

Mitochondrial translocator protein (TSPO) has been a somewhat mysterious player in searching for its true biological functions. TSPO is involved in mitochondrial cholesterol transport and steroids biosynthesis and predominantly expressed in steroid-synthesizing tissues including the central nervous system (CNS). However, recent findings using conditional knockout animal models contradict the long-held view that TSPO plays an essential role in steroidogenesis (Banati et al., 2014; Morohaku et al., 2014). In the CNS, TSPO is upregulated in microglia and reactive astrocytes during injury, serving as a suitable diagnostic biomarker for neuroinflammation. TSPO has been under investigation as a drug target for inflammatory conditions, and several TSPO ligands have been reported to confer neuroprotection in various CNS disorders, including multiple sclerosis (MS) (Da Pozzo et al., 2015).
MS is a common autoimmune demyelinating disease in humans, and experimental allergic (autoimmune) encephalomyelitis (EAE) induced by immunization with myelin antigens is the most thoroughly studied experimental model of MS. We have previously shown that the modulation of TSPO with its synthetic ligand, etifoxine, attenuates severity of EAE in association with a reduction of microglia activation and infiltration of peripheral immune cells (Daugherty et al., 2013). Evidence of increased production of the neurosteroid allopregnanolone has been implicated in etifoxine-induced neuroprotection and tissue repair. It is still not very clear what role TSPO-regulated production of neurosteroids plays in neuroprotection in EAE. In a recent study, Ravikumar et al. (2016) compared the effect of etifoxine to a specific high affinity TSPO agonist, XBD-173, in mouse and rat EAE models. In contrast to etifoxine, XBD-173 failed to provide neuroprotection in EAE, but increased the level of neurosteroids in healthy female rat brain that was not induced by etifoxine. Both compounds upregulated the level of pregnanolone and reduced expression of pro-inflammatory cytokines in LPS-stimulated cultured astrocytes, but only etifoxine was effective to protect cultured neurons from Ca2+ overload-mediated neurotoxicity. This study suggests that etifoxine-induced neuroprotection possibly works through an additional mechanism rather than TSPO alone. Indeed, direct modulation of GABAA receptors by etifoxine is well known and might contribute to neuroprotection. TSPO has been linked to oxidative stress and apoptotic cell death in response to injury (Rupprecht et al., 2010). In our study, improvement of EAE clinical phenotype was also associated with a decrease in TSPO expression in the inflamed spinal cord, suggesting an involvement of TSPO in EAE pathology. The cellular and molecular mechanisms underlying TSPO ligand-mediated neuroprotection in neuroinflammation are still not well defined, and the role of TSPO in glial cell activation in CNS diseases such as MS remains unclear.
Astrocytes play a critical role in CNS immune surveillance and tissue homeostasis. They are positioned at the interface between endothelial cells and the CNS parenchyma, forming direct contact to vascular endothelial cells, neuronal synapses and axonal nodes of Ranvier, performing neurovascular coupling and connecting each other through gap junctions. Astrocytes and astrocyte-derived factors are critical for the development and maintenance of the blood-brain barrier structure and function, limiting the passage of immune molecules and cells into the CNS. Astrocyte ion transporters and water channels regulate interstitial fluid ion composition, pH, water transport and volume of the extracellular space, constituting an essential component of the glymphatic system for clearing the CNS parenchyma from metabolic waste. Astrocytes become reactive in response to CNS injury. Reactive astrogliosis is a prominent feature of neuroinflammation and is found in demyelinated plaques from MS patients and EAE animals. Recent studies with conditional transgenic mice targeting specific astrocyte functions demonstrated that astrocytes play a central role in neuroinflammation, neurodegeneration and demyelination. Reactive astrocytes obtain a hypertrophic morphology and release pro-inflammatory cytokines and chemokines. An enhanced expression of intermediate filament, such as glial fibrillary acidic protein (GFAP) is essential for astrogliosis and glial scar formation (Sofroniew and Vinters, 2010). TSPO expression is also dramatically increased in reactive astrocytes during neuroinflammation.
