

Abstract
Objectives
Epigenetic alterations have been implicated in the development of platinum resistance in ovarian cancer (OC). In this study, we aimed to identify DNA methylation changes in platinum resistant tumors and their functional implications.
Methods
To identify DNA methylation alterations we used the Illumina 450K DNA methylation array and profiled platinum sensitive and resistant OC xenografts. Validation analyses employed RT-PCR and immunohistochemistry (IHC).
Results
Genome-wide DNA methylation analysis of OC xenografts identified 6 genes (SSH3, SLC12A4, TMEM88, PCDHGC3, DAXX, MEST) whose promoters were significantly hypomethylated in resistant compared to sensitive (control) xenografts (p < 0.001). We confirmed that TMEM88 and DAXX mRNA expression levels were increased in platinum resistant compared to control xenografts, inversely correlated with promoter methylation levels. Furthermore treatment of OC cells with guadecitabine, a DNA methyl transferase (DNMT) inhibitor, increased TMEM88 mRNA expression levels, supporting that TMEM88 is transcriptionally regulated by promoter methylation. TMEM88 was detectable by IHC in all histological types of ovarian tumors and its knock-down by using siRNA promoted OC cell proliferation and colony formation and re-sensitized cells to platinum. Furthermore, TMEM88 knock down induced upregulation of cyclin D1 and c-Myc, known Wnt target genes, supporting that TMEM88 inhibits Wnt signaling.
Conclusions
Overall, our results support that OC platinum resistance was correlated with TMEM88 overexpression regulated through decreased promoter methylation. Our data suggest that TMEM88 functions as an inhibitor of Wnt signaling, contributing to the development of platinum resistance.
Keywords: Ovarian cancer, DNA methylation, cisplatin resistance, TMEM88, Wnt
Introduction
Ovarian cancer (OC) remains the most lethal gynecologic malignancy. The American Cancer Society estimates that in 2016, 21,290 new cases of OC will be diagnosed in the United States and 14,180 women will die from the disease [1]. The high mortality rate is due to diagnosis at late stage and almost invariable development of platinum resistance [2]. Adjuvant chemotherapy with combination platinum and taxane has been the standard of care for patients with OC after optimal cytoreductive surgery. This regimen results in clinical complete remissions in greater than 70% of patients with advanced disease; however, over 90% of the original responders relapse within less than two years [3]. Patients who relapse less than six months after their initial treatment (platinum resistant OC) have limited therapeutic options and the worst outcomes. Understanding the biological mechanisms contributing to OC platinum resistance will help identify novel targets which could be blocked to prevent or reverse the process.
Although classic genetic alterations (mutations, deletions, amplifications and translocations) have long been associated with cancer development and drug resistance, epigenetic modifications, including DNA methylation and histone modifications, have also been linked to cancer initiation and progression [4]. The best characterized epigenetic mark remains DNA methylation, which is defined as addition of a methyl group by DNA methyltransferases (DNMTs) to cytosine residues located within CpG islands [5]. Increased methylation of promoter CpG islands suppresses gene expression, while hypomethylation leads to gene over expression [6, 7]. Both increased DNA methylation and hypomethylation have been described in cancer. CpG islands aberrantly methylated in ovarian tumors are associated with silencing of tumor suppressor genes (TSGs) and other genes known to be involved in the regulation of the cell cycle, apoptosis or drug sensitivity [8]. Genes whose promoters are known to be hypermethylated in OC, leading to loss of expression, include the classical TSGs BRCA1 [9], p16 [10], and MLH1 [11], the putative tumor suppressors (RASSF1A, OPCML, SPARC, ANGPTL2, CTGF) [12, 13], and the imprinted genes ARH1 and PEG3 [14]. On the contrary, DNA hypomethylation of oncogenes, such as R-Ras [15] and Maspin [16] has been shown to promote cancer progression. Known hypomethylated genes in OC include IGF2, an imprinted gene [17], and claudin-4, which functions in tight cell junctions [18]. Aberrant promoter methylation (both increased and decreased) has been linked to the development of OC platinum resistance [19, 20]. While the function of genes associated with increased promoter methylation in OC has been studied extensively [21, 22], the mechanism by which hypomethylated genes participate in the development of platinum resistance remains less well characterized.
To identify differentially methylated genes we compared platinum resistant OC cells and xenografts to platinum sensitive controls by using genome-wide DNA methylation profiling. The analysis and subsequent validation studies identified TMEM88 as a DNA methylation-regulated gene associated with cisplatin resistance. We demonstrated that TMEM88 is implicated in platinum resistance by inhibiting the Wnt pathway. Increased TMEM88 expression in recurrent ovarian tumors was associated with shorter time to relapse.
Materials and Methods
Xenograft model
Animal studies were conducted according to the Institutional Animal Care and Use Committee of Indiana University. Female, BALB/c-nu/nu, athymic mice (5-6 weeks old; Harlan) were injected intraperitoneally (IP) with 2 million A2780 cells on day 0. The experimental group (n=3) received weekly carboplatin (Hospira) at 50mg/kg IP starting on day 4 for 3 weeks, while the control group (n=3) received IP PBS, as previously described [23]. Control xenografts were harvested on day 21 from mice not treated with carboplatin. Residual and recurrent xenografts were harvested on day 21 or day 35 respectively from mice treated with weekly carboplatin.
