

Abstract
Alcohol induced hepatic steatosis is a significant risk factor for progressive liver disease. Cyclic adenosine monophosphate (cAMP) signalling has been shown to significantly regulate lipid metabolism; however, the role of altered cAMP homeostasis in alcohol mediated hepatic steatosis has never been studied. Our previous work demonstrated that increased expression of hepatic phosphodiesterase 4 (Pde4), which specifically hydrolyses and decreases cAMP levels, plays a pathogenic role in the development of liver inflammation/injury. The aim of this study was to examine the role of PDE4 in alcohol-induced hepatic steatosis. C57BL/6 wild type and Pde4b knockout (Pde4b−/−) mice were pair-fed control or ethanol liquid diets. One group of wild type mice received rolipram, a PDE4 specific inhibitor, during alcohol feeding. We demonstrate for the first time that an early increase in PDE4 enzyme expression and a resultant decrease in hepatic cAMP levels are associated with the significant reduction in carnitine palmitoyltransferase 1A (Cpt1a) expression. Notably, alcohol fed (AF) Pde4b−/− mice and AF wild type mice treated with Rolipram had significantly lower hepatic free fatty acid content compared to AF wild type mice. Importantly, PDE4 inhibition in alcohol fed mice prevented the decrease in hepatic Cpt1a expression via the Pparα/Sirt1/Pgc1α pathway. These results demonstrate that the alcohol- induced increase in hepatic Pde4, specifically Pde4b expression, and compromised cAMP signalling predisposes the liver to impaired fatty acid oxidation and the development of steatosis. Moreover, these data also suggest that hepatic PDE4 may be a clinically relevant therapeutic target for the treatment of alcohol induced hepatic steatosis.
Keywords: alcohol, cAMP, PDE4, PPARα, PGC1α, SIRT1, CPT1A, hepatic steatosis
Introduction
Alcohol is a leading cause of liver disease and liver-related deaths [1, 2]. Hepatic steatosis (fatty liver) is the initial stage of alcoholic liver disease and the first response to both chronic and acute alcohol consumption. Although alcohol-induced hepatic steatosis is reversible and considered to be benign, it is well-established that steatosis predisposes the liver to more advanced pathologies, such as alcoholic steatohepatitis (ASH), hepatic fibrosis, cirrhosis and even hepatocellular carcinoma [3–5]. Alcohol induced hepatic steatosis is largely mediated by increased de novo lipogenesis and impaired fatty acid beta-oxidation [6]. Several studies have identified the genes involved in alcohol induced dysregulation of lipid metabolism leading to steatosis [7, 8]; however, gaps remain in the understanding of underlying molecular mechanism(s) that contribute to altered expression of genes involved in hepatic steatosis.
Alcohol mediated down-regulation of fatty acid oxidation plays a critical role in the development of alcohol-induced hepatic steatosis. Among the different types of free fatty acid oxidation, mitochondrial free fatty acid β-oxidation has been shown to be significantly impaired by alcohol [9]. Specifically, carnitine palmitoyltransferase-1A (Cpt1a), a rate-limiting step in β-oxidation, has been shown to be down regulated by alcohol [10–12]. Cpt1 expression is regulated by complex transcriptional machinery involving several transcription factors (TF) and co-activators including, Pparα, Pgc1α, Sirt1, Creb, etc. [13–15]. In alcohol induced hepatic steatosis, ethanol exposure has been shown to decrease Pparα and Pgc1α expression/activity, resulting in the reduction of β-oxidation [11, 16–19]. Importantly, regulation of Cpt1a transcriptional activation can be modulated by a critical second messenger, cAMP, via its effectors, protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC), in various cell types including hepatocytes [13, 14, 20, 21]. Despite the evidence of a critical role for cAMP signalling in the regulation of Cpt1a, there are no studies examining whether or not the alcohol effects on transcriptional downregulation of Cpt1a in hepatocytes involves this pathway.
Intracellular levels of cAMP are tightly regulated by the coordinated control of its synthesis via adenylyl cyclase, and its degradation via a large family of phosphodiesterases (PDEs). An increase in cAMP levels triggers a signalling cascade leading to regulation of numerous protein activities and gene expression. The duration and amplitude of cAMP signalling is controlled exclusively by PDEs via cAMP degradation. Thus, any changes in PDE expression will have a significant effect on cAMP signalling. Among the three cAMP specific PDEs (PDE3, PDE4 and PDE7), the PDE4 family is the largest and most ubiquitous, with four genes (PDE4A/B/C/D) encoding over 20 distinct PDE4 isoforms [22, 23].
Previous studies have shown that alcohol affects G protein-coupled receptor stimulated cAMP production in various immune cells through changes in the expression of G protein αs (Gαs) which stimulates adenylyl cyclase to produce cAMP [24]. An effect of alcohol on receptor stimulated cAMP production has also been shown in isolated hepatocytes; however the effect was dependent on alcohol concentration [25, 26]. Specifically, acute exposure (48 h) of hepatocytes with alcohol concentration up to 50 mM had a suppressive effect, whereas high concentrations (50–100 mM) resulted in increased production of cAMP in response to glucagon and adenosine [25, 26]. However, alcohol had no effect on the basal adenylyl cyclase activity [26].
Our earlier work has demonstrated that chronic alcohol exposure can significantly decrease cellular cAMP levels in both receptor-independent and receptor-dependent manners by increasing the expression of cAMP-specific phosphodiesterase 4 (Pde4) in monocytes/macrophages and Kupffer cells [27, 28]. We have also shown that Pde4 enzymes play a pathogenic role in the development of hepatic inflammation and injury in rats [29]. Moreover, PDE inhibitors have been shown to be beneficial in experimental liver injury [29–36], but there have been no studies evaluating the causal role of Pde4 in the development of alcoholic fatty liver. Based on the critical role of cAMP signalling in transcriptional regulation of Cpt1a, and on our previous work, we examined the role of the alcohol mediated increase in hepatic Pde4 expression and a resultant decrease in cAMP signalling in the transcriptional downregulation of Cpt1a. We show for the first time that alcohol induces Pde4 expression in the liver and decreases hepatic cAMP levels. This decrease was associated with the development of hepatic steatosis and a reduction of Cpt1a expression. Using pharmacological and genetic approaches, we examined the causal role of alcohol-induced Pde4 expression/cAMP metabolism in the down-regulation of hepatic Cpt1a expression and consequent development of steatosis. This is the first study demonstrating the contributory role of PDE4 expression to the pathogenesis of alcoholic liver disease (ALD).
