

Abstract
There is a number of drugs demonstrating specific activity towards hereditary cancers. For example, tumors in BRCA1/2 mutation carriers usually arise via somatic inactivation of the remaining BRCA allele, which makes them particularly sensitive to platinum-based drugs, PARP inhibitors (PARPi), mitomycin C, liposomal doxorubicin, etc. There are several molecular assays for BRCA-ness, which permit to reveal BRCA-like phenocopies among sporadic tumors and thus extend clinical indications for the use of BRCA-specific therapies. Retrospective data on high-dose chemotherapy deserve consideration given some unexpected instances of cure from metastatic disease among BRCA1/2-mutated patients. Hereditary non-polyposis colorectal cancer (HNPCC) is characterized by high-level microsatellite instability (MSI-H), increased antigenicity and elevated expression of immunosuppressive molecules. Recent clinical trial demonstrated tumor responses in HNPCC patients treated by the immune checkpoint inhibitor pembrolizumab. There are successful clinical trials on the use of novel targeted agents for the treatment or rare cancer syndromes, e.g. RET inhibitors for hereditary medullary thyroid cancer, mTOR inhibitors for tumors arising in patients with tuberous sclerosis (TSC), and SMO inhibitors for basal-cell nevus syndrome. Germ-line mutation tests will be increasingly used in the future for the choice of the optimal therapy, therefore turnaround time for these laboratory procedures needs to be significantly reduced to ensure proper treatment planning.
Keywords: Hereditary cancer syndromes, Familial cancer, Breast cancer, Ovarian cancer, Colorectal cancer, Cytotoxic therapy, Targeted therapy, Predictive markers, BRCA1, BRCA2
Background
First tumor-predisposing germ-line mutations were discovered a quarter of century ago and were immediately translated into appropriate diagnostic tests [1–5]. Identification of mutation carriers among cancer patients, their yet healthy relatives and, to a lesser extent, some other individuals rapidly entered clinical routine and saved thousands of lives by delivering specific diagnostic and preventive efforts to the subjects at-risk. However, treatment schemes for hereditary and sporadic cancers remained virtually identical until this decade, therefore the genetic testing was usually considered rather as a part of the follow-up than the component of the initial decision-making process. We are currently witnessing a cultural change in clinical perception of hereditary cancers. It is getting increasingly recognized that many germ-line mutation-driven tumors develop via authentic molecular pathways and therefore have a unique spectrum of sensitivity to both conventional cytotoxic compounds and novel targeted drugs [6–9]. Many doctors now request rapid genetic testing at the time of treatment planning, and these attitudes are likely to become mandatory for the good clinical practice in a very near future. Here we review recent advances and controversies in the therapy of hereditary cancers (Table 1).
Table 1.
Examples of cytotoxic and targeted drugs showing promising activity towards hereditary cancers
| Hereditary cancer type | Drug |
|---|---|
| BRCA1/2-driven cancers (breast, ovarian, prostate, pancreatic, stomach, etc.) | Genotoxic agents: platinum compounds, PARP inhibitors, mitomycin C, pegylated doxorubicin, etc.; high dose chemotherapy |
| Hereditary non-polyposis colorectal cancer | Immune checkpoint inhibitors: pembrolizumab |
| Familial adenomatous polyposis | Non-steroidal anti-inflammatory drugs (sulindac) and EGFR inhibitors (erlotinib) |
| Tumors arising in patients with tuberous sclerosis (giant-cell astrocytomas, angiomyolipomas) | mTOR inhibitors: everolimus |
| Tumors associated with the basal-cell nevus syndrome (basal-cell carcinomas, keratocystic odontogenic tumors) | SMO inhibitors (vismodegib), COX2 inhibitors (celecoxib), antifungal drugs with Hedgehog pathway inhibitory activity (itraconazole) |
| Hereditary medullary thyroid cancer | RET inhibitors (vandetanib, cabozantinib) |
Note: See the text for comments and references
BRCA1 and BRCA2
Therapeutic window in BRCA-driven tumors
Breast-ovarian hereditary cancer syndrome is by far more common than other categories of familial cancers, with BRCA1 and BRCA2 being among the most studied genes. BRCA1/2-driven tumors usually arise via 2-hit mechanism: while the involved gene is present in heterozygous but still proficient state in the normal cells of the carrier, cancer cells are characterized by somatic loss of the remaining BRCA allele and therefore demonstrate deficiency in DNA repair by homologous recombination (HR). This opens an elegant therapeutic window by making tumor cells specifically vulnerable to DNA damaging drugs and poly(ADP-ribose) polymerase (PARP) inhibitors (Fig. 1). For example, platinating drugs induce DNA crosslinks, which cannot be effectively repaired in the absence of HR. Similarly, PARP inhibition results in accumulation of single-strand DNA breaks, which are subsequently converted to double-strand DNA breaks and turn out to be lethal for BRCA-deficient cells. This concept was initially confirmed in various laboratory studies and recently received validation in a series of clinical investigations [10–13].
Fig. 1.
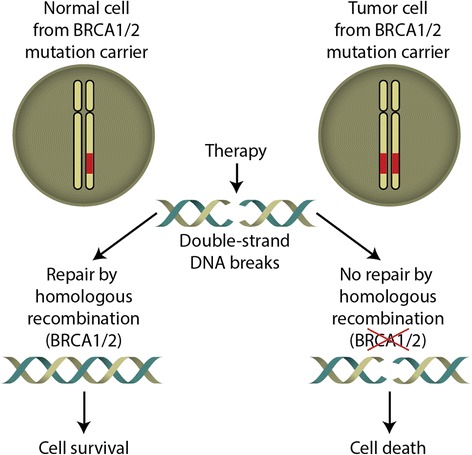
Selective sensitivity of BRCA1/2-associated tumors to genotoxic agents. Normal cells from BRCA1/2 mutation carriers retain full capacity of genome maintenance mechanisms (left). Development of tumors in these patients involves somatic inactivation of the remaining BRCA1/2 allele, therefore malignant cells are unable to cope with double-strand DNA breaks (right)
Platinum-based therapies
High sensitivity of BRCA1-driven tumors to cisplatin was initially demonstrated in a small Polish neoadjuvant breast cancer (BC) study involving mainly patients with small tumors [14]. Given that the pathologic complete response (pCR) was observed in 9 (90%) out of 10 included women [14], cisplatin quickly became a popular therapeutic option for hereditary BC in Poland. Recently Byrski et al. [15] reported an update on their experience of using cisplatin in the neoadjuvant BC setting: pCR was documented in 65/107 (61%) patients. Cisplatin appears to be clearly superior when compared to other schemes of preoperative therapy in BRCA1 mutation carriers [16, 17]. Furthermore, remarkable performance of cisplatin in chemonaive BRCA1-related BC was confirmed in several independent reports [18–20]. The key question is whether these high pCR rates will indeed translate into improved long-term outcomes; pooled analysis of neoadjuvant BC trials provides encouraging information suggesting that the relationship between pCR and survival is particularly strong in patients with the triple-negative BC disease [21].
Data on the use of platinum compounds in metastatic BC are limited. Byrski et al. [22] recruited both chemonaive and pretreated BRCA1 mutation carriers for cisplatin study, and reported the response in 16/20 (80%) cases with median progression-free survival (PFS) 12 months. Isakoff et al. [23] used either cisplatin or carboplatin in 11 patients with BRCA1/2-driven BC, most of whom experienced prior chemotherapy; the response was seen in 6 (54.5%) women, however its duration was short (3.3 months). Tutt et al. [24] compared carboplatin vs. docetaxel in triple-negative metastatic BC; for BRCA1/2 mutation carriers, the response rates were 68% vs. 33%, and PFS was 6.8 months vs. 3.1 months. There are several factors which critically compromise the assessment of platinum compounds in women with metastatic hereditary BC. First, the majority of patients presenting with inoperable disease have a history of prior chemotherapy; this is indeed an important confounding factor for BRCA-driven cancers, given that the development of the drug resistance frequently involves restoration of BRCA function [25]. Second, cisplatin and carboplatin have distinct efficacy and therefore have to be considered separately [26]. Third, there are multiple ongoing trials on PARP inhibitors (PARPi); it is probable that the cohort of patients receiving platinum therapy is somehow enriched by women, who were not included in PARPi studies due to contraindications or some other reasons.
