

Abstract
Non-small-cell lung cancer is the leading cause of cancer death worldwide and is comprised of several histological subtypes, the two most common being adenocarcinoma (AC) and squamous cell carcinoma (SCC). Targeted therapies have successfully improved response rates in patients with AC tumors. However, the majority of SCC tumors lack specific targetable mutations, making development of new treatment paradigms for this disease challenging. In the present study, we used iterative non-negative matrix factorization, an unbiased clustering method, on mRNA expression data from the cancer genome atlas (TCGA) and a panel of 24 SCC cell lines to classify three disease segments within SCC. Analysis of gene set enrichment and drug sensitivity identified an immune-evasion subtype that showed increased sensitivity to nuclear factor-κB and mitogen-activated protein kinase (MAPK) inhibition, a replication-stress associated subtype that showed increased sensitivity to ataxia telangiectasia inhibition, and a neuroendocrine-associated subtype that showed increased sensitivity to phosphoinositide 3-kinase and fibroblast growth factor receptor inhibition. Additionally, each of these subtypes exhibited a unique microRNA expression profile. Focusing on the immune-evasion subtype, bioinformatic analysis of microRNA promoters revealed enrichment for binding sites for the MAPK-driven ETS1 transcription factor. Indeed, we found that knockdown of ETS1 led to upregulation of eight microRNAs and downregulation of miR-29b in the immune-evasion subtype. Mechanistically, we found that miR-29b targets the DNA-demethylating enzyme, TET1, for downregulation resulting in decreased 5-hmC epigenetic modifications. Moreover, inhibition of MAPK signaling by gefitinib led to decreased ETS1 and miR-29b expression with a corresponding increase in TET1 expression and increase in 5-hmC. Collectively, our work identifies three subtypes of lung SCC that differ in drug sensitivity and shows a novel mechanism of miR-29b regulation by MAPK-driven ETS1 expression which leads to downstream changes in TET1-mediated epigenetic modifications.
Introduction
Non-small-cell lung cancer is the most prevalent type of lung cancer in the world and the leading cause of cancer death, accounting for 1.4 million deaths annually.1 Non-small-cell lung cancer consists of several histological subtypes, the two most common being adenocarcinoma (AC) and squamous cell carcinoma (SCC).2 While targeted therapies, such as those that target epidermal growth factor receptor (EGFR), have been successful in improving response rates in patients with AC tumors, the majority of SCC tumors lack specific targetable mutations. One challenge in defining treatment paradigms for SCC is the high level of heterogeneity within this disease. Gene expression profiling has improved our understanding of cancer and led to the development of multigene signatures that predict outcomes and response to therapy.3, 4, 5 However, such signature classifications have not changed treatment for SCC. Therefore, the development of therapies targeted for SCC will depend on gaining a greater understanding of the molecular underpinnings that drive tumorigenesis and progression in this disease setting.
MicroRNAs are small (20–30 nucleotide) non-coding RNAs that can function as either tumor promoters or suppressors during tumorigenesis by exerting post-transcriptional effects on gene expression.6 Additionally, microRNAs are often expressed in a tissue- and disease-specific manner,7 making them ideal candidates as biomarkers.7, 8 In this study, we used global gene expression profiling to define subtypes present within lung SCC. Importantly, we found each of these subtypes to have a unique therapeutic sensitivity and microRNA expression profile. We demonstrate that the ETS1 transcription factor, driven by pathways enriched in the immune-evasion subtype, drives the differential expression of a subset of microRNAs expressed in this subtype. Through this analysis, we identified miR-29b as a microRNA whose expression is driven by ETS1, through activated mitogen-activated protein kinase (MAPK) signaling. Additionally, we found that miR-29b targets the 5-hydroxymethylcytosine dioxygenase, TET1 for downregulation and has downstream effects on TET1-mediated epigenetic modifications.
Results
Iterative non-negative matrix factorization clustering of lung SCC reveals three genomic subtypes with unique drug sensitivity and cell signaling profiles
In order to classify genomically distinct subtypes within lung SCC, we employed iterative non-negative matrix factorization (iNMF), an unbiased clustering technique, chosen for its ability to overcome the limitations of consensus clustering and provide higher resolution than hierarchical clustering.9, 10 iNMF was applied to mRNA expression data from 258 SCC patient samples available from the cancer genome atlas (TCGA), revealing three subtypes, driven by a specific subset of genes (Supplementary Table S1 and Figure 1a). This technique was validated using an independent data set,11 which also resulted in three subtypes, with similar gene enrichment profiles (Supplementary Figure S1 and Supplementary Table S2). Despite the differences in gene expression between the subtypes, there was no statistical difference in survival proportion, tumor stage or smoking status between these subtypes (Supplementary Figure S2). Similar to our work, iNMF iClustering performed by TCGA research network yielded three consensus clusters.12 Additionally, a previous study, using the consensus plus clustering method reported four subtypes of SCC designated as classical, basal, secretory and primitive.13 Comparison of our analysis to these studies indicated a high degree of overlap between the three methods (Figure 1b and Supplementary Figure S3), with cluster 1 most closely aligning with the previously reported basal and secretory subtypes, cluster 2 with the classical subtype and cluster 3 with the primitive subtype. Differences in clustering likely reflect differences in patient sample numbers, patient population and differences in methodology. Having demonstrated that our methods agreed with previously published methods performed on different data sets, we applied hierarchical clustering based on the features identified by iNMF to a panel of 24 lung SCC cell lines (GSE57083) to model these clinically relevant subtypes in cell line models of disease (Figure 1c). Fifteen cell lines closely aligned with patient samples from cluster 1, seven cell lines aligned with cluster 2 and two cell lines aligned with cluster 3 (Figure 1d).
Figure 1.

iNMF clustering to identify subtypes of lung SCC. (a) iNMF was used to uncover three core subtypes of gene expression differences within 258 samples of lung SCC obtained from The Cancer Genome Atlas (TCGA). (b) The map showing overlap of individual TCGA samples clustered by iCluster,12 Subtype13 and iNMF. Cluster 1 is indicated in blue, cluster 2 in red and cluster 3 in green. (c, d) iNMF clusters were applied to a panel of 24 SCC cell lines.
Next, we investigated whether cell lines belonging to each subtype differed in sensitivity to targeted therapies. To do this, we devised a compound sensitivity score where the average negative log GI50 of a compound across all cell lines was subtracted from the average negative log GI50 of cell lines in each cluster. A negative value was indicative that cell lines in a particular cluster were more resistant to a compound than the panel as a whole, while a positive value indicated that cluster cell lines were more sensitive to a compound than the panel as a whole. We found that each cluster exhibited a unique drug response profile (Table 1 and Supplementary Table S3).
Table 1. Compound sensitivity score.