Materials and Methods
Animal model
Twelve-week old male C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). A breeding pair of Pde4b knockout mice generated on C57BL/6 background was a kind gift from Prof. Marco Conti (UCSF). All mice were housed in a pathogen-free, temperature-controlled animal facility with 12 h light/12 h dark cycles. All experiments were carried out according to the criteria outlined in the Guide for Care and Use of Laboratory Animals and with approval of the University of Louisville Institutional Animal Care and Use Committee.
C57BL/6 and Pde4b knockout male mice were pair-fed the Lieber-DeCarli liquid diet (Lieber-DeCarli type, Bioserv, Frenchtown, NJ) containing either alcohol (AF) or isocaloric maltose dextrin (PF) for 4 weeks. Alcohol was gradually increased over a period of one week and then mice were fed the ethanol diet [5% (v/v)] ad libitum for 4 weeks (AF). The control pair-fed (PF) mice were given the isocaloric liquid diet. Additional groups of AF and PF animals were treated with PDE4 specific inhibitor, rolipram, at 5 mg/kg, 3 times a week for 4 weeks. Rolipram (C16H21NO3) (Biomol, Enzo Life Sciences, Farmingdale, NY) was dissolved in sterile DMSO and diluted with sterile phosphate buffered saline just before injection. Mice were sacrificed at 1, 2 and 4 wk after starting 5% alcohol. At sacrifice, mice were anesthetized with intraperitoneal injection of Nembutal, 80mg/kg. There were 5–7 mice in each experimental group. Whole blood was collected and plasma was stored at −80°C for analysis. Liver tissue was cut into smaller pieces, snap-frozen in liquid nitrogen and stored at −80°C. An additional liver piece was fixed in 10% neutral-buffered formalin for immunohistochemical analysis.
Blood alcohol levels
Blood alcohol levels were measured in freshly drawn samples using an Ethanol Assay Kit (Sigma, St. Louis, MO), according to the manufacturer’s instructions. Whole blood was centrifuged at 4°C and plasma was diluted prior to measurement.
Primary hepatocyte culture and treatments
Primary hepatocytes were isolated from livers of male Sprague Dawley rats as described previously [37]. Treatments were performed 24 h after isolation. Cells were treated with a PKA inhibitor (H89), the cAMP analogue dibutyryl cAMP (dbcAMP) and the PKA-selective activators N6-Phenyl adenosine-3′, 5′-cyclic monophosphate (N6-Phenyl-cAMP) and Sp-5,6-DCl-cBIMPS for 24 h. Cells were pre-treated for 30 min with H89 before dbcAMP treatment. Both dbcAMP and H89 were purchased from Enzo Life Sciences, Inc. (Farmingdale, NY) and N6-Phenyl-cAMP and Sp-5,6-DCl-cBIMPS from BioLog (BIOLOG Life Science Institute, Bremen, Germany).
PDE4 Enzymatic Assay
Liver tissue (50 mg) was lysed in 1 ml of lysis buffer containing 20 mM HEPES, 150 mM NaCl, 2 mM EDTA, 5% glycerol and 1% NP-40 supplemented with protease and phosphatase inhibitors. PDE4-specific enzymatic activity was determined using a PDE4 assay kit (FabGennix Inc. International, Frisco, TX) as described previously [38].
Western blot analysis
Liver tissue (50 mg) was lysed using RIPA buffer containing protease and phosphatase inhibitor (Sigma-Aldrich, St. Louis, MO). For nuclear protein extraction, an EpiSeeker Nuclear Extraction Kit from Abcam (Abcam, Cambridge, MA) was used. Proteins (25 μg) were analysed by SDS-polyacrylamide gel electrophoresis using a Bio-Rad (Hercules, CA) electrophoresis system. Western blot membranes were stripped using Restore™ PLUS Stripping Buffer (Thermo Scientific, Rockford, IL) and re-probed using another antibody. Immunoreactive bands were visualized using enhanced chemiluminescence light detection reagents (Amersham, Arlington Heights, IL). Quantification was performed with Image LabTMSoftware (BioRad, Life Science Research, Hercules, CA). PPARα, CPT1A, PGC1, SIRT1, PDE4C, PDE4D, β-actin and Histone 3 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). PDE4B antibody was a kind gift from Dr. Marco Conti; PDE4A antibody was a gift from FabGennix Inc. (FabGennix Inc. International, Frisco, TX). The information about the source and dilution of primary antibodies is presented in Supplementary Table S1.
RNA isolation and Reverse Transcription (RT)-qPCR analysis
Total RNA was isolated from 50mg liver tissue using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and RT-qPCR was performed as described previously [29]. The specific primers listed in Table 1 were purchased from Integrated DNA Technologies (IDT) (Coralville, Iowa).
Table 1.
Primers for RT-qPCR
| Mouse Pde4a | |
| Pde4a_F | 5′-CACAGCCTCTGTGGAGAAGTC-3′ |
| Pde4a_R | 5′-GTGATACCAATCCCGGTTGTC-3′ |
| Mouse Pde4b | |
| Pde4b_F | 5′-GACCGGATACAGGTTCTTCG-3′ |
| Pde4b_R | 5′-CAGTGGATGGACAATGTAGTCA-3′ |
| Mouse/Rat Pde4c | |
| Pde4c_F | 5′-TTTCTCATCAACACCAACTCAGA-3′ |
| Pde4c_R | 5′-CTGCAGGAGCTTGAAGCCTA-3′ |
| Mouse Pde4d | |
| Pde4d_F | 5′-TGTCCACAGTCAACGCCGGGAG-3′ |
| Pde4d_R | 5′-CCAAGACCTGAGCAAACGGGGTCA-3′ |
| Mouse Cpt1a | |
| Cpt1a_F | 5′-GCTGCACTCCTGGAAGAAGA-3′ |
| Cpt1a_R | 5′-GGAGGGGTCCACTTTGGTAT-3′ |
| Mouse Cyp2e1 | |
| Cyp2e1_F | 5′-AGGGGACATTCCTGTGTTCC-3′ |
| Cyp2e1_R | 5′-TTACCCTGTTTCCCCATTCC-3′ |
| Mouse Sirt1 | |
| Sirt1_F | 5′-CAGACCCTCAAGCCATGTTT-3′ |
| Sirt1_R | 5′-ACACAGAGACGGCTGGAACT-3′ |
| Mouse Ppargc1a | |
| Ppargc1a_F | 5′-ACAGCTTTCTGGGTGGATTG-3′ |
| Ppargc1a_R | 5′-CGCTAGCAAGTTTGCCTCAT-3′ |
| Rat Cpt1a | |
| Cpt1a_F | 5′-CTGCATGGAAGATGCTTTGA-3′ |
| Cpt1a_R | 5′-GCCATGACATACTCCCACAA-3′ |
Histopathology, immunohistochemistry and Oil Red O staining
Histological analyses were performed as described previously [10]. CPT1A and pCREB antibodies were purchased from Proteintech Group Inc., (Chicago, IL) and Abcam (Cambridge, MA) respectively (Supplementary Table S1).