DNA extraction, bisulfate conversion and DNA methylation profiling
Genomic DNA was extracted from xenografts by using QIAamp DNA mini kit (QIAGEN, Valencia, CA). Sodium bisulfite conversion was performed by using the EZ DNA Methylation-Gold kit (Zymo Research, Orange, CA), according to the manufacturer's instructions. After bisulfite conversion, methylation of CpG sites was determined by Infinium HumanMethylation450 BeadChips (Illumina, San Diego, CA) following a procedure provided by Illumina, at the University of Chicago Genomics Core, Knapp Center for Biomedical Discovery (Chicago, IL). Data quality verification and levels of methylation of the 485,000 CpG sites included in the array were generated by the Illumina GenomeStudio Data Analysis Software. The Illumina Infinium 450k array was used to analyze DNA methylation in promoter site regions. The method measures the methylation levels over 482,000 CpG probes. The average percentage of methylation levels were expressed as β -values and ranged from 0 (completely unmethylated) to 1 (completely methylated). Data are deposited in GEO (NCBI# pending).
Cell culture
A2780, CP70, PEO1 and PEO4 cell lines (Sigma-Aldrich, Saint Louis, MO) were cultured in RPMI 1640 media (Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) with 1% penicillin and streptomycin. SKOV3 cell lines from American Type Culture Collection were cultured in media containing 1:1 MCBD 105 (Sigma-Aldrich, Saint Louis, MO) and M199 (Cellgro, Manassas, VA) with 10% FBS and 1% penicillin and streptomycin. All cells were grown at 37°C under 5% CO2.
Transfection
Transient knockdown of TMEM88 in CP70 cells was performed using small interfering RNA (siRNA) (Invitrogen, Carlsbad, CA) or scrambled siRNA (control, Invitrogen, Carlsbad, CA) and Dreamfect Gold transfection reagent (OZ biosciences, San Diego, CA). TMEM88 was also stably knocked down in SKOV3 cells by transducing shRNA targeting TMEM88 (OriGene, Rockville, MD) and control shRNA (Origene, Rockville, MD). Stable clones were collected after selection with puromycin at 2ug/ml and TMEM88 knockdown was verified using RT-PCR and western blotting.
Cell proliferation assay
Cell Counting Kit-8 solution (Dojindo Molecular Technologies, Inc., Rockville, MD) was used to measure cell proliferation, according to the manufacturer's protocol. EL800 microplate reader (BioTek Instruments, Inc., Winooski, VT) measured absorbance at 450nm. Treatments included Wnt3A (R & D systems, Minneapolis, MN) at a concentration of 150ng/ml (4hrs), XAV-939 (Selleckchem, Houston, TX) at 250ng/ml (4hrs), cisplatin (Sigma, St Louis, MO) at 5μM, 10μM, and 20μM (48hrs), SGI-110 (Astex Pharmaceuticals Inc, Dublin, CA) at 2.5μM (120hrs). Culture media and various treatments were renewed every 48hrs.
Clonogenic Assay
SKOV3 cells stably transduced with shTMEM88 (OriGene, Rockville, MD) or shControl (OriGene, Rockville, MD) were seeded at 500 cells per well in 6 well plates. After 10 days, plates were washed with PBS, fixed with 10% formalin (Sigma) for 15 min and colonies were stained with crystal violet for 5 min (0.025% w/v, Sigma) and counted.
RNA isolation and RT-PCR
Total RNA was extracted from cultured cells and frozen xenografts by using RNA Stat-60 reagent (Tel-Test Inc., Gainsville, FL). The iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA) was used for reverse transcription and iTaq SYBR Green Supermix with ROX (Bio-Rad, Hercules, CC) was used for real-time PCR. PCR products were resolved by agarose gel electrophoresis and visualized by ethydium bromide staining. Densitometric analysis using the ImageJ software (https://rsb.info.nih.gov/ij/) was used to quantify mRNA amplification relative to GAPDH. The primers used include: TMEM88 forward 5′-GCT GCC TTC AAT CTT CTC CTG-3′ (Tm 56°C) reverse 5′-ATA AAG GCT CGG CTG TAG G-3′ (Tm 54°C); DAXX forward 5′-TCT ACA ACT TTG GCT GCC ACC TC-3′ (Tm 59°C), reverse 5′-GTC TCT TCT GTC TCT CGC CCT-3′ (Tm 58°C); c-Myc forward 5′-CACCAGCAGCGACTCTGA-3′ (Tm 60.4°C), reverse 5′-GAT CCA GAC TCT GAC CTT TTG C (Tm 60.4°C); Cyclin-D1 forward 5′-TGT GTT CGC AGC AAA TGG-3′ (Tm 60.4°C), reverse 5′-GGC ATT TTG GAG AGG AAG TG-3′ (Tm 61°C); SSH3 forward 5′-AGA AGG TCT GAG CCA GGA TG-3′ (Tm 56.4°C), reverse 5′-TTG GTG CAA TAC CTG GAG TG-3′ (Tm 55°C); SLC12A4 forward 5′-GGA CAT CCG CCC AAA GGT-3′ (Tm 58°C), reverse 5′-AGG TCA AGC CGC AGG AAG AG-3′ (Tm 59.7°C); PCDHGC3 forward 5′-GGA GTT TTG CTT CTG CTT GG-3′ (Tm 54.5°C), reverse 5′-TAC AGT GCA AGA GGG CAG TG-3′ (Tm 57.3°C); MEST forward 5′-CGG CCA TGG TGC GCC GAG AT-3′ (Tm 65.6°C), reverse 5′-ACG CAG CAA GCA GGG GCA CG-3′ (Tm 66.2°C); GAPDH forward 5′-AGC CAC ATC GCT CAG ACA C (Tm 56°C), reverse 5′-GCC CAA TAC GAC CAA ATC C3′ (Tm 56°C).
Analysis of OC TCGA data
The RNASeq data from 419 ovarian serous adenocarcinomas were obtained from the TCGA database. The normalized RPKM data was filtered to remove genes with no expression value. Pearson correlation was performed between TMEM88 and the remaining 22,661 genes. Pearson correlation coefficients between TMEM88 and target genes in Wnt pathway were calculated.
