

Keywords: nerve regeneration, brain injuries, permanent ischemic stroke, autophagy; apoptosis, LC3-II, cleaved caspase-3, infarct volume, neurological deficit score, western blotting, immunofluorescence, dynamic variations
Abstract
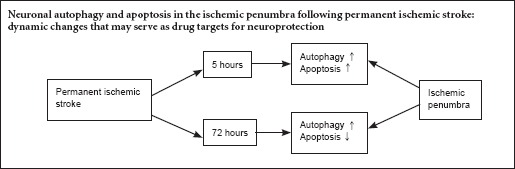
The temporal dynamics of neuronal autophagy and apoptosis in the ischemic penumbra following stroke remains unclear. Therefore, in this study, we investigated the dynamic changes in autophagy and apoptosis in the penumbra to provide insight into potential therapeutic targets for stroke. An adult Sprague-Dawley rat model of permanent ischemic stroke was prepared by middle cerebral artery occlusion. Neuronal autophagy and apoptosis in the penumbra post-ischemia were evaluated by western blot assay and immunofluorescence staining with antibodies against LC3-II and cleaved caspase-3, respectively. Levels of both LC3-II and cleaved caspase-3 in the penumbra gradually increased within 5 hours post-ischemia. Thereafter, levels of both proteins declined, especially LC3-II. The cerebral infarct volume increased slowly 1–4 hours after ischemia, but subsequently increased rapidly until 5 hours after ischemia. The severity of the neurological deficit was positively correlated with infarct volume. LC3-II and cleaved caspase-3 levels were high in the penumbra within 5 hours after ischemia, and after that, levels of these proteins decreased at different rates. LC3-II levels were reduced to a very low level, but cleaved caspase-3 levels remained high 72 hours after ischemia. These results indicate that there are temporal differences in the activation status of the autophagic and apoptotic pathways. This suggests that therapeutic targeting of these pathways should take into consideration their unique temporal dynamics.
Introduction
Cerebral ischemia (stroke) is one of the most serious neurological disorders, with high morbidity and mortality, and is a major cause of permanent disability and death worldwide (Klowka et al., 2010). A thorough understanding of the mechanisms underlying neuronal damage, neuronal survival and neural repair in ischemic stroke is urgently required for the development of effective treatment strategies.
Recent studies have shown that, in addition to necrosis and apoptosis, autophagy is another type of cell death in ischemic stroke (Jiang et al., 2014, 2005; Kroemer et al., 2005; Rami et al., 2008). Cells in the infarct core die within several minutes after stroke (Olsen et al., 1983), whereas in the surrounding region (ischemic penumbra) cell death proceeds slowly over a period of hours or even days post-insult, and the major types of cell death are apoptosis and autophagy (Balduini et al., 2012). Necrosis is considered an irreversible form of cell death. Therefore, the ischemic core is not a target that neurologists and clinicians should focus on. The relatively slow progression of cell death in the penumbra makes it an attractive therapeutic target.
Numerous studies have shown that autophagy and apoptosis are simultaneously activated in the penumbra following ischemic stroke (Lo, 2008; Rami et al., 2008). Autophagy is a cellular process that normally recycles damaged organelles, certain pathogens and long-lived cytoplasmic proteins by lysosomal degradation (Yoshimori, 2007). The role that autophagy plays in cerebral ischemia is controversial. Some studies have suggested that autophagy protects neurons from death under physiological conditions, and insufficient or excessive autophagy accelerates cell death (Komatsu et al, 2006). These observations suggest that autophagy may be a potential target for post-ischemic neuroprotection (Su et al., 2014; Shao and Liu, 2015). Indeed, it has been reported that treatment with inhibitors of autophagy significantly decreases brain damage, and activation of autophagy in cerebral ischemia results in a negative outcome (Degterev et al., 2005; Descloux et al., 2015). Thus, it is reasonable to hypothesize that the extent and time window of autophagy would affect the efficacy of these drugs in ischemic stroke. Apoptosis is also activated in neurons in the penumbra in ischemic stroke, but the correlation between autophagy and apoptosis, and the dynamics of these pathological processes in ischemic stroke are unclear. Studies have demonstrated that autophagy-positive neurons that are also apoptosis-positive are found in the superficial layers of the cortex 24 hours after ischemic stroke. However, at 48–72 hours, there is a switch from apoptosis to necrosis in the same area (Balduini et al., 2012), suggesting that there are time-dependent differences between autophagy, apoptosis and necrosis.