Platinum-based therapy forms a backbone for the standard systemic treatment of ovarian cancer (OC). Multiple studies indicate that BRCA1/2-driven OC are characterized by increased sensitivity to platinating agents as compared to sporadic carcinomas [27–30]. Interstudy comparison of long-term results of the OC therapy is complicated, because OC outcomes are significantly influenced by the quality of cytoreductive surgery [31].
PARP inhibitors
There is a number of PARP inhibitors, including olaparib (Lynparza, AstraZeneca), veliparib (ABT-888, AbbVie), rucaparib (AG-014699, Clovis Oncology), niraparib (MK-4827, Tesaro), talazoparib (BMN-673, BioMarin Pharmaceutical), etc. [13]. Iniparib (Sanofi) was also initially developed as a PARP inhibitor; however recent studies showed that it has limited if any PARP-inhibiting activity and therefore cannot be considered as a drug belonging to PARPi class [32, 33].
Olaparib is the only PARP inhibitor already approved for the clinical use. Its initial trial as monotherapy agent demonstrated its preferential effect towards cancers arising in BRCA1/2 germ-line mutation carriers [34]. Subsequent study involving chemotherapy-pretreated ovarian cancer patients revealed that the efficacy of olaparib is more pronounced in women with platinum-sensitive disease and prolonged platinum-free interval [35]. Overall, there is consistent evidence for activity of olaparib in BRCA1/2-related cancers [36–41]. Olaparib received accelerated approval from the Food and Drug Administration (FDA) for the use in patients with BRCA1/2-driven ovarian cancers who received at least 3 prior lines of chemotherapy. In addition, this compound was assessed as a maintenance treatment in OC patients experiencing response to platinum-based therapy; it prolonged PFS as compared to placebo (11.2 vs. 4.3 months) in cases with germ-line or somatic BRCA1/2 mutations, although overall survival did not differ between these 2 arms [42]. Based on these data, olaparib is now approved for OC maintenance therapy by the European Medicines Agency (EMEA).
Given that neither olaparib nor conventional cytotoxic compounds are capable to achieve high rate of prolonged complete responses while administered separately, there is some interest to the use of these drugs in combination. Preclinical experiments support this concept [43–45]. Combination of olaparib and carboplatin has already been assessed in a Phase 1/1b study involving mixed population of patients with BRCA1/2 germ-line mutations, however only 1 out of 42 evaluable patients achieved complete response [46]. Another Phase I study demonstrated reasonable tolerability of combining olaparib with pegylated liposomal doxorubicin [47]. Oza et al. [48] studied OC patients with recurrent platinum-sensitive disease; while analyzing the subgroup of BRCA1/2 germ-line mutation carriers, they revealed that addition of olaparib to paclitaxel and carboplatin improved PFS as compared to chemotherapy alone.
Only a few studies assessed the efficacy of olaparib against standard cytotoxic treatment. Comparison of olaparib vs. pegylated liposomal doxorubicin (PLD) in recurrent BRCA1/2-associated ovarian cancer produced similar results for both drugs [49]. It has to be mentioned that PLD showed noticeably higher activity in BRCA1/2 mutation carriers as compared to historical non-selected OC series [50].
Data on the use of other PARP inhibitors are less extensive. Veliparib was evaluated in heavily pretreated ovarian cancer patients with BRCA1/2 mutation, and induced tumor responses in 13/50 (26%) women; similarly to experience with olaparib, there was a clear difference in response rates between platinum-resistant and platinum-sensitive disease (20% vs. 35%) [51]. Niraparib was assessed in a Phase I study; it demonstrated responses in 8/20 (40%) ovarian and 2/4 (50%) breast BRCA1/2-related cancers [52]. High disease control rates were also reported in a rucaparib monotherapy trial [53].
It is important to acknowledge, that all published clinical trials on PARP inhibitors involved pretreated BRCA1/2-mutated patients. This could be a critical limitation, as at least some BRCA1/2-driven tumors demonstrate restoration of intratumoral BRCA1/2 function during cytotoxic therapy [25, 29]. Low efficacy of PARPi in platinum-resistant as compared to platinum-sensitive ovarian cancer supports this assumption [51].
Other BRCA-specific therapies
In addition to cisplatin, there are several other non-expensive cytotoxic drugs showing BRCA-specific activity in preclinical experiments [6]. Based on these data, Moiseyenko et al. [54] administered single-agent mitomycin C (10 mg/m2, every 4 weeks) to 12 heavily pretreated ovarian cancer patients and obtained encouraging results: there was 1 complete response, 2 partial responses and 6 instances of the disease stabilization.
Trabectedin (Yondelis, Janssen) is a novel DNA damaging cytotoxic drug approved by FDA and EMEA for the therapy of inoperable soft tissue sarcomas [55]. In addition, it is used in some countries for the treatment of relapsed ovarian cancer in combination with pegylated liposomal doxorubicin [56]. It was assessed as a monotherapy in pretreated metastatic BRCA1/2-mutated breast cancer patients: 6/35 (17%) evaluable women experienced response, and median PFS approached to 3.9 months [57]. Promising activity of trabectedin was also shown in chemotherapy-pretreated hereditary ovarian cancer patients, although similar rates of tumor responses were observed in BRCA1/2 mutation carriers vs. non-carriers [58].
Eribulin (Halaven, Eisai) is a novel microtubule inhibitor, which demonstrated improvement of overall survival in patients with metastatic breast cancer after failure of multiple lines of systemic therapy. It was evaluated in combination with carboplatin in neoadjuvant trial involving triple-negative breast cancer patients. The study included 3 patients with BRCA1/2 mutation; clinical response was observed in all these women, with 2 of them achieving pathologic complete response [59].
BRCA1 is required for the execution of taxane-induced apoptosis, and at least some data indicate that taxane-containing regimens show limited efficacy towards BRCA1-associated breast cancers [16, 17, 60]. It is essential to acknowledge that some breast cancer studies do not support this concept [61]. Furthermore, paclitaxel monotherapy is effective in relapsed ovarian cancer in BRCA1 mutation carriers [62], although this study did not consider possible restoration of BRCA1 function during the prior therapy [25]. Recently Burness et al. [63] communicated 2 cases of BRCA1-associated chemonaive breast cancers, which demonstrated complete response to the paclitaxel monotherapy. These data deserve high level of attention, as they clearly contradict to the current views on the mechanisms of taxane action [7]. It is almost certain, that some aspects of cytotoxic effects of taxanes still remain unrecognized, however at least 2 reservations need to be kept in mind with regard to the report of Burness et al. [63]. First, there could be a publication bias, i.e. unexpected observations have significantly better chances to be published than routine clinical experience. Second, somatic inactivation of the wild-type BRCA1 allele may not be the only mechanism of breast cancer development in BRCA1 mutation carriers, as at least a subset of BRCA1-driven tumors appear to retain BRCA1 function [64, 65]; it is tempting to speculate that the sensitivity to taxanes is preserved in the latter category of BC.
Anthracyclines appear to exert substantial activity against BRCA1/2-driven tumors [6, 16, 17], with the novel formulations of these drugs producing remarkable responses in a subset of patients [49, 66–68]. There are limited data on the use of alkylating cytotoxic drugs in BRCA1/2-mutated cancers [7]. Kummar et al. [69] recently investigated low-dose daily cyclophosphamide in pretreated ovarian cancer patients, and observed 1 complete and 6 partial responses among 38 treated women.