Quantification of phosphorylated CREB immunohistochemistry
The pCREB positive (3,3′-diaminobenzidine-stained in brown) and negative (hematoxylin-stained in blue) cells were counted from five representative liver fields (objective magnification 20x) of each mouse using a freely available image analysis software http://www.cellprofiler.org. The percent ratio of pCREB-positive cells over total cell number was calculated for each experimental group and presented as mean ± standard deviation.
Hepatic Free Fatty Acids
Hepatic tissue (100 mg) was homogenized in 1 ml of 50 mM NaCl and an aliquot (500 μl) mixed with chloroform/methanol (2:1, 4 ml) then incubated overnight at room temperature with gentle shaking. Homogenates were vortexed and centrifuged for 5 min at 3000 x g. The lower lipid phase was collected and concentrated by vacuum. The lipid pellets were dissolved in 1% Triton X100 in phosphate-buffered saline. Liver non-esterified fatty acids (NEFAs) were assayed using a commercially available kit; HR Series NEFA-HR (2) from Wako Chemical USA (Richmond, VA).
Measurements of cAMP levels
Hepatic tissues and hepatocytes were homogenized and cAMP assayed using a cAMP ELISA kit (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer’s instructions. Homogenates were vortexed and centrifuged for 10 min at > 600 x g to pellet the debris. Levels were normalized by protein content and are presented as pmol/mg protein.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism Software. Data are presented as the mean ± standard deviation (SD). Statistical significance was calculated using one-way ANOVA followed by Bonferroni’s post-test. P<0.05 was considered significant.
Results
Alcohol upregulates hepatic Pde4 expression leading to decreased levels of cAMP
To address the potential pathogenic role of PDE4 in the development of alcohol induced hepatic steatosis, hepatic PDE4 expression was initially analysed in mice fed control (pair-fed, PF) and alcohol (alcohol-fed, AF) diet. A significant up-regulation of mRNA levels for all PDE4 subfamilies, Pde4a, b, c and d, was observed as early as following 1 wk of alcohol feeding compared to controls (Figure 1A). The increase in Pde4 mRNA was accompanied by elevated Pde4 enzymatic activity in the liver (Figure 1B). Upregulated Pde4 expression continued for two wk with a subsequent decline to baseline levels at 4 weeks (data not shown). Corresponding to the increased Pde4 expression, a sustained decrease in hepatic cAMP levels was observed in AF mice compared to PF mice (Figure 1C). Analysis of hepatocytes isolated from mice fed control and alcohol diets for 2 wk confirmed that alcohol feeding resulted in decreased hepatocyte cAMP levels (Figure 1D). Additionally, the effect of alcohol on Pde4 expression was examined on primary rat hepatocytes in vitro. Treatment of hepatocytes with 50 mM alcohol for 48 hours led to a significant upregulation of Pde4(a–d) mRNA and protein levels (Figure 1E and F). These results show that alcohol affects hepatocyte Pde4 expression both in vivo and in vitro.
Figure 1.

Significant upregulation of hepatic Pde4 expression in alcohol fed mice. (A) mRNA levels of Pde4a, Pde4b, Pde4c, and Pde4d (n=5–7 mice per group). (B) Hepatic Pde4-specific activity after 1 wk of alcohol feeding (n=5–7). (C) Hepatic cAMP levels after 1 and 2 wk of alcohol feeding (n=5–7). (D) cAMP levels in hepatocytes isolated after 2 wk of alcohol feeding (n=3 mice per group). (E) levels of Pde4a, Pde4b, Pde4c, and Pde4d mRNAs in primary rat hepatocytes after 48 h of alcohol exposure (50 mM) (n=3). (F) Representative Western blot images of Pde4a, b, c and d protein levels in primary rat hepatocytes after 48 h of alcohol exposure (50 mM). Data are presented as mean ± S.D. *P < 0.05, **P<0.01compared to PF and UT.
Pde4 inhibition prevents alcohol mediated fat accumulation in the liver
To examine if increased expression of PDE4 enzymes and a resultant decrease in hepatic cAMP levels play a causal role in the development of alcohol induced hepatic steatosis, we employed both pharmacological (rolipram) and gene-knockout approaches (Pde4−/−) to block the activity of PDE4 and prevent the degradation of cAMP. Alcohol intake was carefully monitored throughout the feeding. No differences were observed in food consumption between the study groups. Moreover, alcohol-inducible hepatic Cyp2e1, as well as blood alcohol levels, were not significantly affected in rolipram-treated or Pde4b−/− mice, indicating that PDE4 inhibition does not influence ethanol metabolism (Supplementary Figure 1A–C).
Histological analysis showed that alcohol feeding led to a significant hepatic fat accumulation in wild type mice, which was significantly attenuated in both Pde4b−/− and rolipram-treated and mice (Figure 2A, B). Further, correspondent to the histological analysis, biochemical evaluation of hepatic free fatty acids also showed a marked reduction in FFA levels by Pde4b inhibition (Figure 2C).
Figure 2.

Pde4 inhibition attenuates alcohol induced lipid accumulation in the liver. (A) H&E staining. (B) Oil red O staining. (C) Hepatic free fatty acids (FFA). Data are presented as the mean ± SD, n=5–7 mice per group. *P<0.05, ** P< 0.01.
Pde4 inhibition prevents alcohol induced decrease in hepatic cAMP levels and Cpt1a expression
Development of alcohol induced hepatic steatosis is significantly mediated by decreased expression of Cpt1a, a rate limiting enzyme in fatty acid β-oxidation [10, 11, 39]. Immunohistochemical staining of Cpt1a in liver sections showed a decrease of Cpt1a in alcohol fed WT mice compared to WT pair fed mice (Fig. 3A). In comparison, alcohol fed WT mice treated with rolipram and Pde4b−/− mice showed no decrease of Cpt1a compared to AF WT mice (Fig. 3A). RT-qPCR analysis of hepatic Cpt1a mRNA levels also demonstrated that PDE4 inhibition prevented an alcohol mediated decrease in Cpt1a mRNA (Figure 3B).