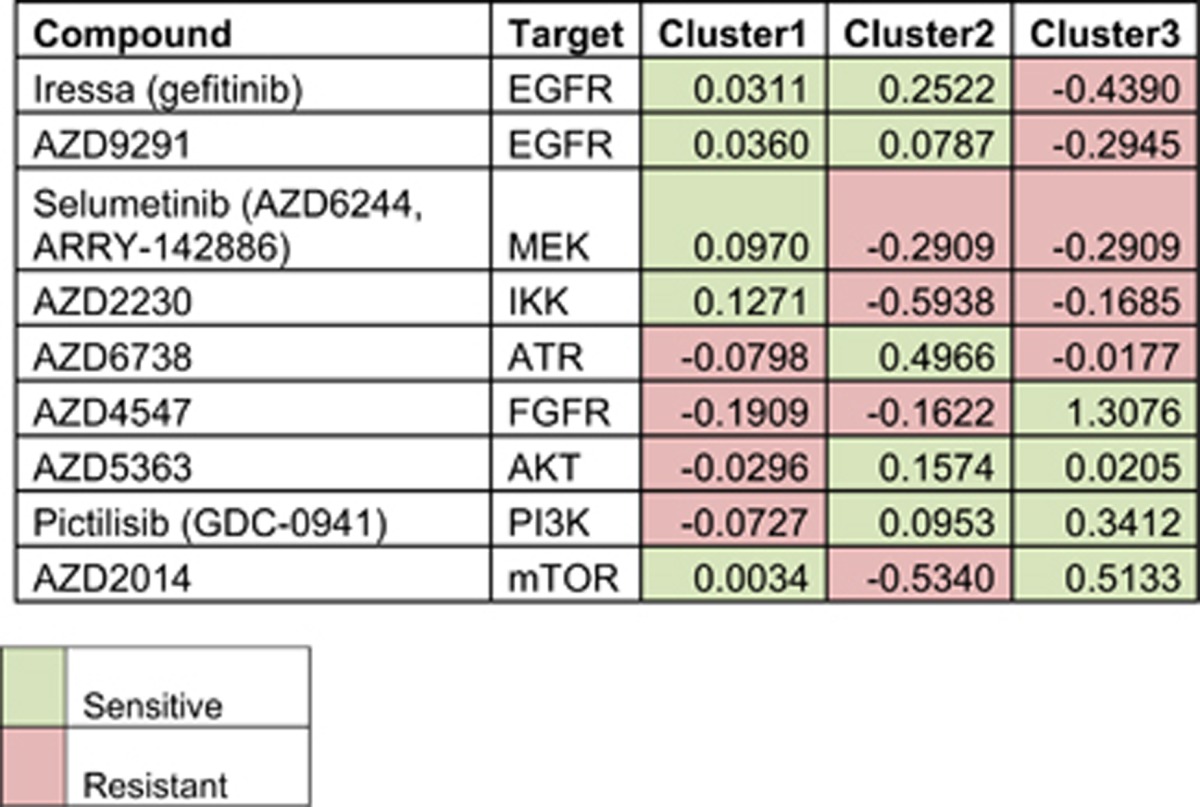
To better understand the underlying molecular pathways driving the differential drug sensitivity between subtypes, we used gene set enrichment analysis (GSEA) to determine the signaling pathways most active in each cluster in both TCGA and cell line samples (Table 2 and Supplementary Table S4). Cluster 1 samples were enriched for EGFR, MAPK and nuclear factor-κB signaling. Nuclear factor-κB signaling is known to subvert the immune system within the tumor micro-environment.14 Moreover, enhanced signaling through this pathway due to EGFR and KRAS mutations has been shown to upregulate programmed death-ligand 1 contributing to tumor immune escape.15, 16, 17, 18 Indeed, both cluster 1 TCGA and cell line samples that had high levels of signaling through EGFR/MAPK pathways showed higher levels of programmed death-ligand 1 (Supplementary Figure S4). Based on this evidence we named cluster 1 the immune-evasion subtype. Interestingly, cell lines from this immune-evasion subtype, which had increased EGFR and nuclear factor-κB signaling also showed enhanced sensitivity to inhibition of EGFR/MAPK signaling pathways by gefitinib (EGFR tyrosine-kinase inhibitor), AZD9291 (EGFR tyrosine-kinase inhibitor) and the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886). In addition, this cell line cluster showed enhanced sensitivity to nuclear factor-κB inhibition by AZD2230 (Table 1).
Table 2. Gene set enrichment analysis (GSEA).
| Gene set | TCGA P-value | Cell line P-value | Description |
|---|---|---|---|
| Cluster 1—Immune-evasion subtype | |||
| EGFR_UP.V1_UP | <0.001 | <0.001 | Genes upregulated in MCF-7 cells (breast cancer) positive for ESR1 and engineered to express ligand-activatable EGFR |
| RAF_UP.V1_UP | <0.001 | <0.001 | Genes upregulated in MCF-7 cells (breast cancer) positive for ESR1 MCF-7 cells (breast cancer) stably over-expressing constitutively active RAF1 |
| HINATA_NFKB_IMMU_INF | <0.001 | 0.004 | Immune or inflammatory genes induced by NF-kappaB in primary keratinocytes and fibroblasts |
| Cluster 2—Replication-stress associated subtype | |||
| REACTOME_ACTIVATION_OF_ATR_IN_RESPONSE_TO_REPLICATION_STRESS | <0.001 | 0.005 | Genes involved in activation of ATR in response to replication stress |
| NFE2L2.V2 | 0.093 | <0.001 | Genes upregulated in MEF cells (embryonic fibroblasts) with knockout of NFE2L2 |
| SIRNA_EIF4GI_UP | 0.432 | <0.001 | Genes upregulated in MCF10A cells vs knockdown of EIF4G1 gene by RNAi |
| Cluster 3—Neuroendocrine subtype | |||
| CAHOY_NEURONAL | <0.001 | 0.036 | Genes upregulated in neurons |
| BCAT_GDS748_DN | 0.137 | — | Genes regulated in HEK293 cells (kidney fibroblasts) expressing constitutively active form of CTNNB1 gene |
| BCAT_GDS748_UP | — | 0.003 | |
| ESC_V6.5_UP_EARLY.V1_UP | 0.033 | <0.001 | Genes upregulated during early stages of differentiation of embryoid bodies from V6.5 embryonic stem cells |
GSEA of cluster 2 indicated that these patient samples and cell lines were enriched for genes regulated by the transcription factor NRF2. Additionally, GSEA identified high expression of genes upregulated by knockdown of eIF4G1, a translation initiation factor linked to nutrient sensing by mammalian target of rapamycin (mTOR),19 suggesting decreased mTOR signaling in cluster 2 (Table 2). Concordantly, cell lines from this subtype showed resistance to mTOR inhibition by AZD2014 (Table 1). Another key feature of cluster 2 samples was enrichment for DNA synthesis and replicative stress (Table 2). Tumors harboring enhanced levels of replication stress are dependent on ataxia telangiectasia-mediated replication responses for their survival.20 Indeed, cluster 2 cell lines showed enhanced sensitivity to ataxia telangiectasia inhibition by AZD6738 (Table 1). Given this evidence, we entitled this cluster the replication-stress-associated subtype.