Wnt pathway analysis
Expression of 84 genes associated with the Wnt pathway was analyzed by real time RT-PCR using RT2 Profiler PCR arrays. The Wnt profiler array, RT2 First Strand Kit and RT2 SYBR Green ROX qPCR master mix were purchased from QIAGEN (Valencia, CA). Real-time thermal cycler 7900HT (Applied Biosystems) was used for PCR amplification. Reference housekeeping genes were used to calculate relative changes in gene expression in CP70 cells transfected with siRNA targeting TMEM88 or control.
Western blotting
Equal amounts of protein lysates were separated by SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked with 5% FBS and probed overnight with TMEM88 (Abcam, Cambridge, MA) antibodies at 4°C. After incubation with HRP-conjugated secondary antibody, the antigen-antibody complexes were visualized using enhanced chemiluminescence detection (Thermo Bioscience) and a luminescent image analyzer (LAS 3000, Fuji Film).
Cell Cycle Analysis
Cell cycle analysis was performed by DNA staining with propidium iodide in cells stably transfected with control or TMEM88 targeting shRNA. Cell cycle profile was determined by flow cytometry with a BD Fortessa analyzer (BD Biosciences, San Jose, CA). The size of sub-G1, G0/G1, S, and G2-M were determined by analyzing the histograms using FlowJo software (FlowJo, Ashland, OR). All experiments were performed in triplicates and data are presented as means +/- standard deviation (SD).
Immunohistochemistry
A tissue microarray from Pantomics (San Francisco, CA) including 47 de-identified human epithelial ovarian tumors of different histological types (see Supplementary Table 2) arrayed in duplicates was immunostained for TMEM88. Another tissue microarray was built using paired de-identified human ovarian tumors (n = 20, primary and recurrent) at the Indiana University Simon Cancer Center. Clinical information for each specimen collected included patient's age, histological type of cancer, type of primary treatment (surgery, type of chemotherapy, timing of chemotherapy), stage, date of surgeries, date of disease recurrence and date of death, when available. Immunostaining for TMEM88 was carried out by using an antibody from Abcam (Cambridge, MA) at 1:50 concentration, at 4°C temperature overnight followed by the avidin-biotin peroxidase technique with DAKO Detection Kit (DAKO, Hamburg, Germany). Scoring used a 0-3+ scale for intensity and the percentage of cells staining was quantified by a board certified pathologist (RE). An H score was calculated as the product between intensity and percentage of cells staining. Paired tumors were compared and classified based on the change in H score (ratio>1, increased or <1, decreased).
Statistical Analysis
Student t-test and ANOVA were used for statistical analyses and p < 0.05 was considered significant. Chi-square test analyzed the clinical relevance of the change in TMEM88 protein expression from paired tumor samples. The genome-wide methylation analysis used the Partek Genomics Suite (version 6.5). Differences in methylation levels between samples were calculated by using a mixed-model ANOVA. The resultant P values < 0.05 denoted significant differential methylation levels at specific sites. Region specific analysis used a novel method to calculate the differential methylation levels between two samples. Since the mixed-model ANOVA was highly dependent on both the CpG site location and the individual sample, a mixed-model Generalized Linear Model (GLM) was devised to correct such biases. A hybrid C++/R script was used to parse the original matrix and calculate the GLM matrix and the resultant P value. Each CpG region contains any number of individual CpG sites. For each CpG region, the individual β-values were gathered and the CpG location and sample covariates were determined. At each CpG site in the region, those samples that contained all β-values less than 10% or greater than 90% were filtered. Average methylation signals within each CpG region were hierarchically clustered using Partek Genomics Suite with Pearson dissimilarity and average linkage as clustering parameters. Heatmap illustrating hierarchical clustering of differentially methylated CpG loci is shown. For the differential methylation analysis, false discovery rates (FDR) was used using an improved Benjamini-Hochberg calculation.
Results
Differential methylation profile in platinum resistant xenografts compared to controls
We [23] and others [24] have shown that treatment with platinum induces effective OC xenograft volume decrease in vivo. However small tumors enriched in cancer stem cells persist [23] and upon discontinuing platinum, tumors rapidly recur. Using this in vivo model, we compared control treated A2780-derived tumors (platinum sensitive) to recurrent (grown 2 weeks after discontinuing therapy) or residual (persistent after 3 weeks of carboplatin treatment) platinum resistant tumors Figure 1A). Changes in methylation were assessed by using 450K DNA methylation arrays (Illumina), which cover 480,000 CpG sites. Distinct methylation profiles between sensitive (control) and resistant tumors (residual or recurrent) were noted by using unsupervised hierarchical clustering (Figure 1B). Additionally, the comparison between sensitive and resistant tumors identified six promoters (SSH3, SLC12A4, TMEM88, PCDHGC3, DAXX, MEST) significantly differentially methylated (defined as p < 0.01, and FDR < 0.09) in the platinum resistant tumors compared to controls (Supplementary Table 1). All six promoters were hypomethylated in resistant tumors. Although other genes were found to be hypermethylated in resistant tumors, none met the preset bar of statistical significance.
Figure 1. Differential DNA methylation in platinum sensitive and resistant OC xenografts.