Figure 3.
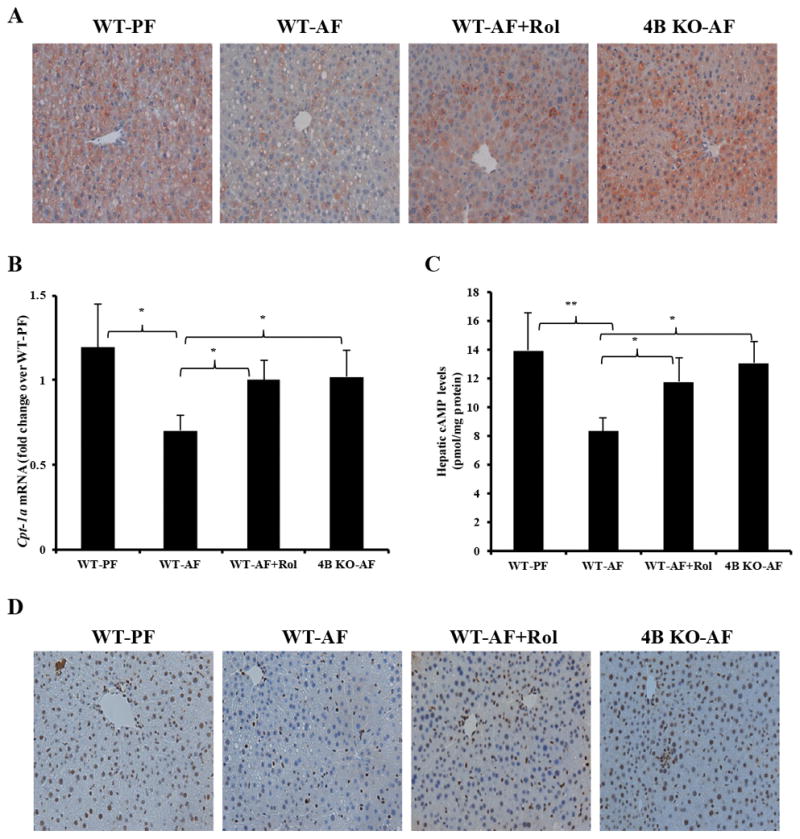
Effect of Pde4 inhibition on hepatic Cpt1a expression and cAMP/pCREB levels. (A) Immunohistochemical staining with anti-CPT-1A antibody (×20 final magnification). (B) Cpt1a mRNA levels after 4 wk of feeding. (C) Hepatic cAMP levels after 2 wk of alcohol feeding. (D) Nuclear pCREB staining of livers after 4 wk of alcohol feeding. Data are presented as the mean ± SD (n=5–7 mice per group). *P<0.05, ** P< 0.01, *** P< 0.001.
Previous work has shown that in hepatocytes, cAMP induces Cpt1a expression and involves cAMP response element binding protein (CREB) [14, 40]. To examine whether the observed decrease in Cpt1a expression by alcohol was a result of cAMP-mediated changes in phosphorylated CREB (pCREB) levels, we examined hepatic levels of pCREB. Indeed, nuclear staining of active pCREB in WT-AF mice livers was significantly lower compared to WT-PF mice (Figure 3C, D and Supplementary Figure S2). By comparison, Pde4b inhibition in alcohol fed mice led to maintained levels of both cAMP and pCREB (Figure 3C, D), indicating that among the Pde4 sub-family of enzymes, Pde4b is primarily involved in the alcohol-inducible decrease in hepatic cAMP levels and consequently Cpt1a expression.
Effect of cAMP signalling on Cpt1a expression in primary hepatocytes
To further examine if cAMP dependent signalling in hepatocytes influences Cpt1a expression, we performed in vitro experiments using primary rat hepatocytes. Specifically, cells were treated with the cAMP-specific protein kinase A (PKA) inhibitor, H89 (to decrease cAMP signalling), or the non-degradable cAMP analogue, dbcAMP (to increase cAMP signalling). Western blot analysis of pCREB levels confirmed that PKA inhibition by H89 significantly decreased CREB phosphorylation (Figure 4A). H89 treatment also lowered Cpt1a mRNA levels (Figure 4B), whereas dbcAMP increased Cpt1a expression (Figure 4B). Moreover, co-treatment of hepatocytes with H89 and dbcAMP mitigated the dbcAMP effect on Cpt1a mRNA, demonstrating that the dbcAMP effect is significantly mediated by PKA (Figure 4B). Consistent with the dbcAMP mediated increase in Cpt1a expression, PKA-selective activators, which do not activate EPAC (N6-Phenyl-cAMP and Sp-5,6-DCl-cBIMPS) significantly increased baseline pCREB levels and consequently Cpt1a mRNA expression (Figure 4C and D). These findings are in agreement with previous work demonstrating that cAMP signalling via PKA/CREB plays a critical role in the transcriptional regulation of Cpt1a in hepatocytes [14]. Taken together, these results demonstrate that Pde4-mediated decreases in hepatic cAMP signalling contribute to alcohol-induced reduction in hepatic Cpt1a expression and development of steatosis.
Figure 4.

Effect of cAMP signalling on hepatocyte Cpt1a mRNA expression. Rat primary hepatocytes were treated with PKA inhibitor, H89 (10 μM) followed by dbcAMP (250 μM) for 24 h. (A) pCREB levels in primary rat hepatocytes are decreased after H89 treatment. (B) Cpt1a mRNA expression in primary rat hepatocytes. (C) Rat primary hepatocytes were treated with PKA-selective activators N6-Phenyl-cAMP (500 μM) and Sp-5,6-DCl-cBIMPS (100 μM) for 24 h. (D) pCREB levels after 24 h treatment of rat primary hepatocytes with N6-Phenyl-cAMP (500 μM) and Sp-5,6-DCl-cBIMPS (100 μM). Data are presented as mean ± SD from 3 independent experiments. **P<0.01, ***P<0.01.