Cluster 3 showed enrichment for signatures associated with a de-differentiated state: stem cell, β-catenin and neuronal signatures (Table 2). Similar to the neuroendocrine subtype of small cell lung cancers, these tumors also displayed upregulation of the neuroendocrine markers chromogranin A and chromogranin B (Supplementary Figure S4), leading us to name this cluster the neuroendocrine subtype. Both cell lines in this neuroendocrine subtype are known to have copy number gains in fibroblast growth factor receptor 1 indicating enhanced fibroblast growth factor receptor 1 signaling and explaining sensitivity to fibroblast growth factor receptor inhibition by AZD4547 in these cells. Additionally, these neuroendocrine cell lines showed sensitivity to AKT, phosphoinositide 3-kinase and mTOR inhibition with AZD5363, GDC0941 and AZD2014 compounds, respectively (Table 1). Neuroendocrine tumors from small cell lung and colorectal cancers also display an enhanced sensitivity to mTOR pathway inhibition, indicating that the phenomenon may apply more broadly to other neuroendocrine cancers.21, 22, 23
SCC genomic subtypes have distinct microRNA expression profiles
To determine if differences in cell signaling in each subtype drive differences in microRNA expression, we used analysis of variance (ANOVA) analysis with a cutoff of P<0.05 to define the microRNAs that are most differentially expressed between all three subtypes in the TCGA data set (Figure 2a) and found 306 differentially expressed microRNAs (Supplementary Table S5). Next, total RNA was extracted from a subset of the cell line panel (15 cell lines) and subjected to microRNA profiling. ANOVA analysis was performed using a cutoff of P<0.05 and we found 92 microRNAs differentially expressed between the subtypes in our representative cell lines (Figure 2b and Supplementary Table S5). By combining these two lists, we determined which microRNAs most clearly defined each subtype in both the TCGA samples and cell lines (Table 3). Overall, these data show that there are molecularly distinct subtypes present in SCC that have distinct differences in microRNA expression, which may be useful for identifying subtypes.
Figure 2.

SCC genomic subtypes have distinct microRNA expression profiles. (a) The most differentially expressed microRNAs between the three iNMF clusters in TCGA (ANOVA, P<0.05). (b) The most differentially expressed microRNAs in the SCC cell lines (ANOVA, P<0.05). Cluster 1 (immune-evasion) is indicated in blue, cluster 2 (replication-stress) is indicated in red and cluster 3 (neuroendocrine) is indicated in green.
Table 3. microRNAs differentially expressed in both TCGA and cell lines.
|
Cell lines |
TCGA |
||||
|---|---|---|---|---|---|
| MicroRNA | Fold change | P-value | MicroRNA | Fold change | P-value |
| Cluster 1—immune-evasion subtype | |||||
| Upregulated miRs | |||||
| hsa-miR-30a | 7.80 | 0.0211 | hsa-miR-30a | 1.64 | 1.58E−07 |
| hsa-miR-29a | 7.37 | 0.0144 | hsa-miR-29a | 1.44 | 5.45E−08 |
| hsa-miR-29b | 4.89 | 0.0166 | hsa-miR-29b-2 | 1.36 | 0.0004605 |
| 0.0166 | hsa-miR-29b-1 | 1.38 | 0.0004026 | ||
| hsa-miR-21 | 3.78 | 0.0049 | hsa-miR-21 | 1.21 | 0.0000343 |
| hsa-miR-29c | 2.36 | 0.0175 | hsa-miR-29c | 1.36 | 0.0015875 |
| hsa-miR-1976 | 1.10 | 0.0232 | hsa-miR-1976 | 1.33 | 0.0018874 |
| Downregulated miRs | |||||
| hsa-miR-1301 | 0.81 | 0.0065 | hsa-miR-1301 | 0.58 | 1.62E−08 |
| hsa-miR-345 | 0.76 | 0.0341 | hsa-miR-345 | 0.60 | 1.52E−06 |
| hsa-miR-454 | 0.72 | 0.0422 | hsa-miR-454 | 0.78 | 0.0011665 |
| hsa-miR-16 | 0.70 | 0.0389 | hsa-miR-16-1 | 0.78 | 0.0022378 |
| 0.0389 | hsa-miR-16-2 | 0.69 | 3.48E−06 | ||
| hsa-miR-421 | 0.65 | 0.0012 | hsa-miR-421 | 0.61 | 9.57E−07 |
| hsa-miR-944 | 0.60 | 0.0263 | hsa-miR-944 | 0.42 | 0.0000337 |
| hsa-miR-105 | 0.57 | 0.0058 | hsa-miR-105-1 | 0.43 | 0.0053822 |
| 0.0058 | hsa-miR-105-2 | 0.40 | 0.0017061 | ||
| hsa-miR-215 | 0.55 | 0.0132 | hsa-miR-215 | 0.77 | 0.0362918 |
| hsa-miR-192 | 0.52 | 0.0139 | hsa-miR-192 | 0.58 | 4.74E−06 |
| hsa-miR-194 | 0.50 | 0.0021 | hsa-miR-194 | 0.71 | 0.0001613 |
| hsa-miR-20a | 0.49 | 0.0261 | hsa-miR-20a | 0.59 | 2.51E−09 |
| hsa-miR-17 | 0.48 | 0.0195 | hsa-miR-17 | 0.61 | 4.22E−11 |
| hsa-miR-20b | 0.46 | 0.0106 | hsa-miR-20b | 0.43 | 8.69E−07 |
| hsa-miR-301b | 0.45 | 0.0200 | hsa-miR-301b | 0.38 | 6.40E−12 |
| hsa-miR-183 | 0.40 | 0.0278 | hsa-miR-183 | 0.52 | 5.04E−11 |
| hsa-miR-301a | 0.36 | 0.0233 | hsa-miR-301a | 0.61 | 0.0000269 |
| hsa-miR-363 | 0.30 | 0.0417 | hsa-miR-363 | 0.65 | 0.0020219 |
| hsa-miR-196b | 0.20 | 0.0098 | hsa-miR-196b | 0.54 | 0.0001893 |
| hsa-miR-9 | 0.16 | 0.0096 | hsa-miR-9-2 | 0.42 | 4.51E−06 |
| 0.0096 | hsa-miR-9-1 | 0.42 | 0.0000041 | ||
| hsa-miR-200a | 0.11 | 0.0189 | hsa-miR-200a | 0.68 | 0.0000433 |
| hsa-miR-429 | 0.09 | 0.0099 | hsa-miR-429 | 0.62 | 0.0000735 |
| hsa-miR-204 | 0.08 | 0.0222 | hsa-miR-204 | 0.31 | 6.08E−07 |
| hsa-miR-200b | 0.08 | 0.0389 | hsa-miR-200b | 0.66 | 0.0000202 |
| hsa-miR-200c | 0.06 | 0.0403 | hsa-miR-200c | 0.77 | 0.000397 |
| Cluster 2—Replication-stress subtype | |||||
| Upregulated miRs | |||||
| hsa-miR-205 | 221.50 | 0.0107 | hsa-miR-205 | 3.35 | 5.56E−11 |
| hsa-miR-203 | 14.99 | 0.0084 | hsa-miR-203 | 2.77 | 5.08E−11 |
| hsa-miR-944 | 3.03 | 0.0000 | hsa-miR-944 | 5.15 | 5.37E−18 |
| hsa-miR-149 | 3.02 | 0.0116 | hsa-miR-149 | 2.34 | 2.92E−11 |
| hsa-miR-663 | 2.53 | 0.0248 | hsa-miR-663 | 1.41 | 0.0004309 |
| hsa-miR-345 | 1.53 | 0.0073 | hsa-miR-345 | 1.43 | 0.0005957 |
| hsa-miR-3652 | 1.53 | 0.0057 | hsa-miR-3652 | 1.31 | 0.0038245 |
| hsa-miR-421 | 1.50 | 0.0389 | hsa-miR-421 | 1.39 | 0.0008246 |
| hsa-miR-513c | 1.36 | 0.0173 | hsa-miR-513c | 1.57 | 0.0000372 |
| hsa-miR-3687 | 1.33 | 0.0389 | hsa-miR-3687 | 1.60 | 0.0001114 |
| hsa-miR-378b | 1.14 | 0.0370 | hsa-miR-378b | 1.28 | 0.0008767 |
| Downregulated miRs | |||||
| hsa-miR-181b | 0.24 | 0.0389 | hsa-miR-181b | 0.68 | 5.2E−08 |
| hsa-miR-181a | 0.18 | 0.0161 | hsa-miR-181a | 0.71 | 2.33E−07 |
| Cluster 3—Neuroendocrine subtype | |||||
| Upregulated miRs | |||||