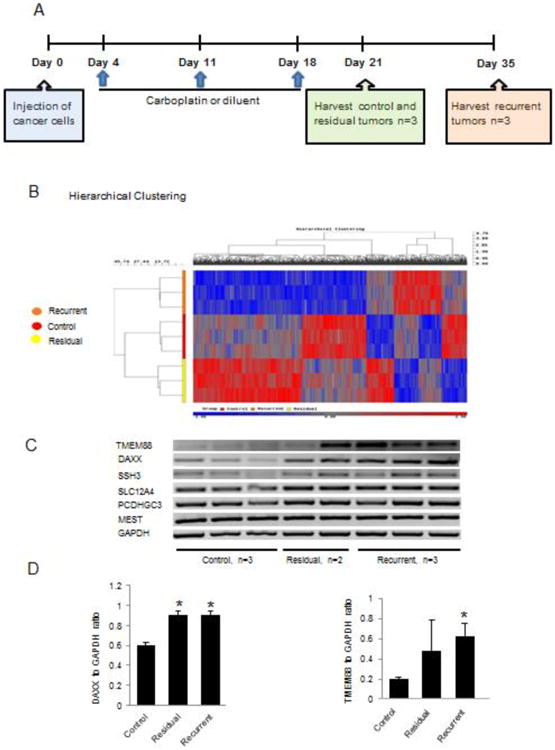
A. Experimental schema depicts the timeline of treatments and tumor harvests, as described in Materials and Methods. B. Hierarchical clustering displays differential DNA methylation profiles of xenografs (control n=3, residual n=2 and recurrent n=3). C. RT-PCR measures mRNA expression levels of 6 genes (DAXX, TMEM88, SSH3, SLC12A4, PCDHGC3 and MEST) identified as being differentially methylated in residual or recurrent tumors vs. control tumors. D. Densitometric analysis of DAXX and TMEM88 mRNA expression levels in control n=3, residual n=2 and recurrent n=3 xenograft tumors. Bars represent means ± SE. * Denotes statistical significance (p < 0.05).
To validate whether the observed differences in DNA methylation were associated with significant changes in gene expression, mRNA expression levels were quantified by using RT-PCR in platinum resistant xenografts versus controls. Increased mRNA expression levels corresponding to significantly hypomethylated promoters in resistant versus control tumors were confirmed for TMEM88 and DAXX (p < 0.05, Figures 1C and 1D), but not for the other genes.
To confirm the relationship between TMEM88 and DAXX gene expression levels with platinum resistance, we evaluated their mRNA expression levels in platinum resistant and sensitive paired isogenic OC cell lines (A2780 vs. CP70 and PEO1 vs. PEO4)[25, 26]. mRNA expression levels of TMEM88, but not of DAXX, were increased in the two platinum resistant OC cell lines (CP70 and PEO4) compared to the corresponding platinum sensitive cells (A2780 and PEO1) (Figure 2A, 2B and 2C). Additionally, increased TMEM88 protein expression level was observed in platinum resistant OC cells (Figure 2D).
Figure 2. TMEM88 and DAXX expression in platinum sensitive and platinum resistant OC cell lines.
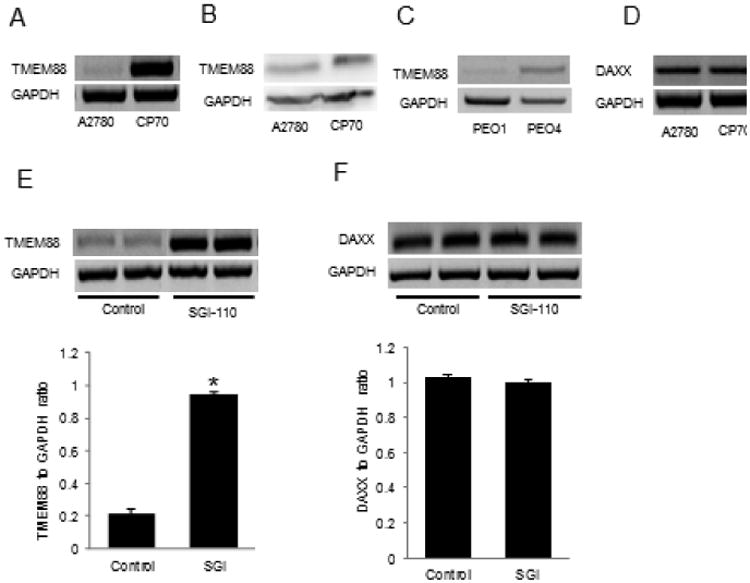
A. RT-PCR measures TMEM88 mRNA expression levels in platinum resistant CP70 compared to platinum sensitive A2780 cells. B. RT-PCR measures TMEM88 mRNA expression level in platinum resistant PEO4 compared to platinum sensitive PEO1 cells. C. RT-PCR measures DAXX mRNA expression level in CP70 compared to A2780 cells. D. Western blot measures TMEM88 protein level in platinum resistant CP70 compared to platinum sensitive A2780 cells. E. RT-PCR and corresponding densitometric analysis measure TMEM88 mRNA expression level in A2780 cells treated with SGI-110 (2.5μM for 120hrs). F. RT-PCR and corresponding densitometric analysis measure DAXX mRNA expression level in A2780 cells treated with SGI-110 (2.5μM for 120hrs). Bars represent means ± SE. * Denotes statistical significance (p < 0.05).
Increased TMEM88 expression levels is associated with DNA hypomethylation
To confirm that increased TMEM88 expression levels in platinum resistant OC xenografts and cells is the result of promoter hypomethylation, SGI-110 (guadecitabine), a new DNA methyltransferase inhibitor, was used [27]. Treatment with SGI-110 increased TMEM88 mRNA expression levels in A2780 cells (Figure 2E, p = 0.00003), but did not alter DAXX expression levels (Figure 2F), further supporting the concept that TMEM88 is regulated by promoter methylation. The hypomethylating effects of SGI-110 was also observed for two CpG islands within the TMEM88 promoter [23], for which the differences in methylation were 9% and 11%, respectively (p < 0.01, FDR < 0.06), as measured by the 450K Illumina arrays in platinum resistant xenografts treated with SGI-110 compared to control.