Effect of Pde4 inhibition on Pparα and Pgc1α
Expression of the Cpt1a gene is critically regulated by the transcription factor Pparα and its coactivator, Pgc1α (Ppar gamma coactivator-1α) [13, 19, 41]. Alcohol feeding has been shown to decrease hepatic Pparα levels [11]. Thus, we first examined whether PDE4 inhibition had any effect on Pparα. Western blot and real time PCR analysis of Pparα expression levels confirmed that alcohol lowered Pparα protein levels in WT-AF compared to WT-PF mice (Figure 5A, B). In contrast, Pparα levels were maintained in alcohol fed Pde4b−/− and rolipram-treated mice (Figure 5A, B). Further, we examined whether Pde4 inhibition had any effect on Pgc1α levels. Alcohol feeding resulted in a modest but statistically non-significant decrease in hepatic PGC1α (Ppargc1a) mRNA levels (Figure 5C), as observed by other studies [17, 42]; however the levels were significantly higher in AF Pde4b−/− and rolipram-treated AF mice compared to wild type AF mice (WT-AF) (Figure 5C). Western blot analysis of nuclear Pgc1α also confirmed the increase in Pgc1α levels in Pde4-inhibited mice fed alcohol when compared to alcohol fed WT mice (Figure 5D).
Figure 5.

Effect of Pde4 inhibition on hepatic Pparα and Pgc1α expression after 4 wk of feeding. (A) Western blot analysis of nuclear Pparα protein levels. (B) Pparα mRNA levels were quantified by RT-qPCR. (C) Pgc1α (Ppargc1a) mRNA levels were quantified by RT-qPCR. (D) Western blot analysis of nuclear PGC1α protein levels. Data are presented as mean ± SD (n = 5–7). *P < 0.05, **P < 0.01. Hepatic nuclear lysates from 3 mice/treatment group were resolved using a gradient gel to allow the simultaneous examination of protein levels of Pparα (55kDa) and Pgc1α (90kDa) on the same membrane. Histone 3 (15kDa) served as a loading control for both Pparα and Pgc1α.
Pde4 inhibition increases Sirt1 expression
Sirt1 deacetylase plays a critical role in the regulation of transcriptional activity of several transcription factors including Pgc1α [13, 42–45]. An increase in hepatic Sirt1 expression has been shown to attenuate alcoholic hepatic steatosis [46]. Importantly, previous studies have shown that agents which increase cAMP levels also increase Sirt1 expression and decrease hepatic steatosis [20, 39, 47]. Hence, we evaluated the effect of Pde4 inhibition on hepatic Sirt1 expression. Examination of hepatic Sirt1 mRNA levels showed that there was a modest increase in Sirt1 mRNA expression in WT-AF mice compared to WT-PF consistent with a previous report [48]; however, Pde4b−/− and rolipram treated mice showed a significant increase in Sirt1 mRNA levels after alcohol feeding when compared to WT-AF mice (Figure 6A). Similar to mRNA levels, Pde4 inhibition also led to increased Sirt1 protein levels (Figure 6B).
Figure 6.

Pde4 inhibition increases hepatic Sirt1 expression. (A) After 4 wk of feeding Sirt1 mRNA levels were quantified by RT-qPCR (n=5–7). (B) Western blot analysis of nuclear Sirt1 protein levels after 4 wk of feeding. Western blot membrane probed with Pparα and Pgc1α was stripped and re-probed using Sirt1 (120kDa) antibody. Data are presented as mean ± SD *P < 0.05, ***P<0.001.
Discussion
Alcoholic fatty liver is the first manifestation of alcoholic liver disease, and it develops in more than 90% of alcohol drinkers. Steatosis is considered to be a benign condition, although it has been demonstrated that the degree of steatosis positively correlates with the progression to more severe forms of ALD, such as alcoholic hepatitis and fibrosis [49–51]. Alcohol induced hepatic steatosis is mediated by dysregulated lipid metabolism, largely involving impaired fatty acid β-oxidation [9]. Several studies have examined the impact of alcohol on β-oxidation and the development of hepatic steatosis, although the underlying mechanistic determinants are not completely understood. Signalling through cAMP has been shown to critically regulate lipid metabolism, including fatty acid β-oxidation [13, 14, 21, 52]. Our earlier work demonstrated that chronic alcohol exposure can significantly induce Pde4 expression and decrease cAMP levels in hepatic Kupffer cells and monocytes/macrophages, affecting LPS-inducible inflammatory gene expression [27, 28]. However, the effect of alcohol on hepatocyte Pde4 as a regulator of cAMP signalling in relation to lipid metabolism has never been investigated. Hence, in the present work, we examined the causal role of alcohol-induced hepatic Pde4 expression and compromised cAMP metabolism in the development of steatosis using pharmacological and genetic approaches.
Our results show that alcohol significantly decreases hepatic cAMP levels early on in alcohol feeding following the upregulation of Pde4 expression. This decline in cAMP levels is accompanied by a decrease in pCREB levels and a reduction in Cpt1a expression (a rate limiting enzyme in mitochondrial fatty acid β-oxidation). Importantly, alcohol effects on hepatic cAMP and Cpt1a levels were abrogated by Pde4 inhibition via both pharmacological and genetic approaches. These findings suggested a pivotal role for Pde4 in decreasing hepatic cAMP levels in response to chronic alcohol feeding. Further, with regard to the involvement of a distinct Pde4 subfamily member, the data from Pde4b−/− animals strongly support the pathogenic involvement of Pde4b expression in the alcohol-induced decline in hepatic cAMP levels. Moreover, the key role of the gut-derived endotoxaemia in ALD [2] also emphasizes the role of PDE4B, which is known to be endotoxin responsive [38, 53, 54].