There are some data on the activation of PTEN/PI3K/AKT/mTOR pathway in cancers arising in BRCA mutation carriers. Two mTOR inhibitors, temsirolimus and everolimus, are already available for the clinical use, however their performance in BRCA-driven cancers has not been assessed yet in preclinical or clinical settings. Recent studies demonstrate that mTOR down-regulation may sensitize cancer cells to PARP inhibitors [70, 71]. There is also some interest to the evaluation of BRCA-specific therapeutic potential of PI3K inhibitors [72–74].
BRCA1/2-deficient tumors are characterized by increased mutational load and therefore appear to be more antigenic than sporadic cancers. In accordance with this, these cancers demonstrate increased lymphocyte infiltration and show distinct pattern of expression of immune-related molecules [75, 76]. These data justify clinical trials involving immune checkpoint inhibitors.
Rare types of BRCA-associated cancers
Carriers of BRCA1 and BRCA2 germ-line mutations usually develop breast and/or ovarian cancers, however there is also some association with other cancer types. Inherited BRCA2 heterozygosity is associated with elevated prostate cancer risk. BRCA2-driven prostate cancers demonstrate good response to platinum-containing therapy and PARP inhibitors [38, 77–79]. Similar experience is obtained with pancreatic cancer [6, 9, 38, 80]. It remains under-recognized that some BRCA heterozygotes develop gastric cancer; causal relationship with BRCA status is supported by the evidence for tumor-specific inactivation of the wild-type BRCA allele. BRCA1-related stomach cancers are characterized by prolonged response to platinum containing therapy [81].
BRCA-ness
Many sporadic tumors share biological characteristics with BRCA1/2-associated hereditary cancers [82, 83]. For example, tumor-specific somatic inactivation of BRCA1 is the frequent cause of BRCA-ness phenotype. It may be manifested by the loss of BRCA1 expression, caused by either BRCA1 promoter hypermethylation or yet unknown reasons, or by mutational inactivation of both alleles of BRCA1 [84, 85]. Disruption of other genes belonging to HR pathway may also lead to similar consequences [78].
While some “BRCA-ness assays” rely on the identification of genetic causes of this phenotype, i.e. the detection of biallelic inactivation of BRCA or similar genes, novel generation of diagnostic tests utilizes characteristic mutational pattern of BRCA1/2-driven (HR-deficient) tumors [86–89]. For example, chromosomal instability results in a specific tumor karyotype, which can be revealed by array comparative genomic hybridization (aCGH) or some other techniques [90, 91]. BRCA1/2-deficient tumors demonstrate increased number of losses of heterozygosity (LOH) of a certain size (more than 15Mb but less than the entire chromosome) [92]. There is also a correlation between BRCA1/2 deficiency and allelic imbalances at telomeres [93]. Furthermore, BRCA1/2 inactivation manifests by large-scale chromosomal rearrangements [94].
In addition to characteristic mutational signatures, HR-deficient tumors demonstrate specific expression profiles [95, 96]. Cells with functional HR form RAD51 foci after DNA damage; there are attempts to establish ex vivo tumor HR testing using the analysis of induced RAD51 response [97].
Clinical data on the role of BRCA-ness demonstrate high level of consistency. BRCA-ness phenotype is associated with higher tumor sensitivity to platinum-containing agents and PARP inhibitors [23, 42, 78, 98, 99] and poor response to taxane-containing regimens [100]. The discovery of BRCA-ness (HR-ness) in a subset of sporadic tumors significantly extends potential indications for the drugs, which were initially considered to be active only in BRCA1/2-related hereditary cancers [82, 83].
It is essential to acknowledge that the participation of BRCA1 in DNA damage repair is not limited to HR, therefore the definitions of BRCA-ness and HR-ness are not necessarily identical. Hill et al. [101] studied triple-negative breast cancer cell lines and revealed no major HR defects. However, these cells exhibited deficiency in the repair of stalled replication forks, yet another feature of BRCA1 inactivation. Importantly, while the disruption of HR is associated with sensitivity to both cisplatin and PARPi, cells with abnormal repair of stalled replication forks are more selective, i.e. they would respond to the former but not to the latter [101]. These subtle differences may partially explain why there is no complete cross-resistance between DNA damaging cytotoxic drugs and PARP inhibitors [40, 51].
Mechanisms of resistance to BRCA-specific therapies
Most of disease-causing BRCA1/2 mutations are represented by relatively small alterations in DNA sequence, which cause frame-shift or generation of premature stop-codon. There is a series of fascinating reports, which demonstrate a somatic restoration of the open reading frame of BRCA genes in the therapy-resistant tumor cells; this is achieved by the occurrence of the second mutation in the proximity to the first one. As expected, second mutations are observed both in platinum- and in PARPi-treated tumors [25, 102–108].
While the above mechanisms represent mutational evolution of tumor genome under the pressure of BRCA1/2-specific therapy, we observed a distinct root of acquiring platinum resistance [29]. We analyzed somatic BRCA1 status in hereditary ovarian cancers undergoing short-term preoperative therapy (on average, 3 cycles of platinum-containing cocktails given with 21-day intervals). Astonishingly, while the chemonaive carcinomas showed LOH characteristic for BRCA1-associated malignancies, the wild-type allele was preserved in the residual tumor masses removed upon surgery. Given that LOH is not an early event in BRCA1-driven tumorigenesis and that tumors from BRCA1 mutation carriers are known to contain a fraction of cells with the intact wild-type allele [109], this phenomenon can be explained by rapid selection of preexisting BRCA1-proficient cells during platinum exposure. The mere fact of selection of treatment-resistant cells upon the therapy is not at all surprising; what is indeed entirely unexpected, is the speed of this transition. On the level of clinical measuring of the changes in tumor size, the process of acquiring drug resistance by initially chemosensitive tumors usually takes several months; our data indicate that truly responsive neoplastic cells die within very first weeks (days?) of treatment, and they are rapidly replaced by the clones with potentially chemorefractory phenotype (Fig. 2). These data cast some doubt on the ideology of neoadjuvant treatment. Preoperative therapy is commonly viewed as a highly informative in vivo test for the actual tumor sensitivity [110]. If pronounced reduction of tumor burden is observed before the surgery, the same schemes are often administered in the adjuvant setting [111]. However, if the turnover of tumor cell populations is indeed so rapid, and the surgically removed cancer mass is biologically distinct from the initial malignancy, the rationale for continuing the same therapy after the surgery may look questionable.
Fig. 2.
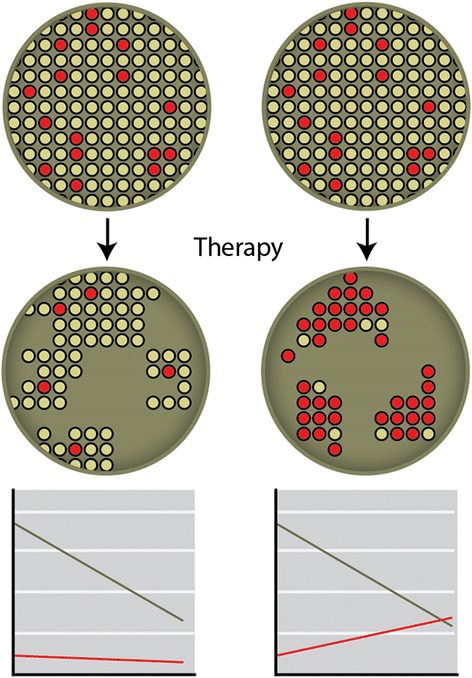
The dynamics of distinct tumor cell populations upon systemic therapy. Many tumors respond well to the initial therapy; although the existence of intratumoral cellular heterogeneity is widely acknowledged, it is generally believed that the evolution of treatment-resistant clones requires additional genetic events and usually takes at least several months (left). Our data indicate that even short-term (neoadjuvant) exposure of BRCA1-driven tumors to platinum therapy results in the replacement of tumor mass by BRCA1-proficient cells [29]. While cells with BRCA1 LOH die almost immediately after beginning of the treatment, clones with retained BRCA1 continue to proliferate during platinum exposure and rapidly repopulate the tumor lump (right)
There are some other proposed mechanisms of the resistance of BRCA1/2-mutated cells to the specific therapy, such as inactivation of TP53BP1 protein, efflux of platinum drugs or PARP inhibitors, or some other molecular events [25, 112–115]. However, they were demonstrated mainly in laboratory models, and their actual clinical relevance remains uncertain.