TMEM88 is expressed in ovarian tumors and regulates cell proliferation
As the expression and function of TMEM88 have not been previously studied in OC, we first investigated whether TMEM88 is expressed in ovarian tumors by using immunohistochemistry (IHC) and a human multi-tissue array including all OC histological subtypes (serous, mucinous, endometrioid, and clear cell). Strong (2+/3+) TMEM88 membranous and cytoplasmic immunoreactivity was noted in 24 out of 47 tumors (Figure 3A and Supplementary Table 2).
Figure 3. TMEM88 is expressed in ovarian cancer tumors and is associated with cell proliferation.
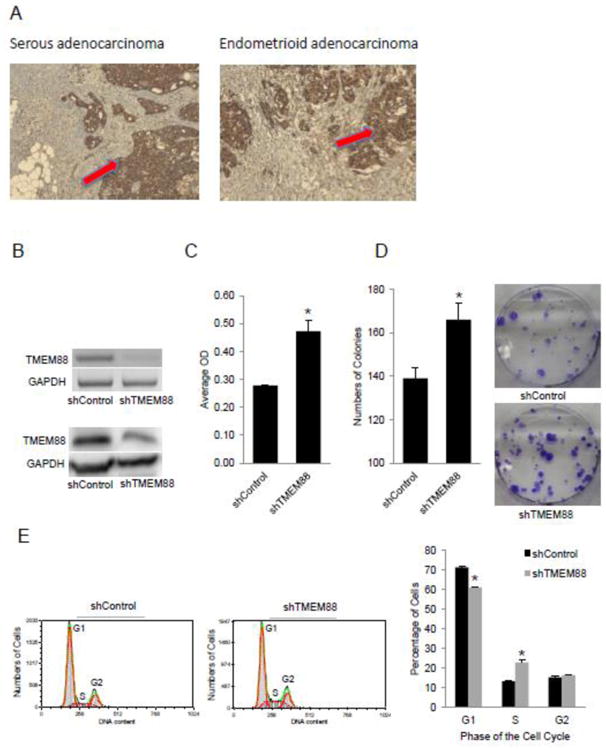
A. Representative immunostaining for TMEM88 in serous and endometrioid OC (100× magnification). B. RT-PCR measures TMEM88 mRNA expression level in SKOV3 cells stably transduced with shRNA targeting TMEM88 (shTMEM88) vs control shRNA (shControl). Western blot measures TMEM88 protein level expression in shTMEM88 and shControl transduced SKOV3 cells. C. Proliferation of SKOV3 cells stably transduced with shTMEM88 or shControl. D. Numbers of colonies formed by SKOV3 cells stably transduced with shTMEM88 or shControl. Bars represent means ± SE. * denotes statistical significance (p < 0.05). E. Cell cycle analysis by flow cytometry in SKOV3 cells transduced with shControl and shTMEM88. Bars represent means ± SD, * denotes statistical difference (p < 0.01).
Next, to understand its function in OC cells, stable knock down was achieved by using shRNA targeting TMEM88. Western blotting and RT-PCR confirmed stable knock down of TMEM88 at mRNA and protein levels (Figure 3B) in SKOV3 cells. TMEM88 knock down induced increased cell proliferation (Figure 3C, p < 0.05), colony formation (Figure 3D, p < 0.05), and percentage of cells in S-phase (22.9% vs. 13.3%, Figure 3E, p <0.05) supporting that TMEM88 inhibits cell proliferation.
TMEM88 regulates cell proliferation by inhibiting the Wnt Pathway
Recent reports have identified TMEM88 as an inhibitor of Wnt signaling through direct interaction with Disheveled [28, 29]. As the Wnt pathway is a known regulator of cell proliferation [30], we sought to determine whether TMEM88 alters OC cell proliferation by blocking Wnt signaling. TMEM88 knock down in the cisplatin resistant CP70 cell line (Figure 4A-B) resulted in significant (p < 0.05) upregulation of c-Myc and cyclin-D1, known Wnt downstream genes. We further examined the expression of 84 Wnt-signaling associated genes, by using a pathway focused RT-PCR based array. Knock down of TMEM88 resulted in the upregulation of other Wnt target genes including JUN, PTIX2, CTNNB1 (β-catenin) (Table 1), further supporting that TMEM88 inhibits this pathway. A significant correlation between TMEM88 and c-Myc mRNA expression levels (r = -0.12081, p=0.01252) as well as between TMEM88 and β-catenin (r = -0.12, p=0.0128) were confirmed (Figure 4C) in the transcriptomic data associated with the ovarian TCGA database [31], supporting that this inverse relationship is also detectable in human ovarian tumors.
Figure 4. Effect of TMEM88 on Wnt signaling.
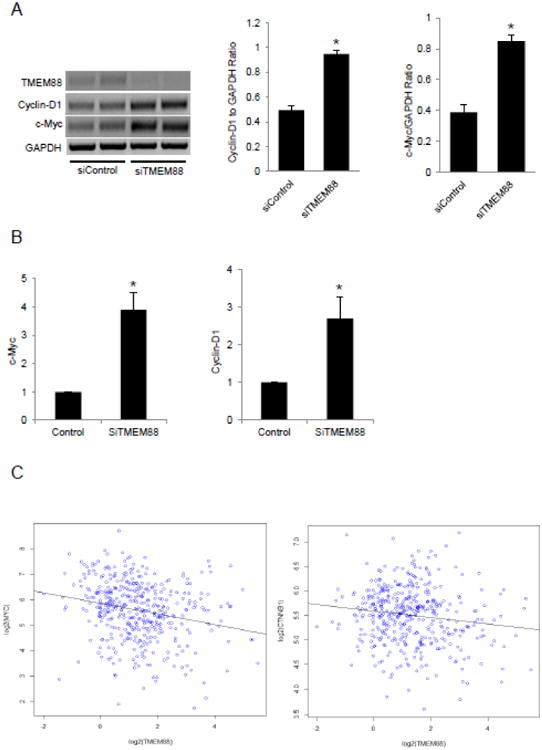
A. RT-PCR and densitometric analysis of cyclin-D1 and c-Myc mRNA expression levels in platinum resistant cell line CP70 after transient TMEM88 knockdown with siRNA. B. QRT-PCR measures c-Myc and cyclin-D1 mRNA expression levels in SKOV3 after transient TMEM88 knockdown with siRNA. C. RNAseq from TCGA data showing negative correlation between TMEM88 and c-Myc and TMEM88 and CTNNB1 (β –catenin). Bars represent means ± SE. * denotes statistical significance (p < 0.05).