Cpt1a expression is regulated by various transcription factors and co-activators [13–15]. In experimental animal models of ALD, development of hepatic steatosis and reduction in Cpt1a expression is attributed to a decreased expression and transcriptional activity of Pparα [11, 55]. Our results show that alcohol significantly decreased Pparα expression and this decline was associated with decreased hepatic cAMP levels. Importantly, the prevention of cAMP degradation by Pde4 inhibition prevented a decrease in Pparα. These results could be explained by previous studies showing that cAMP promotes Pparα expression in hepatocytes [56]. It is well established that optimal transcriptional activity of Pparα requires formation of a complex with its critical co-activator Pgc1α [13, 15]. Pgc1α interacts with and recruits proteins having histone acetylating activities which then results in transcriptionally permissive promoter histone modification allowing increased transcription factor binding [57]. Pgc1α in turn, must be de-acetylated by Sirt1 to function as a co-activator [58, 59]. The roles of Pgc1α and Sirt1 in promoting Pparα mediated fatty acid β-oxidation have been extensively studied in alcohol induced hepatic steatosis [16–18, 39, 46, 60]. Our results show a marked increase in Pgc1α and Sirt1 expression in both rolipram-treated and Pde4b−/− knockout alcohol fed mice when compared to wild type AF mice. These findings are in agreement with previous studies demonstrating that the proximal region of Pgc1α contains functional CREB binding sites which respond to increased cAMP levels by inducing Pgc1α (Ppargc1a) mRNA [14, 56, 58, 61]. There are also several CREB binding sites in the Sirt1 promoter, and CREB has been shown to increase Sirt1 expression [47, 62]. In this regard, our data show that decreased pCREB levels did not have a significant negative effect on Sirt1 mRNA expression in alcohol fed wild type mice. These results suggest that under alcohol feeding conditions, there are other mechanisms which could play a role in Sirt1 transcriptional induction. Indeed, more recently, NFκB has been identified as a significant transcription factor for Sirt1 expression [63]. Therefore, the alcohol mediated increase in hepatic NFκB activation [64, 65] could have contributed to maintaining Sirt1 levels, even in the presence of increased Pde4 and decreased pCREB expression. It is noteworthy that in addition to transcriptional induction, cAMP via PKA and EPAC has been shown to increase Sirt1 activity [20, 66]. Hence, it is possible that although the Sirt1 mRNA levels were not affected in AF-WT mice, its function/activity was negatively impacted by decreased cAMP levels.
Taken together the findings strongly support the role of increased hepatic Pde4 expression and decreased cAMP levels in the alcohol-induced hepatic lipid accumulation by down regulating the expression of Cpt1a, a key rate limiting enzyme in fatty acid β-oxidation. However, increased de novo lipogenesis via increased expression of lipogenic genes also contributes to alcohol-induced hepatic steatosis. In this regard, it has been demonstrated that cAMP signalling can suppress the expression/function of several lipogenic genes and lipid synthesis [67–71]. In view of this, it is possible that besides decreasing β-oxidation, alcohol-induced Pde4 expression also played a role in enhancing hepatic lipogenesis which was targeted in rolipram treated and Pde4b KO animals. Although, due to the scope of the present work, the role of Pde4 expression and cAMP in the regulation of hepatic lipogenesis was not examined and is currently being pursued as a part of ongoing studies.
The present work provides supporting evidence for PDE4 as a potential therapeutic target in the treatment of early ALD. Indeed, PDE4-specific inhibitors have been recently approved by the FDA for the treatment of certain non-hepatic inflammatory diseases [72–75] and could provide therapeutic benefit in patients with ALD. In this regard, Pentoxifylline (PTX), a relatively weak non-selective phosphodiesterase inhibitor has shown efficacy in some initial studies in severe alcoholic hepatitis (AH) [76, 77], and is used by many hepatologists as an alternative to glucocorticoids. With respect to hepatic steatosis, several smaller studies have shown that PTX reduces hepatic steatosis and inflammation in patients with non-alcoholic steatohepatitis (NASH) [78–80]. Moreover, pre-clinical data from our group and others suggest that inhibition of PDE4 activity may not only decrease hepatic steatosis but also attenuate Kupffer cell pro-inflammatory cytokine production and hepatic stellate cell activation and fibrosis [27–29].
In summary, we demonstrate for the first time that alcohol increases hepatic Pde4 expression leading to a decrease in cAMP levels and downstream cAMP/PKA/CREB signalling. Importantly, the data strongly support a predominant pathogenic role for Pde4b (among the Pde4 sub-family members) in the down-regulation of Cpt1a expression and consequent development of alcohol-induced hepatic steatosis. It is noteworthy that the present work also identifies PDE4 as a potential therapeutic target in the treatment of alcoholic fatty liver disease.
Supplementary Material
Figure S1. Effect of Pde4 inhibition on alcohol metabolism
Figure S2. Quantification of immunohistochemical staining for hepatic pCREB levels
Table S1. Information about primary antibodies used in this study
Acknowledgments
We thank Marion McClain for editing the manuscript. This work was supported by grants from National Institute on Alcohol Abuse and Alcoholism (NIAAA) R21AA022189 (LG), U01AA021901 (CJM), R01AA023681 (CJM) and R01AA018869 (CJM). We would like to acknowledge the NIAAA-funded Southern California Research Center for ALPD and Cirrhosis, the Animal Core facilities (P50 AA011999) model and the Non-Parenchymal Liver Cell Core (R24 AA012885) for providing isolated mouse hepatocytes from Lieber-De Carli ALD model.
Footnotes
Authors declare no conflicts of interest
Authorship contributions
LG, CJM and SB designed the study and interpreted the results; DVA, DFB and JZ performed the experiments; DVA, LG performed data analysis; DVA, SB and LG wrote the manuscript.