High-dose chemotherapy
High-dose chemotherapy followed by autologous hematopoietic stem cell transplantation is used for the treatment of germ-cell tumors, several hematological malignancies, childhood cancers etc. It was a popular experimental approach for the treatment of breast cancer in mid 1990s, but failed to demonstrate improved long-term outcomes in unselected BC patients [116–118]. However, retrospective analysis of women with metastatic BC revealed several instances of unexpectedly long remission of the disease; importantly, these survivors are enriched by BRCA1/2 germ-line mutation carriers, which is in good agreement with the data on increased chemosensitivity of BRCA-driven BC [119]. Furthermore, patients with BRCA1-like stage III BC show substantial benefit from adjuvant high-dose therapy [91, 120], and this effect correlates with the molecular status of BRCA1-related pathways [115]. There is also a number of case reports describing patients with germ-line BRCA1/2 mutations, who were treated by high-dose chemotherapy for the metastatic disease and remained tumor-free for years [121, 122]. Given that many BRCA1/2 mutation carriers are diagnosed with metastatic cancer at a relatively young age and therefore retain sufficiently good health status to survive a risky intervention, there is a rationale for considering high-dose therapy trials for this uniquely chemosensitive category of tumors.
Adverse effects of BRCA-specific therapy in the germ-line mutation carriers: potential impact of haploinsufficiency
Although normal cells in BRCA1/2 mutation carriers retain a wild-type copy of the involved gene and therefore are able to cope with DNA damage, some experiments suggest that the loss of even single BRCA1 allele results in some decrease of BRCA1 functional capacity [123]. If the data on BRCA haploinsufficiency are applicable to the individuals with inherited BRCA defects, one may expect distinct pattern and severity of adverse effects of cancer therapy in the mutations carriers vs. non-carriers. The majority of available clinical investigations did not acknowledge unexpected adverse reactions [6, 22, 119], which may be interpreted, with some caution, in favor of normal tolerability of chemotherapy in BRCA1/2 mutation carriers. Furthermore, systematic single-center analysis of BC patients receiving conventional cytotoxic drugs did not reveal increased toxicity in BRCA1/2 heterozygotes vs. other women [124]. Nevertheless, Moon et al. [125] observed increased incidence of hypersensitivity reactions in BRCA1/2-mutated ovarian cancer patients receiving carboplatin and olaparib. There is also a discussion whether therapeutic radiation is associated with increased risk of induced cancers in BRCA1/2 heterozygotes [126, 127].
Novel types of hereditary breast cancer
While BRCA1/2-related cancers have been studied with a high level of comprehension, the available information on clinical behavior of novel categories of hereditary breast cancer remains very limited. Importantly, BC arising in CHEK2, NBN/NBS1 and BLM heterozygotes usually demonstrates retention of the wild-type allele in the tumor, therefore there is no ground to expect selective chemosensitivity in these tumor types [128]. In agreement with mechanistic considerations, Chrisanthar et al. [129] observed several instances of resistance of CHEK2-associated BC to epirubicin. Pfeifer et al. [17] described 8 CHEK2 mutation carriers receiving neoadjuvant therapy, with none of them achieving pathologic complete response; 4 out of these 8 women experienced objective clinical response, however the reduction of the tumor size was observed only in 1 out of 4 patients treated by anthracycline-based therapy without taxanes. While data of Chrisanthar et al. [129] and Pfeifer et al. [17] indicate relative chemoresistance of CHEK2-driven BC at least to anthracyclines, a large study of Kriege et al. [130] provided conflicting evidence. The latter group described 62 CHEK2-mutated metastatic BC cases, and the response rates to cytotoxic and endocrine therapy did not differ between CHEK2 heterozygotes and mutation-free controls [130].
Hereditary colon cancer
There are multiple types of hereditary colon cancer [131], with the so-called hereditary non-polyposis colorectal cancer (HNPCC) syndrome being the most studied type of this disease. HNPCC is caused by germ-line mutations in DNA mismatch repair genes; HNPCC-related tumors are characterized by a unique phenotypic feature, i.e. high-level microsatellite instability (MSI-H). It is beyond the doubt that MSI-H tumors have distinct biological properties. In particular, they have increased level of antigenicity due to accumulation of multiple background mutations, and therefore are characterized by a relatively good prognosis [132–134].
MSI-H carcinomas combine two distinct categories of malignancies. First, there are hereditary cancers obtained from the germ-line mutation carriers; they tend to be associated with younger age of the patients. Second, there are sporadic MSI-H tumors; they often arise in elderly subjects and frequently carry an actionable activating mutation in BRAF oncogene. In addition to genuine biological diversity of MSI-H tumors, there are some debates regarding technical aspects of MSI status determinations [133–136]. Therefore, it is unclear to what extent the data obtained on MSI-H cancers can be extrapolated to HNPCC-related tumors.
High antigenicity of MSI-H tumors makes them recognizable by the immune system. In order to overcome host defense mechanisms, MSI-H carcinomas express some immune checkpoint molecules thus creating local immunosuppressive environment [137]. Le et al. [138] investigated pembrolizumab (Keytruda, Merck), an antibody capable to inhibit PD1 pathway and thus restore peritumoral immune response, for the treatment of MSI-H cancers. This antibody induced objective tumor responses in all 6 patients with sporadic MSI-H tumors, but only in 3 out of 11 patients with the HNPCC syndrome; the reasons for these differences in efficacy in sporadic vs. hereditary MSI-H cancers remain obscure.
MSI-H tumors may have distinct spectrum of chemosensitivity, which is critically influenced by the individual pattern of somatically mutated genes [134, 136, 139]. There is an ongoing research aiming to develop synthetic lethal strategy for the targeting of MSI-H cancers [140].
There are studies aimed to develop systemic therapy for the familial adenomatous polyposis (FAP). Sulindac, a non-steroidal anti-inflammatory drug, was shown to be effective against colorectal adenomatous polyps. However, it has limited efficacy against duodenal carcinogenesis; duodenal carcinomas are the main cause of death in those FAP patients, who have already undergone colectomy. Synergistic interaction between COX2 and EGFR inhibitors was observed in murine FAP models. Combination of sulindac with erlotinib was assessed in a clinical trial involving 46 FAP patients in the experimental group and 46 subjects treated by placebo. Use of the above drugs resulted in clinically significant reduction of the number and size of duodenal polyps [141].
Novel targeted agents for therapy of rare hereditary cancers
Medullary thyroid cancer is often driven by inherited activating mutation in RET oncogene. There are RET kinase inhibitors, vandetanib (Caprelsa, AstraZeneca) and cabozantinib (Cometriq, Exelixis), which demonstrate clinical activity against both hereditary and sporadic medullary thyroid carcinomas [142–145].
Everolimus (Afinitor, Novartis), an inhibitor of mTOR kinase, has been approved for the treatment of several tumor types. mTOR pathway is specifically activated in tumors arising in patients with tuberous sclerosis (TSC). Everolimus was systematically evaluated in TSC-related giant-cell astrocytomas and angiomyolipomas, and showed remarkable efficacy in these malignancies [146–148].
Vismodegib (Erivedge, Roche/Genentech) inhibits Hedgehog pathway via interaction with SMO protein. Its antitumor activity was examined in patients with basal-cell nevus (Gorlin) syndrome [149]. This drug reduced the size of existing basal-cell carcinomas and prevented the appearance of new lesions; however, 14 out of 26 subjects decided to discontinue the treatment due to adverse events. Vismodegib also showed some activity against keratocystic odontogenic tumors arising in patients with Gorlin syndrome [150]. There is evidence that this hereditary tumor disease may somehow be controlled with some other drugs. For example, a COX2 inhibitor, celecoxib slowed the increase of tumor burden in the treated patients as compared to placebo control [151]. Well-known triazol antifungal drugs (itraconazole, ketoconazole, posaconazole, etc.) turned out to have substantial inhibitory activity against Hedgehog pathway [152, 153]. They are capable to induce regression of basal cell carcinomas at least in a subset of patients [154, 155].