To investigate the role of TSPO in astrocytes in EAE and MS, we developed a conditional TSPO knockout mouse by utilizing the Cre-LoxP system (Daugherty et al., 2016). We first generated a TSPO floxed mouse that was then crossed with a human GFAP (hGFAP)-driven Cre recombinase expressing mouse. The resultant offspring produced a neuroectoderm lineage specific TSPO knockout, where all cells derived from the neuroectoderm undergo a GFAP-expression stage and have conditionally knocked-out of TSPO during development, while the only actively Cre expressing cells are GFAP positive astrocytes and neural precursor cells. Thus, in the resultant mouse TSPO is deleted in astrocytes, oligodendrocytes and ependymal cells lining brain ventricles and the central canal of the spinal cord (Zhuo et al., 2001). Mature neurons do not express TSPO although there are several studies reporting the presence of TSPO in cultured primary cortical and cerebellar granule neurons (Rupprecht et al., 2010), possibly due to their immature stage. Hence, our hGFAP-driven TSPO knockout model primarily represent TSPO knockout in glial cells with the exception of microglia. The knockout animals did not display any developmental or behavioral abnormalities supporting an observation previously reported in global TSPO knockout mice (Banati et al., 2014). Induction of EAE revealed a dramatic amelioration of disease clinical symptoms in TSPO-hGFAP knockout mice compared to TSPO floxed control animals. Importantly, knockout of TSPO in astrocytes resulted in a significant reduction in the expression of GFAP and consequently reduced astrogliosis later in EAE (Figure 1). This correlation highlights the critical involvement of TSPO in reactive astrogliosis during neuroinflammtion.
Figure 1.
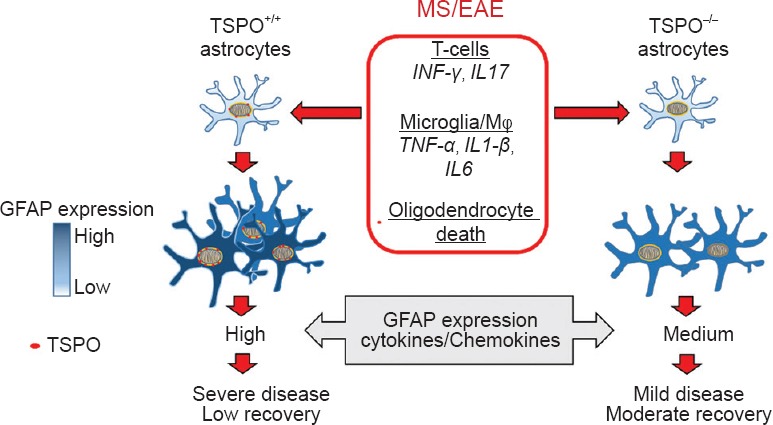
TSPO in astrocytes regulates astrogliosis and severity of autoimmune neuroinflammation.
In multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), cytokines released by infiltrating peripheral immune cells, such as T-cells and macrophages, and activated microglia, and also presence of dying oligodendrocytes cause astrocyte activation and increase in expression of glial fibrillary acidic protein (GFAP). Expression of mitochondrial translocator protein (TSPO) is also upregulated in reactive astrocytes during EAE and MS. Knockout of TSPO in astrocytes reduces reactive astrogliosis, and level of inflammatory mediators in the spinal cord tissue of EAE mice resulting in attenuation of disease clinical symptoms.
Chronic inflammation results in a pro-inflammatory state, and an increased level of pro-inflammatory cytokines brings negative consequences to tissue function and homeostasis. We found a decrease of TNFα and CXCL10 mRNA levels in the TSPO knockout group during chronic EAE phase. It is important and interesting to note that although TSPO knockout decreased clinical manifestation at the peak, during recovery and during chronic EAE phase, an earlier onset of disease was observed in TSPO-hGFAP group. This observation indicates a possible suppressive role of TSPO in astrocytes in the regulation of microglia/macrophages reactivity early in disease, suggesting a dual function of TSPO in reactive astrocytes that may be dependent on the degree of astrocytes activation in relation to EAE clinical phase.