References
- 1.Dugum M, McCullough A. Diagnosis and management of alcoholic liver disease. J Clin Transl Hepatol. 2015;3:109–116. doi: 10.14218/JCTH.2015.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148:30–36. doi: 10.1053/j.gastro.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An L, Wang X, Cederbaum AI. Cytokines in alcoholic liver disease. Arch Toxicol. 2012;86:1337–1348. doi: 10.1007/s00204-012-0814-6. [DOI] [PubMed] [Google Scholar]
- 4.Mantena SK, King AL, Andringa KK, et al. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdelmegeed MA, Banerjee A, Jang S, et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol Med. 2013;65:1238–1245. doi: 10.1016/j.freeradbiomed.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco MP, Marco C, Segovia JL. Chronic ingestion of ethanol stimulates lipogenic response in rat hepatocytes. Life Sci. 2001;68:1295–1304. doi: 10.1016/s0024-3205(00)01035-3. [DOI] [PubMed] [Google Scholar]
- 8.Carrasco MP, Jimenez-Lopez JM, Segovia JL, et al. Comparative study of the effects of short- and long-term ethanol treatment and alcohol withdrawal on phospholipid biosynthesis in rat hepatocytes. Comp Biochem Physiol B Biochem Mol Biol. 2002;131:491–497. doi: 10.1016/s1096-4959(02)00006-4. [DOI] [PubMed] [Google Scholar]
- 9.Wanders RJ, Ruiter JP, LIJ, et al. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis. 2010;33:479–494. doi: 10.1007/s10545-010-9104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirpich I, Zhang J, Gobejishvili L, et al. Binge ethanol-induced HDAC3 down-regulates Cpt1alpha expression leading to hepatic steatosis and injury. Alcohol Clin Exp Res. 2013;37:1920–1929. doi: 10.1111/acer.12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang X, Zhong W, Liu J, et al. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–1250. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alenghat T, Meyers K, Mullican SE, et al. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature. 2008;456:997–1000. doi: 10.1038/nature07541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugden MC, Caton PW, Holness MJ. PPAR control: it’s SIRTainly as easy as PGC. J Endocrinol. 2010;204:93–104. doi: 10.1677/JOE-09-0359. [DOI] [PubMed] [Google Scholar]
- 14.Louet JF, Hayhurst G, Gonzalez FJ, et al. The coactivator PGC-1 is involved in the regulation of the liver carnitine palmitoyltransferase I gene expression by cAMP in combination with HNF4 alpha and cAMP-response element-binding protein (CREB) J Biol Chem. 2002;277:37991–38000. doi: 10.1074/jbc.M205087200. [DOI] [PubMed] [Google Scholar]
- 15.Song S, Attia RR, Connaughton S, et al. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol Cell Endocrinol. 2010;325:54–63. doi: 10.1016/j.mce.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Liu Y, Gao R, et al. Ethanol suppresses PGC-1alpha expression by interfering with the cAMP-CREB pathway in neuronal cells. PLoS One. 2014;9:e104247. doi: 10.1371/journal.pone.0104247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaung WW, Jacob A, Ji Y, et al. Suppression of PGC-1alpha by Ethanol: Implications of Its Role in Alcohol Induced Liver Injury. Int J Clin Exp Med. 2008;1:161–170. [PMC free article] [PubMed] [Google Scholar]
- 18.Lieber CS, Leo MA, Wang X, et al. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun. 2008;370:44–48. doi: 10.1016/j.bbrc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 19.You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- 20.Park SJ, Ahmad F, Philp A, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148:421–433. doi: 10.1016/j.cell.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lazennec G, Canaple L, Saugy D, et al. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol Endocrinol. 2000;14:1962–1975. doi: 10.1210/mend.14.12.0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Mochly-Rosen D, Chang FH, Cheever L, et al. Chronic ethanol causes heterologous desensitization of receptors by reducing alpha s messenger RNA. Nature. 1988;333:848–850. doi: 10.1038/333848a0. [DOI] [PubMed] [Google Scholar]
- 25.Nagy LE. Role of adenosine A1 receptors in inhibition of receptor-stimulated cyclic AMP production by ethanol in hepatocytes. Biochem Pharmacol. 1994;48:2091–2096. doi: 10.1016/0006-2952(94)90509-6. [DOI] [PubMed] [Google Scholar]
- 26.Nagy LE, DeSilva SE. Ethanol increases receptor-dependent cyclic AMP production in cultured hepatocytes by decreasing G(i)-mediated inhibition. Biochem J. 1992;286(Pt 3):681–686. doi: 10.1042/bj2860681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gobejishvili L, Barve S, Joshi-Barve S, et al. Chronic ethanol-mediated decrease in cAMP primes macrophages to enhanced LPS-inducible NF-kappaB activity and TNF expression: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2006;291:G681–688. doi: 10.1152/ajpgi.00098.2006. [DOI] [PubMed] [Google Scholar]
- 28.Gobejishvili L, Barve S, Joshi-Barve S, et al. Enhanced PDE4B expression augments LPS-inducible TNF expression in ethanol-primed monocytes: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2008;295:G718–724. doi: 10.1152/ajpgi.90232.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gobejishvili L, Barve S, Breitkopf-Heinlein K, et al. Rolipram attenuates bile duct ligation-induced liver injury in rats: a potential pathogenic role of PDE4. J Pharmacol Exp Ther. 2013;347:80–90. doi: 10.1124/jpet.113.204933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer W, Schudt C, Wendel A. Protection by phosphodiesterase inhibitors against endotoxin-induced liver injury in galactosamine-sensitized mice. Biochem Pharmacol. 1993;45:2399–2404. doi: 10.1016/0006-2952(93)90219-m. [DOI] [PubMed] [Google Scholar]
- 31.Gantner F, Kusters S, Wendel A, et al. Protection from T cell-mediated murine liver failure by phosphodiesterase inhibitors. J Pharmacol Exp Ther. 1997;280:53–60. [PubMed] [Google Scholar]
- 32.Matsuhashi T, Otaka M, Odashima M, et al. Specific type IV phosphodiesterase inhibitor ameliorates thioacetamide-induced liver injury in rats. J Gastroenterol Hepatol. 2005;20:135–140. doi: 10.1111/j.1440-1746.2004.03512.x. [DOI] [PubMed] [Google Scholar]
- 33.Taguchi I, Oka K, Kitamura K, et al. Protection by a cyclic AMP-specific phosphodiesterase inhibitor, rolipram, and dibutyryl cyclic AMP against Propionibacterium acnes and lipopolysaccharide-induced mouse hepatitis. Inflamm Res. 1999;48:380–385. doi: 10.1007/s000110050475. [DOI] [PubMed] [Google Scholar]
- 34.Tukov FF, Luyendyk JP, Ganey PE, et al. The role of tumor necrosis factor alpha in lipopolysaccharide/ranitidine-induced inflammatory liver injury. Toxicol Sci. 2007;100:267–280. doi: 10.1093/toxsci/kfm209. [DOI] [PubMed] [Google Scholar]
- 35.Windmeier C, Gressner AM. Pharmacological aspects of pentoxifylline with emphasis on its inhibitory actions on hepatic fibrogenesis. Gen Pharmacol. 1997;29:181–196. doi: 10.1016/s0306-3623(96)00314-x. [DOI] [PubMed] [Google Scholar]
- 36.Xiang M, Zaccone P, Di Marco R, et al. Prevention by rolipram of concanavalin A-induced T-cell-dependent hepatitis in mice. Eur J Pharmacol. 1999;367:399–404. doi: 10.1016/s0014-2999(98)00901-7. [DOI] [PubMed] [Google Scholar]