Conclusions and perspectives
The number of known hereditary cancer syndromes will rapidly grow within next several years, thanks to the invention of whole exome sequencing [131]. Many of already identified hereditary cancer types are represented by exceptionally rare instances of the disease, and future investigations are likely to reveal even more uncommon cancer syndromes. In addition, unlike many other medical conditions, genetic diseases are often population-specific, i.e. their spread is limited by a few ethnic groups. It is unrealistic to expect that each hereditary cancer type will be subjected to systematic laboratory investigations and comprehensive clinical trials. There are some approaches which may facilitate search for novel treatment strategies for orphan and/or hereditary cancer types. For instance, collection and analysis of biological material from these patients deserve to be encouraged. In addition, some ex vivo testing for tumor drug sensitivity may turn out to be particularly practical in this clinical setting [156]. There are also some bioinformatic tools pretending to predict drug sensitivity of the tumor based on its molecular characteristics [157]. Finally, several potentially practice-changing investigations in this field became possible due to availability of large clinical databases [16, 27, 120]. Well-designed retrospective studies may help to significantly improve the use of existing cancer therapies.
Abbreviations
aCGH, array comparative genomic hybridization; BC, breast cancer; EMEA, European Medicines Agency; FAP, familial adenomatous polyposis; FDA, Food and Drug Administration; HNPCC, hereditary non-polyposis colorectal cancer; HR, homologous recombination; LOH, loss of heterozygosity; MSI-H, high-level microsatellite instability; OC, ovarian cancer; PARP, poly(ADP-ribose) polymerase; PARPi, PARP inhibitors; pCR, pathologic complete response; PFS, progression-free survival; PLD, pegylated liposomal doxorubicin; TSC, tuberous sclerosis
ᅟ
ᅟ
Funding
This work has been supported by the Russian Scientific Fund (grant number 14-15-00528).
Availability of data and supporting materials section
Not applicable
Authors’ contributions
The authors contributed equally to the writing of the manuscript and approved the final version.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable
Ethics approval and consent to participate
Not applicable
Contributor Information
Aglaya G. Iyevleva, Email: aglayai@inbox.ru
Evgeny N. Imyanitov, Phone: +7-812-4399528, Email: evgeny@imyanitov.spb.ru
References
- 1.Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 2.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 3.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-S. [DOI] [PubMed] [Google Scholar]
- 4.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 5.Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 6.Imyanitov EN, Moiseyenko VM. Drug therapy for hereditary cancers. Hered Cancer Clin Pract. 2011;9(1):5. doi: 10.1186/1897-4287-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayraktar S, Glück S. Systemic therapy options in BRCA mutation-associated breast cancer. Breast Cancer Res Treat. 2012;135(2):355–366. doi: 10.1007/s10549-012-2158-6. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell KN, Domchek SM. Cancer treatment according to BRCA1 and BRCA2 mutations. Nat Rev Clin Oncol. 2012;9(9):520–528. doi: 10.1038/nrclinonc.2012.123. [DOI] [PubMed] [Google Scholar]
- 9.Imyanitov EN, Byrski T. Systemic treatment for hereditary cancers: a 2012 update. Hered Cancer Clin Pract. 2013;11(1):2. doi: 10.1186/1897-4287-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner NC, Tutt AN. Platinum chemotherapy for BRCA1-related breast cancer: do we need more evidence? Breast Cancer Res. 2012;14(6):115. doi: 10.1186/bcr3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pothuri B. BRCA1- and BRCA2-related mutations: therapeutic implications in ovarian cancer. Ann Oncol. 2013;24(Suppl 8):viii22–viii27. doi: 10.1093/annonc/mdt307. [DOI] [PubMed] [Google Scholar]
- 12.Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25(1):32–40. doi: 10.1093/annonc/mdt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonnenblick A, de Azambuja E, Azim HA, Jr, Piccart M. An update on PARP inhibitors--moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12(1):27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- 14.Byrski T, Huzarski T, Dent R, Gronwald J, Zuziak D, Cybulski C, et al. Response to neoadjuvant therapy with cisplatin in B RCA1-positive breast cancer patients. Breast Cancer Res Treat. 2009;115(2):359–363. doi: 10.1007/s10549-008-0128-9. [DOI] [PubMed] [Google Scholar]
- 15.Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat. 2014;147(2):401–405. doi: 10.1007/s10549-014-3100-x. [DOI] [PubMed] [Google Scholar]
- 16.Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28(3):375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 17.Pfeifer W, Sokolenko AP, Potapova ON, Bessonov AA, Ivantsov AO, Laptiev SA, et al. Breast cancer sensitivity to neoadjuvant therapy in BRCA1 and CHEK2 mutation carriers and non-carriers. Breast Cancer Res Treat. 2014;148(3):675–683. doi: 10.1007/s10549-014-3206-1. [DOI] [PubMed] [Google Scholar]
- 18.Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28(7):1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kołacińska A, Chałubińska J, Błasińska-Morawiec M, Dowgier-Witczak I, Fendler W, Kordek R, et al. Pathological complete response in younger and older breast cancer patients. Arch Med Sci. 2012;8(2):310–315. doi: 10.5114/aoms.2012.28559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moiseyenko VM, Dolmatov GD, Moiseyenko FV, Ivantsov AO, Volkov NM, Chubenko VA, et al. High efficacy of cisplatin neoadjuvant therapy in a prospective series of patients carrying BRCA1 germ-line mutation. Med Oncol. 2015;32(4):89. doi: 10.1007/s12032-015-0514-1. [DOI] [PubMed] [Google Scholar]
- 21.Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–172. doi: 10.1016/S0140-6736(13)62422-8. [DOI] [PubMed] [Google Scholar]
- 22.Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012;14(4):R110. doi: 10.1186/bcr3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2015;33(17):1902–1909. doi: 10.1200/JCO.2014.57.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tutt A, Ellis P, Kilburn L, Gilett C, Pinder S, Abraham J, et al. The TNT trial: A randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). San Antonio Breast Cancer Symposium. 2014:abstr S3-01.