To date, the diverse function and specific phenotypes of astrocytes in inflammation versus immune suppression have not been well characterized. Barres and colleagues recently reported the upregulation of distinct sets of genes in reactive astrocytes depending on the type of inducing injury (Zamanian et al., 2012). The authors provoked reactive astrogliosis by either peripherally administering the bacterial endotoxin LPS or inducing an ischemic stroke and used fluorescence activated cell sorting and gene chip analysis to show an approximately 50% difference in the gene expression profiles of isolated astroglia between the two conditions. These results provide a paradigm for understanding astrocytic activation and heterogeneity - with some forms being beneficial (e.g., in the case of ischemic stroke), and other types, deleterious (e.g., after LPS challenge). The conceptual framework developed by these studies is of high importance both from basic science and translational biology perspectives towards defining the role of astrocytes in CNS diseases, such as MS. Our study using TSPO-hGFAP knockout model revealed the positive correlation between reactive astrogliosis and severity of EAE clinical symptoms with TSPO being involved (Figure 1). Because multicellular system works in the orchestrated manner, further characterization is needed to clarify whether astrocytic TSPO deletion induces direct changes in the production of pro-inflammatory cytokines and chemokines or causes an indirect effect on the reduction of inflammatory infiltrating cells recruitment. Reactive astroglia in the inflamed CNS are active participants in CNS innate immune responses, but they may also play an important role in CNS myelin maintenance and remyelination. In fact, the most profound neuroprotective effect of TSPO-hGFAP knockout was evident after the peak and during recovery phase of EAE. Also, the degree of oligodendrocyte death and myelin loss needs to be carefully studied. Whether TSPO is involved in oligodendrocyte survival, proliferation and axonal remyelination needs to be examined.
Based on the observation that demyelinating lesions in MS are characterized by infiltrates of peripheral immune cells, therapeutic approaches have been developed to control inflammation and immune responses in the periphery. Such strategies are effective in reducing disease severity in relapsing-remitting MS patients, but are not curative as the neurodegeneration proceeds and neurological disability remains in these patients. Currently, no treatments exist for the progressive course of the disease that is characterized by the neurodegenerative process. Thus, both immunomodulation and neuroprotection are the goals in the development of therapeutics for MS. By using TSPO-hGFAP knockout mice, we determined the specific role of TSPO in astrocytes in neuroinflammation, providing insights into therapeutic approaches to the treatment of MS. These findings represent a novel conceptual advance for the understanding of how mitochondrial TSPO is critically involved in CNS function, opening up a new avenue by identifying a specific target for MS.
This work was in part supported by grants from the National Institutes of Health [R01NS061983, R01ES015988 and R01HD087566)] and Shriners Hospitals for Children to WD.
References
- 1.Banati RB, Middleton RJ, Chan R, Hatty CR, Kam WW, Quin C, Graeber MB, Parmar A, Zahra D, Callaghan P, Fok S, Howell NR, Gregoire M, Szabo A, Pham T, Davis E, Liu GJ. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun. 2014;5:5452. doi: 10.1038/ncomms6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Da Pozzo E, Giacomelli C, Barresi E, Costa B, Taliani S, Passetti Fda S, Martini C. Targeting the 18-kDa translocator protein: recent perspectives for neuroprotection. Biochem Soc Trans. 2015;43:559–565. doi: 10.1042/BST20150028. [DOI] [PubMed] [Google Scholar]
- 3.Daugherty DJ, Chechneva O, Mayrhofer F, Deng W. The hGFAP-driven conditional TSPO knockout is protective in a mouse model of multiple sclerosis. Sci Rep. 2016;6:22556. doi: 10.1038/srep22556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daugherty DJ, Selvaraj V, Chechneva OV, Liu XB, Pleasure DE, Deng W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med. 2013;5:891–903. doi: 10.1002/emmm.201202124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology. 2014;155:89–97. doi: 10.1210/en.2013-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravikumar B, Crawford D, Dellovade T, Savinainen A, Graham D, Liere P, Oudinet JP, Webb M, Hering H. Differential efficacy of the TSPO ligands etifoxine and XBD-173 in two rodent models of multiple sclerosis. Neuropharmacology. 2016;108:229–237. doi: 10.1016/j.neuropharm.2016.03.053. [DOI] [PubMed] [Google Scholar]
- 7.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 8.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]