- 37.Song Z, Zhou Z, Song M, et al. Alcohol-induced S-adenosylhomocysteine accumulation in the liver sensitizes to TNF hepatotoxicity: possible involvement of mitochondrial S-adenosylmethionine transport. Biochem Pharmacol. 2007;74:521–531. doi: 10.1016/j.bcp.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gobejishvili L, Avila DV, Barker DF, et al. S-adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via cAMP/protein kinase A pathway. J Pharmacol Exp Ther. 2011;337:433–443. doi: 10.1124/jpet.110.174268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ajmo JM, Liang X, Rogers CQ, et al. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–842. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon JC, Puigserver P, Chen G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 41.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 42.You M, Considine RV, Leone TC, et al. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–577. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 44.Rodgers JT, Lerin C, Gerhart-Hines Z, et al. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 46.You M, Jogasuria A, Taylor C, et al. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2015;4:88–100. doi: 10.3978/j.issn.2304-3881.2014.12.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noriega LG, Feige JN, Canto C, et al. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011;12:1069–1076. doi: 10.1038/embor.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leung TM, Lu Y, Yan W, et al. Argininosuccinate synthase conditions the response to acute and chronic ethanol-induced liver injury in mice. Hepatology. 2012;55:1596–1609. doi: 10.1002/hep.25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Day CP, James OF. Hepatic steatosis: innocent bystander or guilty party? Hepatology. 1998;27:1463–1466. doi: 10.1002/hep.510270601. [DOI] [PubMed] [Google Scholar]
- 50.Reeves HL, Burt AD, Wood S, et al. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis. J Hepatol. 1996;25:677–683. doi: 10.1016/s0168-8278(96)80238-8. [DOI] [PubMed] [Google Scholar]
- 51.Sorensen TI, Orholm M, Bentsen KD, et al. Prospective evaluation of alcohol abuse and alcoholic liver injury in men as predictors of development of cirrhosis. Lancet. 1984;2:241–244. doi: 10.1016/s0140-6736(84)90295-2. [DOI] [PubMed] [Google Scholar]
- 52.Mandal S, Mukhopadhyay S, Bandhopadhyay S, et al. 14-Deoxyandrographolide alleviates ethanol-induced hepatosteatosis through stimulation of AMP-activated protein kinase activity in rats. Alcohol. 2014;48:123–132. doi: 10.1016/j.alcohol.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Odashima M, Otaka M, Jin M, et al. Rolipram, a specific type IV phosphodiesterase inhibitor, ameliorates aspirin-induced gastric mucosal injury in rats. Dig Dis Sci. 2005;50:1097–1102. doi: 10.1007/s10620-005-2711-9. [DOI] [PubMed] [Google Scholar]
- 54.Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci U S A. 2002;99:7628–7633. doi: 10.1073/pnas.122041599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crabb DW, Galli A, Fischer M, et al. Molecular mechanisms of alcoholic fatty liver: role of peroxisome proliferator-activated receptor alpha. Alcohol. 2004;34:35–38. doi: 10.1016/j.alcohol.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Manmontri B, Sariahmetoglu M, Donkor J, et al. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J Lipid Res. 2008;49:1056–1067. doi: 10.1194/jlr.M800013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282:647–672. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 60.Yin H, Hu M, Liang X, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 62.Li J, Dou X, Li S, et al. Nicotinamide ameliorates palmitate-induced ER stress in hepatocytes via cAMP/PKA/CREB pathway-dependent Sirt1 upregulation. Biochim Biophys Acta. 2015;1853:2929–2936. doi: 10.1016/j.bbamcr.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katto J, Engel N, Abbas W, et al. Transcription factor NFkappaB regulates the expression of the histone deacetylase SIRT1. Clin Epigenetics. 2013;5:11. doi: 10.1186/1868-7083-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu H, Beier JI, Arteel GE, et al. Transient receptor potential vanilloid 1 gene deficiency ameliorates hepatic injury in a mouse model of chronic binge alcohol-induced alcoholic liver disease. Am J Pathol. 2015;185:43–54. doi: 10.1016/j.ajpath.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerhart-Hines Z, Dominy JE, Jr, Blattler SM, et al. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+) Mol Cell. 2011;44:851–863. doi: 10.1016/j.molcel.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto T, Shimano H, Inoue N, et al. Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver. J Biol Chem. 2007;282:11687–11695. doi: 10.1074/jbc.M611911200. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Yin L, Hillgartner FB. SREBP-1 integrates the actions of thyroid hormone, insulin, cAMP, and medium-chain fatty acids on ACCalpha transcription in hepatocytes. J Lipid Res. 2003;44:356–368. doi: 10.1194/jlr.M200283-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Yellaturu CR, Deng X, Cagen LM, et al. Posttranslational processing of SREBP-1 in rat hepatocytes is regulated by insulin and cAMP. Biochem Biophys Res Commun. 2005;332:174–180. doi: 10.1016/j.bbrc.2005.04.112. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Chen ML, Zhou Y, et al. Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol Nutr Food Res. 2015;59:1443–1457. doi: 10.1002/mnfr.201500016. [DOI] [PubMed] [Google Scholar]
- 71.Foretz M, Pacot C, Dugail I, et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martinez A, Gil C. cAMP-specific phosphodiesterase inhibitors: promising drugs for inflammatory and neurological diseases. Expert Opin Ther Pat. 2014;24:1311–1321. doi: 10.1517/13543776.2014.968127. [DOI] [PubMed] [Google Scholar]
- 73.Fala L. Otezla (Apremilast), an Oral PDE-4 Inhibitor, Receives FDA Approval for the Treatment of Patients with Active Psoriatic Arthritis and Plaque Psoriasis. Am Health Drug Benefits. 2015;8:105–110. [PMC free article] [PubMed] [Google Scholar]
- 74.Felquer ML, Soriano ER. New treatment paradigms in psoriatic arthritis: an update on new therapeutics approved by the U.S. Food and Drug Administration. Curr Opin Rheumatol. 2015;27:99–106. doi: 10.1097/BOR.0000000000000151. [DOI] [PubMed] [Google Scholar]
- 75.Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax. 2014;69:616–622. doi: 10.1136/thoraxjnl-2013-204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637–1648. doi: 10.1053/gast.2000.20189. [DOI] [PubMed] [Google Scholar]
- 77.De BK, Gangopadhyay S, Dutta D, et al. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol. 2009;15:1613–1619. doi: 10.3748/wjg.15.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Satapathy SK, Sakhuja P, Malhotra V, et al. Beneficial effects of pentoxifylline on hepatic steatosis, fibrosis and necroinflammation in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:634–638. doi: 10.1111/j.1440-1746.2006.04756.x. [DOI] [PubMed] [Google Scholar]
- 79.Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54:1610–1619. doi: 10.1002/hep.24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Du J, Ma YY, Yu CH, et al. Effects of pentoxifylline on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol. 2014;20:569–577. doi: 10.3748/wjg.v20.i2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of Pde4 inhibition on alcohol metabolism
Figure S2. Quantification of immunohistochemical staining for hepatic pCREB levels
Table S1. Information about primary antibodies used in this study