- 25.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- 26.Chalasani P, Livingston R. Differential chemotherapeutic sensitivity for breast tumors with “BRCAness”: a review. Oncologist. 2013;18(8):909–916. doi: 10.1634/theoncologist.2013-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vencken PM, Kriege M, Hoogwerf D, Beugelink S, van der Burg ME, Hooning MJ, et al. Chemosensitivity and outcome of BRCA1- and BRCA2-associated ovarian cancer patients after first-line chemotherapy compared with sporadic ovarian cancer patients. Ann Oncol. 2011;22(6):1346–1352. doi: 10.1093/annonc/mdq628. [DOI] [PubMed] [Google Scholar]
- 28.Alsop K, Fereday S, Meldrum C, de Fazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30(21):2654–2663. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorodnova TV, Sokolenko AP, Ivantsov AO, Iyevleva AG, Suspitsin EN, Aleksakhina SN, et al. High response rates to neoadjuvant platinum-based therapy in ovarian cancer patients carrying germ-line BRCA mutation. Cancer Lett. 2015;369(2):363–367. doi: 10.1016/j.canlet.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 30.Mahdi H, Gockley A, Esselen K, Marquard J, Nutter B, Yang B, et al. Outcome of neoadjuvant chemotherapy in BRCA1/2 mutation positive women with advanced-stage Müllerian cancer. Gynecol Oncol. 2015;139(3):407–412. doi: 10.1016/j.ygyno.2015.07.101. [DOI] [PubMed] [Google Scholar]
- 31.Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384(9951):1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- 32.Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin Cancer Res. 2012;18(6):1655–1662. doi: 10.1158/1078-0432.CCR-11-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mateo J, Ong M, Tan DS, Gonzalez MA, de Bono JS. Appraising iniparib, the PARP inhibitor that never was--what must we learn? Nat Rev Clin Oncol. 2013;10(12):688–696. doi: 10.1038/nrclinonc.2013.177. [DOI] [PubMed] [Google Scholar]
- 34.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 35.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 36.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 37.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 38.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Noll R, Marchetti S, Steeghs N, Beijnen JH, Mergui-Roelvink MW, Harms E, et al. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer. 2015;113(3):396–402. doi: 10.1038/bjc.2015.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016;140(2):199–203. doi: 10.1016/j.ygyno.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multi-study analysis of response rates and safety. Ann Oncol. 2016 (in press). [DOI] [PubMed]
- 42.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomized phase 2 trial. Lancet Oncol. 2014;15(8):852–861. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 43.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karginova O, Siegel MB, Van Swearingen AE, Deal AM, Adamo B, Sambade MJ, et al. Efficacy of Carboplatin Alone and in Combination with ABT888 in Intracranial Murine Models of BRCA-Mutated and BRCA-Wild-Type Triple-Negative Breast Cancer. Mol Cancer Ther. 2015;14(4):920–930. doi: 10.1158/1535-7163.MCT-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ávila-Arroyo S, Nuñez GS, García-Fernández LF, Galmarini CM. Synergistic Effect of Trabectedin and Olaparib Combination Regimen in Breast Cancer Cell Lines. J Breast Cancer. 2015;18(4):329–338. doi: 10.4048/jbc.2015.18.4.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106(6):dju089. doi: 10.1093/jnci/dju089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Conte G, Sessa C, von Moos R, Viganò L, Digena T, Locatelli A, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111(4):651–659. doi: 10.1038/bjc.2014.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomized phase 2 trial. Lancet Oncol. 2015;16(1):87–97. doi: 10.1016/S1470-2045(14)71135-0. [DOI] [PubMed] [Google Scholar]
- 49.Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(4):372–379. doi: 10.1200/JCO.2011.36.9215. [DOI] [PubMed] [Google Scholar]
- 50.Konstantinopoulos PA, Cannistra SA. Comparing poly (ADP-ribose) polymerase inhibitors with standard chemotherapy in BRCA-mutated, recurrent ovarian cancer: lessons learned from a negative trial. J Clin Oncol. 2012;30(4):347–350. doi: 10.1200/JCO.2011.40.1489. [DOI] [PubMed] [Google Scholar]
- 51.Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015;137(3):386–391. doi: 10.1016/j.ygyno.2015.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–892. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 53.Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(7):723–730. doi: 10.1038/bjc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moiseyenko VM, Chubenko VA, Moiseyenko FV, Zhabina AS, Gorodnova TV, Komarov YI, et al. Evidence for clinical efficacy of mitomycin C in heavily pretreated ovarian cancer patients carrying germ-line BRCA1 mutation. Med Oncol. 2014;31(10):199. doi: 10.1007/s12032-014-0199-x. [DOI] [PubMed] [Google Scholar]
- 55.D’Incalci M. Trabectedin mechanism of action: what’s new? Future Oncol. 2013;9(12 Suppl):5–10. doi: 10.2217/fon.13.207. [DOI] [PubMed] [Google Scholar]
- 56.Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J Clin Oncol. 2010;28(19):3107–3114. doi: 10.1200/JCO.2009.25.4037. [DOI] [PubMed] [Google Scholar]
- 57.Delaloge S, Wolp-Diniz R, Byrski T, Blum JL, Gonçalves A, Campone M, et al. Activity of trabectedin in germline BRCA1/2-mutated metastatic breast cancer: results of an international first-in-class phase II study. Ann Oncol. 2014;25(6):1152–1158. doi: 10.1093/annonc/mdu134. [DOI] [PubMed] [Google Scholar]
- 58.Lorusso D, Scambia G, Pignata S, Sorio R, Amadio G, Lepori S, et al. Prospective phase II trial of trabectedin in BRCA-mutated and/or BRCAness phenotype recurrent ovarian cancer patients: the MITO 15 trial. Ann Oncol. 2016;27(3):487–493. doi: 10.1093/annonc/mdv608. [DOI] [PubMed] [Google Scholar]
- 59.Kaklamani VG, Jeruss JS, Hughes E, Siziopikou K, Timms KM, Gutin A, et al. Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer ( NCT01372579) Breast Cancer Res Treat. 2015;151(3):629–638. doi: 10.1007/s10549-015-3435-y. [DOI] [PubMed] [Google Scholar]
- 60.Kriege M, Jager A, Hooning MJ, Huijskens E, Blom J, van Deurzen CH, et al. The efficacy of taxane chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer. 2012;118(4):899–907. doi: 10.1002/cncr.26351. [DOI] [PubMed] [Google Scholar]
- 61.Arun B, Bayraktar S, Liu DD, Gutierrez Barrera AM, Atchley D, Pusztai L, et al. Response to neoadjuvant systemic therapy for breast cancer in BRCA mutation carriers and noncarriers: a single-institution experience. J Clin Oncol. 2011;29(28):3739–3746. doi: 10.1200/JCO.2011.35.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan DS, Yap TA, Hutka M, Roxburgh P, Ang J, Banerjee S, et al. Implications of BRCA1 and BRCA2 mutations for the efficacy of paclitaxel monotherapy in advanced ovarian cancer. Eur J Cancer. 2013;49(6):1246–1253. doi: 10.1016/j.ejca.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 63.Burness ML, Obeid EI, Olopade OI. Triple negative breast cancer in BRCA1 mutation carriers with a complete radiologic response to neoadjuvant paclitaxel: a case report. Clin Breast Cancer. 2015;15(2):e155–e158. doi: 10.1016/j.clbc.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 64.King TA, Li W, Brogi E, Yee CJ, Gemignani ML, Olvera N, et al. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann Surg Oncol. 2007;14(9):2510–2518. doi: 10.1245/s10434-007-9372-1. [DOI] [PubMed] [Google Scholar]
- 65.Curtit E, Benhamo V, Gruel N, Popova T, Manie E, Cottu P, et al. First description of a sporadic breast cancer in a woman with BRCA1 germline mutation. Oncotarget. 2015;6(34):35616–35624. doi: 10.18632/oncotarget.5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Safra T, Borgato L, Nicoletto MO, Rolnitzky L, Pelles-Avraham S, Geva R, et al. BRCA mutation status and determinant of outcome in women with recurrent epithelial ovarian cancer treated with pegylated liposomal doxorubicin. Mol Cancer Ther. 2011;10(10):2000–2007. doi: 10.1158/1535-7163.MCT-11-0272. [DOI] [PubMed] [Google Scholar]
- 67.Safra T, Rogowski O, Muggia FM. The effect of germ-line BRCA mutations on response to chemotherapy and outcome of recurrent ovarian cancer. Int J Gynecol Cancer. 2014;24(3):488–495. doi: 10.1097/IGC.0000000000000086. [DOI] [PubMed] [Google Scholar]