| hsa-miR-9 | 15.51 | 0.0014 | hsa-miR-9-2 | 3.55 | 4.31E−08 |
| 0.0014 | hsa-miR-9-1 | 3.53 | 4.47E−08 | ||
| hsa-miR-335 | 6.67 | 0.0109 | hsa-miR-335 | 1.62 | 0.0000279 |
| hsa-miR-19b | 5.70 | 0.0012 | hsa-miR-19b-1 | 1.34 | 0.0136969 |
| 0.0012 | hsa-miR-19b-2 | 1.33 | 0.004951 | ||
| hsa-miR-301b | 4.50 | 0.0000 | hsa-miR-301b | 1.75 | 0.0019509 |
| hsa-miR-17 | 3.78 | 0.0002 | hsa-miR-17 | 1.46 | 0.0000667 |
| hsa-miR-20a | 3.67 | 0.0004 | hsa-miR-20a | 1.48 | 0.0004789 |
| hsa-miR-18a | 3.50 | 0.0164 | hsa-miR-18a | 1.67 | 0.0000272 |
| hsa-miR-20b | 3.49 | 0.0005 | hsa-miR-20b | 1.89 | 0.0031859 |
| hsa-miR-18b | 2.89 | 0.0060 | hsa-miR-18b | 1.47 | 0.0030779 |
| hsa-miR-592 | 2.74 | 0.0024 | hsa-miR-592 | 1.48 | 0.0266718 |
| hsa-miR-92a | 2.47 | 0.0063 | hsa-miR-92a-2 | 1.63 | 0.0000072 |
| 0.0063 | hsa-miR-92a-1 | 1.73 | 0.0000134 | ||
| hsa-miR-105 | 2.38 | 0.0003 | hsa-miR-105-2 | 4.31 | 0.0000456 |
| 0.0003 | hsa-miR-105-1 | 5.36 | 0.0000057 | ||
| hsa-miR-194 | 2.13 | 0.0174 | hsa-miR-194-2 | 1.38 | 0.0044099 |
| 0.0174 | hsa-miR-194-1 | 1.36 | 0.00697 | ||
| hsa-miR-181c | 1.74 | 0.0311 | hsa-miR-181c | 1.44 | 0.0000283 |
| hsa-miR-1301 | 1.29 | 0.0138 | hsa-miR-1301 | 1.38 | 0.0088744 |
| Downregulated miRs | |||||
| hsa-miR-424 | 0.23 | 0.0198 | hsa-miR-424 | 0.80 | 0.040225 |
| hsa-miR-222 | 0.18 | 0.0023 | hsa-miR-222 | 0.64 | 7.04E−06 |
| hsa-miR-221 | 0.18 | 0.0016 | hsa-miR-221 | 0.68 | 0.0000452 |
| hsa-miR-23a | 0.14 | 0.0001 | hsa-miR-23a | 0.75 | 6.79E−06 |
| hsa-miR-27a | 0.13 | 0.0002 | hsa-miR-27a | 0.70 | 0.0000629 |
| hsa-miR-21 | 0.10 | 5.27E-07 | hsa-miR-21 | 0.73 | 1.12E−08 |
| hsa-miR-22 | 0.08 | 0.000038 | hsa-miR-22 | 0.71 | 7.96E−08 |
| hsa-miR-29b | 0.05 | 0.0000397 | hsa-miR-29b-2 | 0.68 | 0.0002611 |
| 0.0000397 | hsa-miR-29b-1 | 0.66 | 0.0001657 | ||
| hsa-miR-29a | 0.02 | 0.0000187 | hsa-miR-29a | 0.74 | 0.0003523 |
Transcription factors predicted to modulate microRNA expression are differentially expressed across the three subtypes
MicroRNA gene sequences are located in various genomic contexts, such as introns, non-coding or coding transcripts, and exonic regions. While much work has been done in recent years to characterize microRNA-target regulation, how the majority of microRNAs are regulated at the level of transcription is only beginning to be elucidated. To better understand how the enriched signaling pathways might be driving differences in microRNA expression, we interrogated the predicted promoter regions of microRNAs differentially expressed between the three clusters and compiled a ‘master list' of transcription factors predicted to regulate these microRNAs (Supplementary Table S6). We then examined the expression of these transcription factors in both TCGA data and cell lines. In the immune-evasion subtype ETS1 was the only predicted transcription factor that was significantly upregulated compared with the other subtypes (Figure 3a) at the level of mRNA expression in both TCGA (Figure 3b) and cell line (Figure 3c) data. Moreover, ETS1 protein expression was higher in cell lines belonging to the immune-evasion subtype (Figure 3d). ETS1 expression has previously been shown to be driven by EGFR and MAPK signaling,24, 25 suggesting that the enhanced EGFR/MAPK signaling in the immune-evasion subtype drives expression of ETS1, which may then drive differences in microRNA expression in this subtype. Consistent with the replication-stress-associated subtype having an enrichment for genes regulated by NRF2 (Table 2), NFE2L2 (NRF2) was the only predicted transcription factor significantly upregulated compared with the other two subtypes (Figure 3e) at the level of mRNA expression in both TCGA (Figure 3f) and cell line (Figure 3g) data. However, protein expression of NRF2 was relatively high in cell lines from all three subtypes (Figure 3h). In the neuroendocrine subtype, the transcription factor INSM1, known to be highly expressed in tumors of neuroendocrine orgin,26 was the only predicted transcription factor significantly upregulated compared with both of the other two subtypes (Figure 3i) at the level of mRNA in both TCGA (Figure 3j) and cell line (Figure 3k) data. Additionally, protein expression of INSM1 (53kda) was high in cell lines belonging to this subtype (Figure 3l).
Figure 3.

Transcription factors predicted to modulate microRNA expression are differentially expressed across the three subtypes. (a) Venn diagram depicting the overlap of transcription factors predicted to bind the promoters of microRNAs differentially regulated between subtypes (TF master list), transcription factors significantly upregulated in cluster 1 TCGA samples and transcription factors significantly upregulated in cluster 1 cell lines compared with the other two clusters. (b) Expression of ETS1 in TCGA samples grouped by subtype. (c) Expression of ETS1 in cell lines grouped by subtype. (d) Immunoblotting of detergent-solubilized whole-cell extracts with ETS1 antibody or GAPDH antibody. (e) Venn diagram depicting the overlap of the TF master list, transcription factors significantly upregulated in cluster 2 TCGA samples, and transcription factors significantly upregulated in cluster 2 cell lines compared with the other two clusters. (f) Expression of NRF2 in TCGA samples grouped by subtype. (g) Expression of NRF2 in cell lines grouped by subtype. (h) Immunoblotting of detergent-solubilized whole-cell extracts with NRF2 antibody or GAPDH antibody. (i) Venn diagram depicting the overlap of the TF master list, transcription factors significantly upregulated in cluster 3 TCGA samples and transcription factors significantly upregulated in cluster 3 cell lines compared with the other two clusters. (j) Expression of INSM1 in TCGA samples grouped by subtype. (k) Expression of INSM1 in cell lines grouped by subtype. (l) Immunoblotting of detergent-solubilized whole-cell extracts with INSM1 antibody or GAPDH antibody. *P<0.05, Student's t-test and all western blots are a representative image from three independent experiments.
