li W, Zhao J, Yu F, Wang L, Meng Y-L, Liu N-N, et al.. Nuclear factor of activated T cells 5 maintained by Hotair suppression of miR-568 upregulates S100 calcium binding protein A4 to promote breast cancer metastasis. Breast Cancer Res 2014; 16:454; PMID:25311085; http://dx.doi.org/ 10.1186/s13058-014-0454-2 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [119].Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr‐1‐mediated upregulation of Snail. EMBO J 2006; 25:3534-45; PMID:16858414; http://dx.doi.org/ 10.1038/sj.emboj.7601213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. [α]-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol 2010; 12:533-42; PMID:20453849; http://dx.doi.org/ 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
- [121].Borghi N, Sorokina M, Shcherbakova OG, Weis WI, Pruitt BL, Nelson WJ, Dunn AR. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell–cell contacts upon externally applied stretch. Proc Natl Acad Sci U S A 2012; 109:12568-73; PMID:22802638; http://dx.doi.org/ 10.1073/pnas.1204390109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998; 392:933-6; PMID:9582075 [DOI] [PubMed] [Google Scholar]
- [123].Tomida T, Hirose K, Takizawa A, Shibasaki F, Iino M. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J 2003; 22:3825-32; PMID:12881417; http://dx.doi.org/ 10.1093/emboj/cdg381 [DOI] [PMC free article] [PubMed] [Google Scholar]