- 68.Hamad L, Khoury T, Vona K, Nestico J, Opyrchal M, Salerno KE. A Case of Metaplastic Breast Cancer with Prolonged Response to Single Agent Liposomal Doxorubicin. Cureus. 2016;8(1):e454. doi: 10.7759/cureus.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kummar S, Oza AM, Fleming GF, Sullivan DM, Gandara DR, Naughton MJ, et al. Randomized Trial of Oral Cyclophosphamide and Veliparib in High-Grade Serous Ovarian, Primary Peritoneal, or Fallopian Tube Cancers, or BRCA-Mutant Ovarian Cancer. Clin Cancer Res. 2015;21(7):1574–1582. doi: 10.1158/1078-0432.CCR-14-2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia. 2014;16(1):43–72. doi: 10.1593/neo.131694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mo W, Liu Q, Lin CC, Dai H, Peng Y, Liang Y, et al. mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin Cancer Res. 2016;22(7):1699–1712. doi: 10.1158/1078-0432.CCR-15-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang D, Wang M, Jiang N, Zhang Y, Bian X, Wang X, et al. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget. 2016;7((11):13153–13166. doi: 10.18632/oncotarget.7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jóhannsson OT, Idvall I, Anderson C, Borg A, Barkardóttir RB, Egilsson V, et al. Tumour biological features of BRCA1-induced breast and ovarian cancer. Eur J Cancer. 1997;33(3):362–371. doi: 10.1016/S0959-8049(97)89007-7. [DOI] [PubMed] [Google Scholar]
- 76.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016 (in press). doi:10.18632/oncotarget.7277. [DOI] [PMC free article] [PubMed]
- 77.Vesprini D, Narod SA, Trachtenberg J, Crook J, Jalali F, Preiner J, et al. The therapeutic ratio is preserved for radiotherapy or cisplatin treatment in BRCA2-mutated prostate cancers. Can Urol Assoc J. 2011;5(2):E31–E35. doi: 10.5489/cuaj.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373(18):1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chedgy EC, Annala M, Beja K, Warner EW, Gleave ME, Chi KN, et al. Moving Toward Personalized Care: Liquid Biopsy Predicts Response to Cisplatin in an Unusual Case of BRCA2-Null Neuroendocrine Prostate Cancer. Clin Genitourin Cancer. 2016;14(2):e233–e236. doi: 10.1016/j.clgc.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 80.Luo G, Lu Y, Jin K, Cheng H, Guo M, Liu Z, et al. Pancreatic cancer: BRCA mutation and personalized treatment. Expert Rev Anticancer Ther. 2015;15(10):1223–1231. doi: 10.1586/14737140.2015.1086271. [DOI] [PubMed] [Google Scholar]
- 81.Moiseyenko VM, Volkov NM, Suspistin EN, Yanus GA, Iyevleva AG, Kuligina ES, et al. Evidence for predictive role of BRCA1 and bTUBIII in gastric cancer. Med Oncol. 2013;30(2):545. doi: 10.1007/s12032-013-0545-4. [DOI] [PubMed] [Google Scholar]
- 82.De Summa S, Pinto R, Sambiasi D, Petriella D, Paradiso V, Paradiso A, et al. BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol. 2013;24(Suppl 8):viii13–viii21. doi: 10.1093/annonc/mdt306. [DOI] [PubMed] [Google Scholar]
- 83.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–120. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 84.Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, 2nd, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–3576. doi: 10.1200/JCO.2009.27.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stefansson OA, Villanueva A, Vidal A, Martí L, Esteller M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics. 2012;7(11):1225–1229. doi: 10.4161/epi.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stecklein SR, Sharma P. Tumor homologous recombination deficiency assays: another step closer to clinical application? Breast Cancer Res. 2014;16(4):409. doi: 10.1186/s13058-014-0409-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16(6):475. doi: 10.1186/s13058-014-0475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014;16(3):211. doi: 10.1186/bcr3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Picciotto N, Cacheux W, Roth A, Chappuis PO, Labidi-Galy SI. Ovarian cancer: Status of homologous recombination pathway as a predictor of drug response. Crit Rev Oncol Hematol. 2016;101:50–59. doi: 10.1016/j.critrevonc.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 90.Lips EH, Laddach N, Savola SP, Vollebergh MA, Oonk AM, Imholz AL, et al. Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011;13(5):R107. doi: 10.1186/bcr3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Wesseling J, Vd Vijver MJ, et al. Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014;16(3):R47. doi: 10.1186/bcr3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2(4):366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72(21):5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 95.Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–3561. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Severson TM, Peeters J, Majewski I, Michaut M, Bosma A, Schouten PC, et al. BRCA1-like signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol Oncol. 2015;9(8):1528–1538. doi: 10.1016/j.molonc.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shah MM, Dobbin ZC, Nowsheen S, Wielgos M, Katre AA, Alvarez RD, et al. An ex vivo assay of XRT-induced Rad51 foci formation predicts response to PARP-inhibition in ovarian cancer. Gynecol Oncol. 2014;134(2):331–337. doi: 10.1016/j.ygyno.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schouten PC, Marmé F, Aulmann S, Sinn HP, van Essen HF, Ylstra B, et al. Breast cancers with a BRCA1-like DNA copy number profile recur less often than expected after high-dose alkylating chemotherapy. Clin Cancer Res. 2015;21(4):763–770. doi: 10.1158/1078-0432.CCR-14-1894. [DOI] [PubMed] [Google Scholar]
- 99.Telli ML, Timms KM, Reid JE, Hennessy B, Mills GB, Jensen KC, et al Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple Negative Breast Cancer. Clin Cancer Res. 2016 (in press). [DOI] [PMC free article] [PubMed]
- 100.Akashi-Tanaka S, Watanabe C, Takamaru T, Kuwayama T, Ikeda M, Ohyama H, et al. BRCAness predicts resistance to taxane-containing regimens in triple negative breast cancer during neoadjuvant chemotherapy. Clin Breast Cancer. 2015;15(1):80–85. doi: 10.1016/j.clbc.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 101.Hill SJ, Clark AP, Silver DP, Livingston DM. BRCA1 pathway function in basal-like breast cancer cells. Mol Cell Biol. 2014;34(20):3828–3842. doi: 10.1128/MCB.01646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 104.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581–2586. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69(16):6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–429. doi: 10.1002/path.4140. [DOI] [PubMed] [Google Scholar]
- 108.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 109.Martins FC, De S, Almendro V, Gönen M, Park SY, Blum JL, et al. Evolutionary pathways in BRCA1-associated breast tumors. Cancer Discov. 2012;2(6):503–511. doi: 10.1158/2159-8290.CD-11-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bardia A, Baselga J. Neoadjuvant therapy as a platform for drug development and approval in breast cancer. Clin Cancer Res. 2013;19(23):6360–6370. doi: 10.1158/1078-0432.CCR-13-0916. [DOI] [PubMed] [Google Scholar]
- 111.Kehoe S, Hook J, Nankivell M, Jayson GC, Kitchener H, Lopes T, et al. Primary chemotherapy vs. primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet. 2015;386(9990):249–257. doi: 10.1016/S0140-6736(14)62223-6. [DOI] [PubMed] [Google Scholar]
- 112.Sun CK, Zhang F, Xiang T, Chen Q, Pandita TK, Huang Y, et al. Phosphorylation of ribosomal protein S6 confers PARP inhibitor resistance in BRCA1-deficient cancers. Oncotarget. 2014;5(10):3375–3385. doi: 10.18632/oncotarget.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP, et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc Natl Acad Sci U S A. 2015;112(27):8409–8414. doi: 10.1073/pnas.1500223112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, et al. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015;29(5):489–494. doi: 10.1101/gad.256214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schouten PC, Vollebergh MA, Opdam M, Jonkers M, Loden M, Wesseling J, et al. High XIST and Low 53BP1 Expression Predict Poor Outcome after High-Dose Alkylating Chemotherapy in Patients with a BRCA1-like Breast Cancer. Mol Cancer Ther. 2016;15(1):190–198. doi: 10.1158/1535-7163.MCT-15-0470. [DOI] [PubMed] [Google Scholar]
- 116.Lake DE, Hudis CA. High-dose chemotherapy in breast cancer. Drugs. 2004;64(17):1851–1860. doi: 10.2165/00003495-200464170-00001. [DOI] [PubMed] [Google Scholar]