The transcription factor ETS1 modulates expression of microRNAs in the immune-evasion subtype
To test the supposition that ETS1, NRF2 and INSM1 were modulating the differential expression of microRNAs in each subtype, we depleted each of these transcription factors using siRNA, then measured changes in microRNA expression in cell lines from each subtype. Depletion of NRF2 and INSM1did not result in consistent microRNA changes between cell lines (data not shown), suggesting that additional co-factors or transcription factors contribute to regulation of microRNA expression in these clusters. For this reason, we focused our attention on microRNA changes induced by ETS1 in immune-evasion subtype cell lines. We depleted ETS1, using four separate siRNAs which resulted in a significant decrease in transcript levels across six cell lines from the immune-evasion subtype (Figure 4a). Next, we measured fold change in microRNA expression resulting from this ETS1 depletion. In general, there was a trend towards microRNAs that had low expression in the original signature being upregulated (Figure 4b). Indeed, ETS1 depletion resulted in significant upregulation of miR-16, miR-17, miR-194, miR-20a, miR-20b, miR-301a, miR-301b and miR-421, suggesting that ETS1 represses expression of these microRNAs (Figure 4b and Supplementary Table S7). Of the four microRNAs that were shown to be highly expressed in the immune-evasion subtype as compared with the other subtypes (miR-21, miR-30a, miR-29a and miR-29b), miR-29b was significantly decreased by ETS1 depletion (Figure 4b and Supplementary Table S7), suggesting that ETS1 drives the expression of miR-29b.
Figure 4.

ETS1 modulates expression of microRNAs in the immune-evasion subtype. (a) Control (non-targeting) or ETS1 specific (ETS1 siRNA #1, #2, #3 and #4) siRNAs were transfected into indicated cell lines and after 72 h, decreased expression of ETS1 was confirmed by semiquantitative real-time PCR (data are the mean±s.e.m. of n=3, *P<0.05, Student's t-test). (b) Fold change in microRNA expression after ETS1 knockdown compared with control siRNA was measured by fluidigm chip PCR (n=3). microRNAs with low expression in the original signature are indicated with an ‘L', while those with high expression are indicated in an ‘H' under the heatmap. Percent knockdown under each experimental condition is indicated to the left of the heatmap. Only microRNAs that were significantly (P<0.05) changed in at least one cell line are shown (see Supplementary Table S7 for complete statistical results).
MiR-29b targets TET1 for downregulation
Given the inverse expression of miR-29b observed between the immune-evasion and the neuroendocrine subtypes (Table 3) and the modulation in expression of this microRNA by ETS1 (Figure 4b), we chose this microRNA for further investigation. To identify mRNA targets of miR-29b, we used Ingenuity Pathway Analysis software to compare predicted targets of miR-29b that showed decreased expression in the immune-evasion subtype, where miR-29b levels are high, and increased expression in the neuroendocrine subtype where miR-29b levels are low in both the TCGA and cell line data sets. This identified 15 possible mRNA targets of miR-29b (Figure 5a). Of these, the 5-methylcytosine dioxygenease TET1 enzyme that facilitates DNA demethylation by converting 5-methylcytosine (5mC) bases to 5-hydroxymethylcytosine (5-hmC)27 was the most significantly inversely correlated to miR-29b (Figure 5a). MiR-29b is transcribed from two genomic loci (miR-29b-1 and miR-29b-2), both of which showed inverse correlation with TET1 expression in TCGA tumors (Figures 5b and c). Additionally, TET1 expression was inversely correlated with miR-29b expression in SCC cell lines (Figures 5a and d). Transfection of an Anti-miR-29b into immune-evasion subtype cell lines, EBC1 and SKMES1, where miR-29b levels are high, led to an increase in TET1 protein levels (Figure 5e). Conversely, transfection of an miR-29b mimic into neuroendocrine subtype cells LK2 and NCIH520, where miR-29b levels are low led to decreased expression of TET1 protein (Figure 5e). Examination of the TET1 3′untranslated region (UTR) revealed that it contains five miR-29b binding sites (Figure 5f). To determine if miR-29b-mediated downregulation of TET1 occurs through miR-29b binding to the TET1 3′UTR, we measured luciferase reporter activity using constructs containing either a wild-type TET1 3′UTR or a mutated TET1 3′UTR (Figure 5f) and compared activity with a construct with no 3′UTR. The high basal levels of miR-29b expression in immune-evasion subtype cell lines EBC1 and SKMES1 directly correlated with greater decreases in TET1 3′UTR-luciferase reporter activity compared with neuroendocrine subtype cells LK2 and NCIH520, which contain low levels of miR-29b (Figure 5g). This correlation was not observed when the miR-29b binding sites in the TET1 3′UTR-luciferase reporter were mutated (Figures 5f and g). Additionally, co-transfection of Anti-miR-29b with the TET1 3′UTR-luciferase reporter in EBC1 cells led to increased expression of luciferase containing the TET1 3′UTR. Importantly, mutating the miR-29b seed sequence-binding region of the TET1 3′UTR abrogated this increase in luciferase expression (Figure 5h). Taken together, these results indicate that miR-29b targets TET1 for downregulation through an interaction with the TET1 3′UTR.
Figure 5.

miR-29b targets TET1 for downregulation. (a) Regression analysis of 15 mRNA targets of miR-29b identified by Ingenuity Pathway Analysis. R2 and P-values for inverse correlation between miR-29b and each gene are shown for both TCGA samples and cell lines (significant=P<0.05). (b) Regression plot of TET1 mRNA expression compared with hsa-miR-29b-1 expression in TCGA samples indicates significant inverse correlation P<0.0001. (c) Regression plot of TET1 mRNA expression compared with hsa-miR-29-2 expression in TCGA samples indicates significant inverse correlation P<0.0001. (d) Regression plot of TET1 mRNA expression compared with hsa-miR-29b expression in cell line samples indicates a significant degree of inverse correlation P=0.0007. (e) Immunoblotting of detergent-solubilized whole-cell extracts with TET1 antibody 48 h after transfection with either Anti-miR-29b (or non-targeting Anti-miR control) or miR-29b mimic (or control mimic). Differences in protein loading were monitored by reprobing stripped membranes with antibody against GAPDH. Shown is a representative image from three independent experiments. (f) Alignment of has-miR-29b to the wild type and mutant TET1 3′UTR sequences. Asterisks indicate mutated miR-29b seed sequence-binding bases. (g) Cells were transiently transfected with a luciferase reporter construct containing no 3′UTR, wild-type TET1 3′UTR or a mutated TET1 3′UTR sequence. Luciferase signal was normalized to the construct with no 3′UTR and the fold decrease in signal was plotted against the miR-29b expression levels of each cell line indicating a significant (P=0.03) correlation between miR-29b expression and decrease in wild-type TET1 luciferase reporter, but not the mutated reporter (P=0.29) (n=3). (h) EBC1 cells were transiently co-transfected with Anti-miR-29b (or Anti-miR control) and either a wild-type TET1 3′UTR reporter or a mutated TET1 3′UTR reporter (TET1 3′UTR mut) and luciferase signal was normalized to Anti-miR control transfected cells. Data are the mean±s.e.m. of n=3, *P<0.05, Student's t-test.