Table 1.
Changes (>1.5 fold) in expression of genes associated with the Wnt pathway after TMEM88 knockdown
| Genea | Fold increase in mRNA expression relative to control | Geneb | Fold decrease in mRNA expression relative to control |
|---|---|---|---|
| Wnt 9A | 3.97 | Wnt5B | -7.2 |
| FZD8 | 2.61 | VANGL2 | -2.62 |
| DVL1 | 2.57 | MMP7 | -2.27 |
| KREMEN1 | 2.53 | DAB2 | -2.2 |
| NKD1 | 2.51 | LEF1 | -1.77 |
| LRP5 | 2.36 | SOX17 | -1.77 |
| JUN | 2.23 | ||
| PITX2 | 2.20 | ||
| CTNNB1 | 1.99 | ||
| CCND1 | 1.81 |
JUN, PITX2, CCND1 - Wnt signaling target genes
SOX17 - Wnt signaling negative regulator
TMEM88 is associated with platinum resistance
Given our initial observations that TMEM88 is upregulated in platinum resistant OC xenografts and cell lines and the newly established functional link between TMEM88 and Wnt signaling [29], partly validated in our OC model, we sought to determine how the Wnt pathway affects response to platinum in ovarian tumors. We used platinum sensitive and resistant OC xenografts and measured the expression of Wnt associated genes using the pathway specific RT-PCR array. The expression of several wnt target genes, including PITX2 and FOSL1 was decreased in platinum resistant xenografts compared to controls (Supplementary Table 3).
We then analyzed the response of OC cells to platinum after modulating the Wnt pathway. We used Wnt3A to activate the pathway or XAV939 [32] to inhibit it, prior to exposure of OC cells to cisplatin. Activation of the Wnt pathway increased OC cells sensitivity to cisplatin (Figure 5A-B, p < 0.05) while the Wnt inhibitor partly blocked response to platinum (Figure 5C-D, p < 0.05).
Figure 5. Inhibition of Wnt pathway may lead to cell dormancy and development to cisplatin resistance.
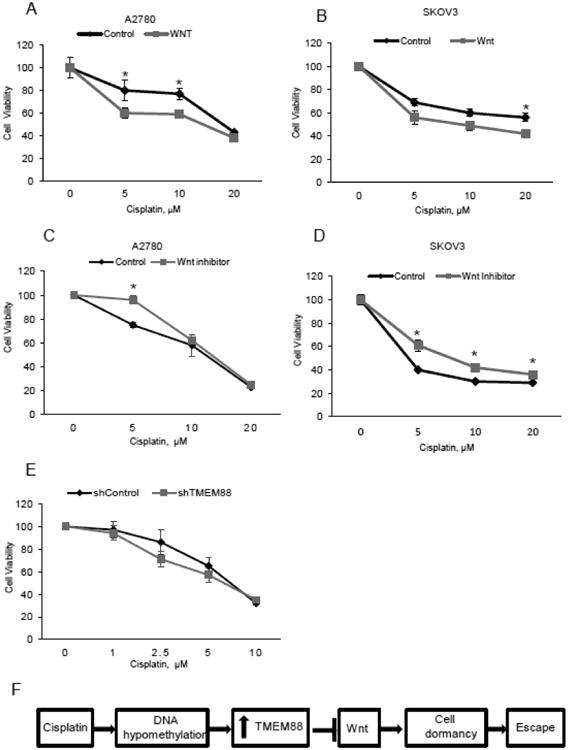
A-B.Proliferation assay measures the number of viable A2780 and SKOV3 cells treated with cisplatin (48hrs) at the indicated concentrations after 4 hour pretreatment with control or Wnt3A (150ng/ml). C-D. Proliferation assay measures the number of viable A2780 and SKOV3 cells treated with cisplatin (48hrs) at the indicated concentrations after 4 hour pretreatment with the inhibitor XAV-939 (250ng/ml). Bars represent means ± SE. * denotes statistical significance (p < 0.05) E. Proliferation assay measures the number of viable SKOV3 cells stably transduced with shTMEM88 vs shControl and treated with cisplatin at the indicated concentrations. F. Model illustrating the potential role of TMEM88 in the development of OC platinum resistance.
As TMEM88 acts as an endogenous inhibitor of Wnt signaling, we next measured the response of OC cells to cisplatin treatment after shRNA mediated TMEM88 knockdown in CP70 platinum resistant cells. Knock down of TMEM88 resulted in increased sensitivity to cisplatin treatment (Figure 5E, p < 0.05), supporting the link between TMEM88 and cisplatin resistance. A proposed model illustrating the mechanism by which TMEM88 affects response to platinum in included in Figure 5F.