- 117.Pedrazzoli P, Ledermann JA, Lotz JP, Leyvraz S, Aglietta M, Rosti G, European Group for Blood and Marrow Transplantation (EBMT) Solid Tumors Working Party et al. High dose chemotherapy with autologous hematopoietic stem cell support for solid tumors other than breast cancer in adults. Ann Oncol. 2006;17(10):1479–1488. doi: 10.1093/annonc/mdl044. [DOI] [PubMed] [Google Scholar]
- 118.Banna GL, Simonelli M, Santoro A. High-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation for the treatment of solid tumors in adults: a critical review. Curr Stem Cell Res Ther. 2007;2(1):65–82. doi: 10.2174/157488807779316964. [DOI] [PubMed] [Google Scholar]
- 119.Vollebergh MA, Nederlof PM, Wessels LF, Schmidt MK, Joosse SA, van Beers E, et al. Predicting response to alkylating chemotherapy in breast cancer patients using array comparative genomic hybridization. Cancer Res. 2009;69(Suppl 1):abstract 6050. doi: 10.1158/0008-5472.SABCS-6050. [DOI] [Google Scholar]
- 120.Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Schmidt MK, van Beers EH, et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann Oncol. 2011;22:1561–1570. doi: 10.1093/annonc/mdq624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang F, Kushner YB, Langleben A, Foulkes WD. Medscape: Eleven years disease-free: role of chemotherapy in metastatic BRCA2-related breast cancer. Nat Rev Clin Oncol. 2009;6:488–492. doi: 10.1038/nrclinonc.2009.90. [DOI] [PubMed] [Google Scholar]
- 122.Steenbruggen TG, Linn SC, Rodenhuis S, Sonke GS. Ongoing Remission Nineteen Years after High-dose Chemotherapy for Oligometastatic Breast Cancer; What Can We Learn from this Patient? Cureus. 2015;7(12):e433. doi: 10.7759/cureus.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S, et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci U S A. 2011;108(43):17773–17778. doi: 10.1073/pnas.1110969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Drooger JC, Heemskerk-Gerritsen BA, Smallenbroek N, Epskamp C, Seynaeve CM, Jager A. Toxicity of (neo)adjuvant chemotherapy for BRCA1- and BRCA2-associated breast cancer. Breast Cancer Res Treat. 2016;156(3):557–566. doi: 10.1007/s10549-016-3777-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moon DH, Lee JM, Noonan AM, Annunziata CM, Minasian L, Houston N, et al. Deleterious BRCA1/2 mutation is an independent risk factor for carboplatin hypersensitivity reactions. Br J Cancer. 2013;109(4):1072–1078. doi: 10.1038/bjc.2013.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bernier J, Poortmans P. Clinical relevance of normal and tumour cell radiosensitivity in BRCA1/BRCA2 mutation carriers: a review. Breast. 2015;24(2):100–106. doi: 10.1016/j.breast.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 127.Drooger JC, Hooning MJ, Seynaeve CM, Baaijens MH, Obdeijn IM, Sleijfer S, et al. Diagnostic and therapeutic ionizing radiation and the risk of a first and second primary breast cancer, with special attention for BRCA1 and BRCA2 mutation carriers: a critical review of the literature. Cancer Treat Rev. 2015;41(2):187–196. doi: 10.1016/j.ctrv.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 128.Suspitsin EN, Yanus GA, Sokolenko AP, Yatsuk OS, Zaitseva OA, Bessonov AA, et al. Development of breast tumors in CHEK2, NBN/NBS1 and BLM mutation carriers does not commonly involve somatic inactivation of the wild-type allele. Med Oncol. 2014;31(2):828. doi: 10.1007/s12032-013-0828-9. [DOI] [PubMed] [Google Scholar]
- 129.Chrisanthar R, Knappskog S, Løkkevik E, Anker G, Østenstad B, Lundgren S, et al. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS One. 2008;3(8):e3062. doi: 10.1371/journal.pone.0003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kriege M, Jager A, Hollestelle A, Berns EM, Blom J, Meijer-van Gelder ME, et al. Sensitivity to systemic therapy for metastatic breast cancer in CHEK2 1100delC mutation carriers. J Cancer Res Clin Oncol. 2015;141(10):1879–1887. doi: 10.1007/s00432-015-1981-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sokolenko AP, Suspitsin EN, Kuligina ES, Bizin IV, Frishman D, Imyanitov EN. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015;369(2):274–288. doi: 10.1016/j.canlet.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 132.De Smedt L, Lemahieu J, Palmans S, Govaere O, Tousseyn T, Van Cutsem E, et al. Microsatellite instable vs stable colon carcinomas: analysis of tumour heterogeneity, inflammation and angiogenesis. Br J Cancer. 2015;113(3):500–509. doi: 10.1038/bjc.2015.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. 2015;16(7):30. doi: 10.1007/s11864-015-0348-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stadler ZK. Diagnosis and management of DNA mismatch repair-deficient colorectal cancer. Hematol Oncol Clin North Am. 2015;29(1):29–41. doi: 10.1016/j.hoc.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 135.Perucho M. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248-57. Cancer Res. 1999;59:249–56. [PubMed] [Google Scholar]
- 136.Devaud N, Gallinger S. Chemotherapy of MMR-deficient colorectal cancer. Fam Cancer. 2013;12(2):301–306. doi: 10.1007/s10689-013-9633-z. [DOI] [PubMed] [Google Scholar]
- 137.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Laghi L, Malesci A. Microsatellite instability and therapeutic consequences in colorectal cancer. Dig Dis. 2012;30(3):304–309. doi: 10.1159/000337003. [DOI] [PubMed] [Google Scholar]
- 140.Guillotin D, Martin SA. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp Cell Res. 2014;329(1):110–115. doi: 10.1016/j.yexcr.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 141.Samadder NJ, Neklason DW, Boucher KM, Byrne KR, Kanth P, Samowitz W, et al. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA. 2016;315(12):1266–1275. doi: 10.1001/jama.2016.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wells SA, Jr, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28(5):767–772. doi: 10.1200/JCO.2009.23.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639–3646. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fox E, Widemann BC, Chuk MK, Marcus L, Aikin A, Whitcomb PO, et al. Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res. 2013;19(15):4239–4248. doi: 10.1158/1078-0432.CCR-13-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 147.Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 148.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol. 2014;15(13):1513–1520. doi: 10.1016/S1470-2045(14)70489-9. [DOI] [PubMed] [Google Scholar]
- 149.Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366(23):2180–2188. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ally MS, Tang JY, Joseph T, Thompson B, Lindgren J, Raphael MA, et al. The use of vismodegib to shrink keratocystic odontogenic tumors in patients with basal cell nevus syndrome. JAMA Dermatol. 2014;150(5):542–545. doi: 10.1001/jamadermatol.2013.7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tang JY, Aszterbaum M, Athar M, Barsanti F, Cappola C, Estevez N, et al. Basal cell carcinoma chemoprevention with nonsteroidal anti-inflammatory drugs in genetically predisposed PTCH1+/- humans and mice. Cancer Prev Res (Phila) 2010;3(1):25–34. doi: 10.1158/1940-6207.CAPR-09-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen B, Trang V, Lee A, Williams NS, Wilson AN, Epstein EH Jr, et al. Posaconazole, a second-generation triazole antifungal drug, inhibits the Hedgehog signaling pathway and progression of basal cell carcinoma. Mol Cancer Ther. 2016 (in press). [DOI] [PMC free article] [PubMed]
- 154.Kim DJ, Kim J, Spaunhurst K, Montoya J, Khodosh R, Chandra K, et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J Clin Oncol. 2014;32(8):745–751. doi: 10.1200/JCO.2013.49.9525. [DOI] [PubMed] [Google Scholar]
- 155.Ally MS, Ransohoff K, Sarin K, Atwood SX, Rezaee M, Bailey-Healy I, et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016;152(4):452–456. doi: 10.1001/jamadermatol.2015.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Coombes RC. Drug testing in the patient: toward personalized cancer treatment. Sci Transl Med. 2015;7(284):284ps10. doi: 10.1126/scitranslmed.aab1214. [DOI] [PubMed] [Google Scholar]
- 157.Menden MP, Iorio F, Garnett M, McDermott U, Benes CH, Ballester PJ, et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One. 2013;8(4):e61318. doi: 10.1371/journal.pone.0061318. [DOI] [PMC free article] [PubMed] [Google Scholar]