miR-29b-mediated downregulation of TET1 leads to changes in the epigenetic modification 5-hmC
TET1 is highly expressed in embryonic stem cells where it plays an important role in pluripotency, self renewal and differentiation28 by preventing both aberrant methylation spreading and stochastic hypermethylation.29, 30 Given the high expression of TET1 and the stem-like features in the neuroendocrine subtype compared with the other two subtypes, we investigated if there were differences in DNA methylation between subtypes. While we did not observe any difference in genome-wide methylation between subtypes (data not shown), we observed lower levels of DNA methylation in the genes identified by iNMF in the neuroendocrine subtype compared with the other two subtypes (Supplementary Figure S5 and Supplementary Table S8), suggesting that high levels of TET1 in this subtype may contribute to decreased methylation of genes driving the subtypes. To examine how changes in TET1 expression might contribute to gene expression signatures driving the subtypes, we compared our iNMF gene signature (Supplementary Table S1) with TET1 knockdown signatures from previously published studies.29, 30, 31 We then compiled a list of 39 genes that showed differential expression between the immune-evasion subtype and the neuroendocrine cell lines and that had been previously reported to be modulated by TET1 depletion (Supplementary Figure S6). Knockdown of TET1 in LK2 and NCIH520 cells where TET1 expression is high (Figure 6a) resulted in a trend towards increased expression of a subset of the genes that were low, and a decrease in a subset of genes that were high after 72 h. Although we did observe a very slight increase in expression of EGFR, this small increase was unable to reprogram and resensitize these resistant cell lines to gefitinib (Supplementary Figure S7 and Supplementary Table S9). Since TET1 is known to modulate DNA demethylation patterns by converting 5mC into 5-hmC,27 we confirmed that knockdown of TET1 (Figure 6a) led to decreased 5-hmC (Figure 6b). Next, we tested if miR-29b expression altered 5-hmC levels within the nucleus of cells. In line with miR-29b leading to downregulation of TET1, overexpression of miR-29b led to significantly decreased 5-hmC levels in the nucleus of EBC1, SKMES1 and LK2 cells (Figures 6c and d).
Figure 6.

miR-29b-mediated downregulation of TET1 leads to changes in the epigenetic modification 5-hmC. (a) Cells were transiently transfected with siRNA against TET1 or control siRNA and knockdown was determined by semi-quantitative real-time PCR, where individual signals were normalized to 18S. (b) After transfection with siRNA against TET1 or control siRNA cells were immunostained for 5-hmC and Hoescht and an algorithm measuring nuclear fluorescence intensity of 5-hmC was used to quantitate the percent of cells with 5-hmC in the nucleus and plotted as fold change to siRNA control (c) Cells were transiently transfected with miR-29b mimic or control mimic, after 48 h cells were immunostained for 5-hmC and Hoescht and quantitated as in (b). (d) Representative images of immunostaining after transfections with control mimic or miR-29b mimic. All experiments are the mean±s.e.m. n=3, *P<0.05, Student's t-test.
MAPK signaling regulates miR-29b-mediated downregulation of TET1and leads to changes in 5-hmC levels
Given the well-established role of ETS1 as an effector of MAPK signaling24, 25 and our evidence that ETS1 upregulates miR-29b expression (Figure 4b), we hypothesized that EGFR/MAPK signaling in the immune-evasion subtype would drive miR-29b expression through ETS1 leading to suppression of TET1. To test this hypothesis, we treated EBC1 and SKMES1 immune-evasion subtype cell lines with the EGFR inhibitor gefitinib, which led to a reduction in MAPK signaling, indicated by decreased phosphorylation of ERK1/2 (Figure 7a). Concomitant with this decrease in MAPK signaling, we observed a decrease in ETS1 and miR-29b and a corresponding increase in TET1 levels (Figure 7a). Additionally, gefitinib treatment of EBC1 cells also led to increased 5-hmC levels in the nucleus (Figure 7b). Interestingly, gefitinib treatment of EBC1 and SKMES1 also resulted in gene expression changes in the opposite direction to TET1 knockdown for a subset of genes (Supplementary Figure S6 and Supplementary Table S10). Collectively, these results indicate ETS1 and miR-29b expression are driven by MAPK signaling and that abrogation of miR-29b-mediated suppression of TET1 by MAPK pathway inhibition leads to increased levels of 5-hmC and may impact downstream methylation and gene expression.
Figure 7.

MAPK signaling regulates miR-29b-mediated downregulation of TET1and leads to changes in 5-hmC levels. (a) Cell lines were treated with 5 μm gefitinib or DMSO for 72 h. Afterwards, miR-29b levels were monitored by semiquantitative real-time PCR (±s.e.m., n=3) and immunoblotting was performed to measure differences in phospho-ERK, ETS1 and TET1. Differences in protein loading were monitored by reprobing stripped membranes with antibody against GAPDH. Shown is a representative image from three independent experiments. (b) Cells were treated with increasing concentrations of gefitinib for 48 h after which point, cells were immunostained for 5-hmC and Hoescht and visualized using a x10 objective on the cellomics Cell Insight. An algorithm measuring the nuclear fluorescence intensity of 5-hmC was used to analyze the cells. Data are the mean±s.e.m. of n=3,*P<0.05, Student's t-test. (c) High levels of EGFR/MAPK signaling in the immune-evasion subtype leads to an upregulation of ETS1 which drives increased expression of miR-29b. miR-29b targets TET1 for downregulation through interacting with its 3′UTR, which leads to decreased 5-hmC levels indicative of increased DNA methylation.
Discussion
Here, we demonstrate that lung SCC is composed of three subtypes that are driven by diverse cell signaling pathways, exhibit differential microRNA expression and differential drug sensitivity profiles. Additionally, we found that the immune-evasion subtype is enriched for EGFR signaling, which drives the expression of the transcription factor ETS1. In turn, ETS1 drives the upregulation of miR-29b, leading to the downregulation of TET1 and downstream decreases in 5-hmC epigenetic modifications (Figure 7c).
Targeted therapies against EGFR have been one of the most successful therapeutic strategies in lung AC patients with activating mutations in EGFR.2 In contrast, lung SCC generally lack activating mutations in EGFR32 and while a small fraction of SCC patients, without activating EGFR mutations, do respond to EGFR targeted therapies, a predictive biomarker for response to EGFR targeted therapies in SCC has yet to be identified.33 Interestingly, we have found that while very few immune-evasion subtype patients and cell lines have mutations in EGFR, there is an overall enrichment for signaling through the EGFR and MAPK signaling pathways. Additionally, We have uncovered a novel role for the MAPK-driven transcription factor ETS1 in regulating the expression of miR-29b, which suggests that miR-29b could be a biomarker for ERK/MAPK pathway activation.