To determine whether the association between TMEM88 expression and platinum resistance is clinically relevant, we evaluated by IHC 20 clinically annotated and de-identified paired human OC specimens (primary and recurrent) on a custom built multi-tissue array. Clinical information linked to these specimens included stage, histological type, type of treatment, platinum-free interval after initial treatment and survival. All patients received platinum based therapy after initial surgery. Median age at diagnosis in this set was 57 (range 41 to 77), 19 of 20 tumors were high grade serous OC (one was mixed mullerian tumor), 18/20 were stage IIIC at diagnosis and 2 were stage IV. TMEM88 expression in recurrent vs. primary tumors was quantified, dichotomized (increased or unchanged/decreased) and correlated with progression free survival (PFS). The group of patients whose tumors displayed increased TMEM88 expression in recurrent compared to primary tumors had shorter PFS compared to patients whose tumors had unchanged/decreased TMEM88 expression, specifically 13 months (n=8) vs. 19 months (n=12, p = 0.06, Supplementary Figure 1).
Discussion
Recent studies have linked alterations in DNA methylation to the development of platinum resistance in OC, a uniformly fatal event. By and large, gains in CpG island methylation have been associated with chemotherapy resistance and the reversal of methylation led to re-sensitization of OC cells to platinum [23, 33]. However, other studies have also shown that hypomethylation of certain genes including synuclein-gamma [34], and brother of regulator of imprinted sites (BORIS) [35] is connected to chemoresistance. Here we identified TMEM88 as a hypomethylated gene in tumors residual or recurrent after platinum treatment. Our study has several implications.
First, this is the first report linking TMEM88 with platinum resistance in OC. We show that TMEM88 was upregulated in platinum resistant xenografts, cell lines and recurrent ovarian human tumors and that TMEM88 knockdown re-sensitized cancer cells to platinum. Although not achieving statistical significance in our small tumor set, the data presented suggest that increased TMEM88 expression in recurrent tumors may be correlated with decreased benefit to platinum based therapy. While the mechanism linking TMEM88 and platinum resistance remains incompletely elucidated here, one potential link is TMEM88 mediated inhibition of Wnt signaling. Our data demonstrating inhibition of Wnt target genes by TMEM88 knock down in OC cells and inverse correlations between TMEM88 and c-Myc or β-catenin in the ovarian TCGA database are consistent with recent reports identifying TMEM88 as an inhibitor of Wnt pathway in lung and breast cancer cells [36, 37]. The C-terminal VWV (Val-Trp-Val) sequence of TMEM88 directly interacts with the PDZ domain of Dishevelled-1 (Dvl-1) in Xenopus embryos leading to inhibition of the canonical Wnt/β-catenin signaling [28].
Second, our manuscript links the Wnt pathway to response to platinum. Our data using activators and inhibitors of Wnt signaling support an inverse correlation between Wnt activation and platinum sensitivity. Our data are consistent with reports suggesting inhibition of Wnt target genes after platinum therapy [38], but dissimilar to studies supporting Wnt inhibition as a modality or re-sensitizing OC to platinum [39]. The distinct cell models and experimental conditions (selection of platinum resistant tumors in vivo vs. in vitro, different doses of inhibitors) may account for the observed differences. We propose that inhibition of Wnt blocks cell proliferation and induces cell dormancy. Dormant cells have been shown to escape the lethal effects of chemotherapy [40] and give raise to recurrent tumors. We suggest that chemotherapy eliminates actively proliferating tumor cells, leaving behind dormant chemo-resistant cells, which have the potential to grow and repopulate tumors when the conditions are favorable.
Third, this is the first report demonstrating that TMEM88 expression is regulated through promoter methylation. The findings correlating increased TMEM88 expression in resistant tumors with promoter hypomethylation and the observations that treatment with a DNMT inhibitor increases expression of this transcript support this new concept. Interestingly, we did not observe significantly increased CpG island methylation, possibly due to the short timeframe of platinum exposure (∼3 weeks) in our model, contrasting to longer treatment durations and repeated exposure to chemotherapy commonly observed in human samples. Other genes were found to by hypomethylated in residual platinum resistant xenografts included the death domain associated protein DAXX, the phosphatase SSH3, the prothocadherin PCDHGC3, and the imprinted mesoderm specific transcript MEST. Their functional relevance remains unknown, however upregulation of DAXX may be related to the apoptotic pathway elicited by cisplatin. MEST has been previously associated also with platinum resistance [7].
In summary our study shows that TMEM88 promoter hypomethylation is associated with cisplatin resistance in ovarian cancer. We propose that platinum-based therapy leads to TMEM88 promoter hypomethylation, which in turn causes increased TMEM88 expression. Upregulated TMEM 88 inhibits Wnt signaling and cell proliferation, inducing cell dormancy and the eventual escape of cancer cells from the cytotoxic effects of platinum. Identification of TMEM88 as an epigenetically modified gene associated with platinum resistance points to a new mechanism predicting tumor recurrence. Blockade of this newly uncovered pathway may provide a future strategy to improve overall survival of patients with OC.
Supplementary Material
Highlights.
TMEM88 is associated with platinum resistance in ovarian cancer.
TMEM88 expression is regulated by promoter methylation
TMEM88 blocks Wnt signaling in ovarian cancer cells.
Acknowledgments
This work was supported by the National Cancer Institute (CA182832 to DM and KN), the V Foundation (to DM and KN), and a US Department of Veterans Affairs Merit Award (DM).