It is well established that epigenetic changes can lead to promotion of tumor initiation and progression.34 The role of DNA methyltransferases, which promote DNA methylation, in driving cancer progression have been well characterized.34 However, until the recent discovery of the TET family of proteins, the role of DNA demethylation in cancer was less well characterized. The TET family is thought to act as tumor suppressors by maintaining other tumor suppressor genes in their unmethylated state.31, 35 Indeed, loss of function mutations in TET2 are frequently found in hematological malignancies and have been implicated in promoting tumor progression.35 Recently, analysis of microRNA-target mRNA expression correlations across 11 human cancer types in TCGA showed that TET1 was significantly inversely correlated with miR-29b across all cancer types.36 We provide mechanistic evidence that miR-29b directly downregulates TET1 expression (Figure 5). At present, there are ~80 validated targets of miR-29b37, 38 with one of the most well characterized being the DNA methyltransferases DNMT3A and DNMT3B.39 Taken together this suggests that miR-29b may act to fine tune DNA methylation status by protecting against aberrant changes in both methylating and demethylating enzymes.
Previous work has shown that suppression of TET1 expression is essential for KRAS-induced DNA hypermethylation and that this occurs through MAPK signaling.31 We provide further evidence that TET1 is downregulated through MAPK signaling and that this occurs through the novel mechanism of ETS1-mediated upregulation of miR-29b, which targets TET1 for downregulation through an interaction with its 3′UTR (Figures 4 and 5), leading to downstream effects on 5-hmC levels (Figures 6 and 7). Several studies have shown synergy between histone deacetylase inhibition and MAPK inhibition40, 41 as well as synergy between DNA-hypomethylating agents like 5-azacytidine and MAPK inhibitors,42, 43 suggesting that DNA demethylation enhances the efficacy of MAPK inhibitors. More recently TET upregulation has been shown to be essential for active demethylation induced by 5-azacytidine.44 This suggests that drug treatments that upregulate TET1 may synergize with MAPK suppression. However, given our finding that neuroendocrine subtype cell lines are not sensitive to EGFR/MAPK inhibition, high TET1 expression in the absence of MAPK signaling pathway activation is not likely to indicate sensitivity to MAPK inhibition.
Materials and methods
Cell culture and constructs
Cell lines were grown in RPMI 1640 media+10% fetal calf serum+2 mm glutamine at 37 °C 5% carbon dioxide and authenticated at AstraZeneca cell banking using DNA fingerprinting short-tandem repeat assays. MiRIDIAN miR-29b microRNA mimic (50 nm final concentration) or hairpin inhibitor (100 nm final concentration) were obtained from Thermo Scientific (Waltham, MA, USA) and ON-TARGETplus siRNA constructs were obtained from Dharmacon (Lafayette, CO, USA). All were transiently transfected using RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's recommendations.
iNMF
RNA-seq data in RSEM format from the 258 sample TCGA lung SCC gene expression data set12 where read data were collapsed down to the gene level was analyzed in R using the package NMF.45 Prior to NMF, genes that were expressed at log2<0 across all samples were removed. Factorization rank estimation was performed, estimating the approximate matrix for up to a maximum of 10 metagenes (or clusters) using 50 runs and benchmarked against randomized data to prevent over fitting. With a cophenetic correlation score of ~0.94, the three cluster solution was selected for the full NMF run using the brunet NMF algorithm and 200 runs. The genes and samples associated with each of the metagenes were extracted. The associated genes were used to separate the cell line data (GSE57083) into clusters using the heatmap.2 function from the R package gplots. For the validation data set,11 only SCC sample data were extracted for analysis, data were RMA normalized and NMF was performed on the probeset level and mapped to genes following analysis.
microRNA expression
Total RNA was purified from cell lines using the miRNeasy kit (Qiagen, Valencia, CA, USA), and 100 ng was labeled and hybridized to the array using the miRNA complete labeling and hyb kit (Agilent, Santa Clara, CA, USA) and spike-in kit (Agilent). Data were generated on a GeneChip Scanner 3000 (Agilent), Human microRNA Microarray Release 16.0, 8x60K arrays (Agilent). Data were imported into R, RMA normalized using the package AgiMicroRna,46 summarized to the log2 scale and returned for further association analysis as a gene by sample matrix. The miR expression data are MIAME compliant and have been submitted to the Gene Expression Omnibus (GSE73774). Differences in microRNA expression were determined with Omics Explorer (Qlucore, Lund, Sweden), using ANOVA on 549 miRs from TCGA12 and 1368 miRs from cell lines using using a cutoff of P<0.05 and false discovery rate of 0.09 and 0.73, respectively, calculated using the Benjamini–Hochberg method. Fold change of miRs found to be significant (P<0.05) in both TCGA and cell lines was then determined by comparing expression in each cluster with the other two clusters (Table 3).
GSEA and transcription factor predictions
High-throughput mRNA sequencing data from 258 lung SCC cancer patients were downloaded from the TCGA data portal. GSEA was performed using software (v2.1.0) obtained from the Broad Institute using the c2 and c6 databases from the MsigDB. P-values, enrichment scores and q-values were computed by permuting the sample labels (cluster number) 1000 times.47, 48 The ACTViewer program (Qu lab at Sun Yat-sen University) was used to identify transcription factor-binding sites in the promoter regions of differentially expressed microRNAs.
Real-time PCR
Total RNA was purified using the supplementary microRNA protocol for the RNeasy Plus Mini Kit (Qiagen). Expression of mRNA targets was measured using QuantiTect one-step reverse transcriptase-PCR reagents (Qiagen) and primer/probe sets from Life Technologies on a LightCycler 480 (Roche, Basel, Switzerland). For microRNA, cDNAs were synthesized using miScript II Reverse Transcription Kit (Qiagen) and diluted fivefold in RNase-free water prior to preamplification using the miScript Microfluidics PreAMP kit (Qiagen) and oligonucleotide primers from Life Technologies. Real-time PCR was carried out using reagents from the Microfluidics qPCR kit (Qiagen) on a Fluidigm 48 × 48 chip or carried out from 10-fold diluted cDNA following the miScript PCR System protocol (Qiagen) on a LightCycler480 (Roche). Primers and probes are listed in Supplementary Table S11.
Luciferase assays
Cells were seeded into 96-well plates and co-transfected using TransIT-X2 (Mirus, Madison, WI, USA) with microRNA mimic (Thermo Scientific) or Anti-miR (Thermo Scientific) along with 50 ng of pMirTarget (Origene, Rockville, MD, USA) housing the 3′UTR sequence of TET1 that contained either a wild-type or mutant version of the miR-29b binding site. Luciferase and RFP expression were measured using the Steady Glo Luciferase kit (Promega, Madison, WI, USA) and a Tecan microplate reader.
In vitro assays
Western blotting was performed as previously described.49 Antibodies are listed in Supplementary Table S12. Proliferation and dose response assays were performed by plating cells into a 96-well plate. Confluence was measured every 4 h using the IncuCyte (Essen Bioscience, Ann Arbor, MI, USA) for 6 days and data were analyzed in Graph Pad Prism 6.
Immunofluorescence
Cells were seeded into a clear bottom, black wall 96-well plate (then transfected with microRNA mimic (Thermo Scientific) Anti-miR (Thermo Scientific), or 20 nm siRNA (Dharmacon) using RNAiMAX (Invitrogen) or treated with drug. Cells were fixed with 3.7% formaldehyde, permeabilized in 0.5% Triton-X-100, denatured with 2 n HCl and neutralized with 100 mm Tris-HCl (pH8.5), then blocked with 5% BSA/0.1% Triton X-100 before probing with an antibody against 5-hydroxymethylcytosine (5-hmC) (Active Motif, Carlsbad, CA, USA) overnight at 4 °C. Following washing in phosphate-buffered saline-Tween 0.05%, cells were incubated with a secondary antibody conjugated to Alexa-Fluor-488 (Invitrogen) and Hoescht (Invitrogen) before washing and imaging using a × 10 objective on the Cellomics Cell Insight. An algorithm measuring the nuclear fluorescence intensity was used for analysis.
Methylation analysis
Methylation by mean β-values were downloaded from TCGA and Omics Explorer (Qlucore) was used to perform ANOVA to determine genes that were differentially methylated.
Statistics
Statistical values were defined using ANOVA or a two-tailed Student's t-test as specified in the text, P<0.05 was considered significant and distribution was assumed to be normal for all in vitro experiments.
Acknowledgments
We thank the Targets Biology group at AstraZeneca for critical comments of the manuscript, and Henry Brown and Hayley Campbell for their scientific expertise on fluidigm experiments. Funding was provided by the AstraZeneca Postdoc Programme.
Footnotes
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
All authors are employees of AstraZeneca.
Supplementary Material
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- Nana-Sinkam SP, Powell CA. Molecular biology of lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013; 143(5 Suppl): e30S–e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999; 286: 531–537. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al. Molecular portraits of human breast tumours. Nature 2000; 406: 747–752. [DOI] [PubMed] [Google Scholar]
- Hayes DN, Monti S, Parmigiani G, Gilks CB, Naoki K, Bhattacharjee A et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol 2006; 24: 5079–5090. [DOI] [PubMed] [Google Scholar]
- Taylor MA, Schiemann WP. Therapeutic opportunities for targeting microRNAs in cancer. Mol Cell Ther [Review] 2014; 2: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D et al. MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–838. [DOI] [PubMed] [Google Scholar]
- Liu A, Tetzlaff MT, Vanbelle P, Elder D, Feldman M, Tobias JW et al. MicroRNA expression profiling outperforms mRNA expression profiling in formalin-fixed paraffin-embedded tissues. Int J Clin Exp Pathol 2009; 2: 519–527. [PMC free article] [PubMed] [Google Scholar]
- Devarajan K. Nonnegative matrix factorization: an analytical and interpretive tool in computational biology. PLoS Comput Biol 2008; 4: e1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senbabaoglu Y, Michailidis G, Li JZ. Critical limitations of consensus clustering in class discovery. Sci Rep 2014; 4: 6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ES, Son DS, Kim SH, Lee J, Jo J, Han J et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin Cancer Res 2008; 14: 7397–7404. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489: 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR et al. Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin Cancer Res 2010; 16: 4864–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete A, Allavena P, Santoro G, Fumarulo R, Corsi MM, Mantovani A. Molecular pathways in cancer-related inflammation. Biochem Med (Zagreb) 2011; 21: 264–275. [DOI] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26: 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013; 3: 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 2014; 25: 1935–1940. [DOI] [PubMed] [Google Scholar]
- D'Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer 2015; 112: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol 2008; 181: 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 2014; 149: 124–138. [DOI] [PubMed] [Google Scholar]
- Wojtalla A, Fischer B, Kotelevets N, Mauri FA, Sobek J, Rehrauer H et al. Targeting the phosphoinositide 3-kinase p110-alpha isoform impairs cell proliferation, survival, and tumor growth in small cell lung cancer. Clin Cancer Res 2013; 19: 96–105. [DOI] [PubMed] [Google Scholar]
- Dhillon T, Mauri FA, Bellezza G, Cagini L, Barbareschi M, North BV et al. Overexpression of the mammalian target of rapamycin: a novel biomarker for poor survival in resected early stage non-small cell lung cancer. J Thorac Oncol 2010; 5: 314–319. [DOI] [PubMed] [Google Scholar]
- Marinov M, Ziogas A, Pardo OE, Tan LT, Dhillon T, Mauri FA et al. AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res 2009; 15: 1277–1287. [DOI] [PubMed] [Google Scholar]
- Liu S, Liang Y, Huang H, Wang L, Li Y, Li J et al. ERK-dependent signaling pathway and transcriptional factor Ets-1 regulate matrix metalloproteinase-9 production in transforming growth factor-beta1 stimulated glomerular podocytes. Cell Physiol Biochem 2005; 16: 207–216. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis 2010; 31: 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan MS, Breslin MB. Structure, expression, and biological function of INSM1 transcription factor in neuroendocrine differentiation. FASEB J 2009; 23: 2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Gribben JG. Loss of 5-hydroxymethylcytosine in cancer: Cause or consequence? Genomics 2014; 104: 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell 2011; 42: 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Lu Y, Jelinek J, Liang S, Estecio MR, Barton MC et al. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res 2014; 42: 6956–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011; 473: 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BK, Brenner C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep 2014; 9: 1827–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008; 359: 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao RG, Watanabe H, Meyerson M, Hammerman PS. Targeted therapy for squamous cell lung cancer. Lung Cancer Manag 2012; 1: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150: 12–27. [DOI] [PubMed] [Google Scholar]
- Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet 2014; 30: 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen A, Silber J, Harinath G, Huse JT, Schultz N, Sander C. Analysis of microRNA-target interactions across diverse cancer types. Nat Struct Mol Biol 2013; 20: 1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res 2014; 42(Database issue): D78–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res 2015; 43(Database issue): D153–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 2007; 104: 15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzese F, Leone A, Rocco M, Carbone C, Piro G, Caraglia M et al. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. J Cell Physiol 2011; 226: 2378–2390. [DOI] [PubMed] [Google Scholar]
- Lin YC, Shih JY, Huang WJ, Chao SW, Chang YL, Chen CC. DUSP1 expression induced by HDAC1 inhibition mediates gefitinib sensitivity in non-small cell lung cancers. Clin Cancer Res 2015; 21: 428–438. [DOI] [PubMed] [Google Scholar]
- Li XY, Wu JZ, Cao HX, Ma R, Wu JQ, Zhong YJ et al. Blockade of DNA methylation enhances the therapeutic effect of gefitinib in non-small cell lung cancer cells. Oncol Rep 2013; 29: 1975–1982. [DOI] [PubMed] [Google Scholar]
- Lou YF, Zou ZZ, Chen PJ, Huang GB, Li B, Zheng DQ et al. Combination of gefitinib and DNA methylation inhibitor decitabine exerts synergistic anti-cancer activity in colon cancer cells. PLoS One 2014; 9: e97719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadian SO, Ehnert S, Vakilian H, Koutsouraki E, Damm G, Seehofer D et al. Induction of active demethylation and 5hmC formation by 5-azacytidine is TET2 dependent and suggests new treatment strategies against hepatocellular carcinoma. Clin Epigenet 2015; 7: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 2010; 11: 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Romero P. Pre-processing and differential expression analysis of Agilent microRNA arrays using the AgiMicroRna Bioconductor library. BMC Genomics 2011; 12: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011; 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014; 4: 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.