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Ries LAG, et al. Patient and tumor characteristics. Bethesda, MD: US Department of Health and Human Services, National Institutes of Health, National Cancer Institute; 2007. Cancer survival among adults: US SEER program, 1988-2001. [Google Scholar]
- 3.Alberts DS. Treatment of refractory and recurrent ovarian cancer. Semin Oncol. 1999;26(1 Suppl 1):8–14. [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PA. DNA methylation and cancer. Oncogene. 2002;21(35):5358–60. doi: 10.1038/sj.onc.1205597. [DOI] [PubMed] [Google Scholar]
- 6.Chang X, et al. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 2010;70(7):2870–9. doi: 10.1158/0008-5472.CAN-09-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeller C, et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene. 2012;31(42):4567–76. doi: 10.1038/onc.2011.611. [DOI] [PubMed] [Google Scholar]
- 8.Fang R, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell. 2010;39(2):222–33. doi: 10.1016/j.molcel.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang J, et al. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr Biol. 2002;12(13):1086–99. doi: 10.1016/s0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- 10.Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6(6):579–89. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrlich M, et al. Quantitative analysis of associations between DNA hypermethylation, hypomethylation, and DNMT RNA levels in ovarian tumors. Oncogene. 2006;25(18):2636–45. doi: 10.1038/sj.onc.1209145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai X, et al. Clinicopathological significance and prognostic value of DNA methyltransferase 1, 3a, and 3b expressions in sporadic epithelial ovarian cancer. PloS one. 2012;7(6):e40024. doi: 10.1371/journal.pone.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, et al. ALDH1A1 is a novel EZH2 target gene in epithelial ovarian cancer identified by genome-wide approaches. Cancer Prev Res (Phila) 2012;5(3):484–91. doi: 10.1158/1940-6207.CAPR-11-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo J, et al. EZH2 regulates expression of p57 and contributes to progression of ovarian cancer in vitro and in vivo. Cancer Sci. 2011;102(3):530–9. doi: 10.1111/j.1349-7006.2010.01836.x. [DOI] [PubMed] [Google Scholar]
- 15.Nishigaki M, et al. Discovery of aberrant expression of R-RAS by cancer-linked DNA hypomethylation in gastric cancer using microarrays. Cancer Res. 2005;65(6):2115–24. doi: 10.1158/0008-5472.CAN-04-3340. [DOI] [PubMed] [Google Scholar]
- 16.Bettstetter M, et al. Elevated nuclear maspin expression is associated with microsatellite instability and high tumour grade in colorectal cancer. J Pathol. 2005;205(5):606–14. doi: 10.1002/path.1732. [DOI] [PubMed] [Google Scholar]
- 17.Murphy SK, et al. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol Cancer Res. 2006;4(4):283–92. doi: 10.1158/1541-7786.MCR-05-0138. [DOI] [PubMed] [Google Scholar]
- 18.Litkouhi B, et al. Claudin-4 overexpression in epithelial ovarian cancer is associated with hypomethylation and is a potential target for modulation of tight junction barrier function using a C-terminal fragment of Clostridium perfringens enterotoxin. Neoplasia. 2007;9(4):304–14. doi: 10.1593/neo.07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee PS, et al. Elevated MAL expression is accompanied by promoter hypomethylation and platinum resistance in epithelial ovarian cancer. International journal of cancer, Journal international du cancer. 2010;126(6):1378–89. doi: 10.1002/ijc.24797. [DOI] [PubMed] [Google Scholar]
- 20.Li M, et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics. 2009;2:34. doi: 10.1186/1755-8794-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matei D, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Research. 2012;72(9):2197–205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balch C, et al. The epigenetics of ovarian cancer drug resistance and resensitization. Am J Obstet Gynecol. 2004;191(5):1552–72. doi: 10.1016/j.ajog.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74(17):4922–36. doi: 10.1158/0008-5472.CAN-14-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah DK, et al. Evaluation of combined bevacizumab and intraperitoneal carboplatin or paclitaxel therapy in a mouse model of ovarian cancer. Cancer Chemother Pharmacol. 2011;68(4):951–8. doi: 10.1007/s00280-011-1566-3. [DOI] [PubMed] [Google Scholar]
- 25.Lewis AD, Hayes JD, Wolf CR. Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: intrinsic differences and cell cycle effects. Carcinogenesis. 1988;9(7):1283–7. doi: 10.1093/carcin/9.7.1283. [DOI] [PubMed] [Google Scholar]
- 26.Perez RP, et al. In vitro interactions between platinum analogues in human ovarian-carcinoma cell lines. Cancer Chemother Pharmacol. 1992;29(6):430–4. doi: 10.1007/BF00684842. [DOI] [PubMed] [Google Scholar]
- 27.Yoo CB, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67(13):6400–8. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- 28.Lee HJ, et al. Identification of transmembrane protein 88 (TMEM88) as a dishevelled-binding protein. J Biol Chem. 2010;285(53):41549–56. doi: 10.1074/jbc.M110.193383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palpant NJ, et al. Transmembrane protein 88: a Wnt regulatory protein that specifies cardiomyocyte development. Development. 2013;140(18):3799–808. doi: 10.1242/dev.094789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 31.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 33.Matei D, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9):2197–205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Czekierdowski A, et al. The role of CpG islands hypomethylation and abnormal expression of neuronal protein synuclein-gamma (SNCG) in ovarian cancer. Neuro Endocrinol Lett. 2006;27(3):381–6. [PubMed] [Google Scholar]
- 35.Woloszynska-Read A, et al. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 36.Yu X, et al. Cytosolic TMEM88 promotes triple-negative breast cancer by interacting with Dvl. Oncotarget. 2015;6(28):25034–45. doi: 10.18632/oncotarget.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, et al. Cytosolic TMEM88 Promotes Invasion and Metastasis in Lung Cancer Cells by Binding DVLS. Cancer Res. 2015;75(21):4527–37. doi: 10.1158/0008-5472.CAN-14-3828. [DOI] [PubMed] [Google Scholar]
- 38.Koussounadis A, et al. Chemotherapy-induced dynamic gene expression changes in vivo are prognostic in ovarian cancer. Br J Cancer. 2014;110(12):2975–84. doi: 10.1038/bjc.2014.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagaraj AB, et al. Critical role of Wnt/beta-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget. 2015;6(27):23720–34. doi: 10.18632/oncotarget.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreso A, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339(6119):543–8. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.