In the present study, we investigate the relationship between neuronal autophagy and apoptosis in the penumbra at 1, 2, 3, 4, 5, 6, 12, 24, 48 and 72 hours post-permanent ischemic stroke. We anticipate that our findings should help in the development of novel and effective treatments for stroke.
Materials and Methods
Animals
Male Sprague-Dawley rats (specific-pathogen-free, 7 weeks of age, 250–280 g) were from Beijing HFK Bio-Technology Co., Ltd (Animal license number: SCXK (Beijing) 2014-0004, Beijing, China). The animals were managed using animal welfare practices and were housed under a 12:12 hour light cycle at 22 ± 1 °C. Water and food were provided ad libitum. All animal experiments were approved by the animal committee of Kunming University of Science and Technology (Kunming, China). All surgeries were performed under anesthesia (10% chloral hydrate, 0.4 mL/100 g), and all efforts were made to minimize pain and distress to the experimental animals.
A total of 132 rats were randomly divided into 11 groups (n = 12 per group) as follows: 1-, 2-, 3-, 4-, 5-, 6-, 12-, 24-, 48- and 72-hour groups, and a sham (control) group. Six rats from each group were used to evaluate autophagy and apoptosis by western blot assay and immunofluorescence staining, and the remaining six rats were used for infarct volume and neurological deficit score assessments.
Preparation of the rat models of ischemic stroke
A rat model of ischemic stroke using permanent middle cerebral artery occlusion (MCAO) was prepared as described previously (Hoehn et al., 2005). Rats were first anesthetized with 10% chloral hydrate (i.p., 360 mg/kg). Then, the left common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA) were separated away from adjacent muscles and nerves. A 4-0 nylon monofilament with a polylysine tip (diameter 0.36 mm, Beijing Shadong Biotechnology Co., Ltd, Beijing, China) was inserted from a mini-incision on the ECA and inserted into the middle cerebral artery (MCA). The nylon monofilament was kept in place over the duration of the experiments. Rats in the control group were subjected to the same experiments, except that insertion of the nylon monofilament was not performed. Laser Doppler flowmetry was used to monitor regional cerebral blood flow during surgery to guarantee the robustness of the experimental stroke model.
Neurological deficit score
Neurological deficit score was assessed by an assistant blinded to experimental design, as previously described (Rogers et al., 1997) at 1, 2, 3, 4, 5, 6, 12, 24, 48 and 72 hours after MCAO: 0, no neurological symptoms; 1, failure to extend right paw completely; 2, the strength of the right fore-limb is noticeably reduced; 3, rotating and crawling towards the right side; 4, unable to walk spontaneously.
Measurement of cerebral infarct volume
1, 2, 3, 4, 5, 6, 12, 24, 48 and 72 hours after MCAO, the animals were sacrificed with 10% chloral hydrate (i.p., 400 mg/kg), the brain tissues were quickly removed and frozen for 20 minutes at −20°C, and then sliced into sections of 2 mm thickness. The slices were stained with 0.5% triphenyltetrazolium chloride (TTC) solution (Sh-haling Biotechnology Co., Ltd, Shanghai, China) for 15 minutes at 37°C. The infarct area was visibly pale. Infarct size (%) = A° / A’ × 100%, where A° represents the infarct volume, A‘ represents the volume of the homolateral hemisphere.
Western blot assay
The rats were sacrificed at 1, 2, 3, 4, 5, 6, 12, 24, 48 and 72 hours after MCAO. The penumbral cortex on the occluded side was dissected out and placed on ice. The tissue was homogenized in RIPA lysis buffer (Beijing BLKW Biotechnology Co., Ltd, Beijing, China), followed by centrifugation at 13,000 × g for 15 minutes at 4 °C. The resulting supernatants were retained. The proteins were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA), and then blocked with 10% nonfat milk for 1.5 hours at room temperature. After a wash step with TPBS (PBS containing 0.1% polysorbate 20), the membranes were labeled with rabbit antibodies against rat LC3B (Cell Signaling Technology, Danvers, MA, USA; 1:1,000), cleaved caspase-3 (Cell Signaling Technology; 1:1,500) or beta actin (Sigma, St. Louis, MO, USA, 1:10,000) overnight at 4°C. After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Beijing Tiangen Bio-Technology, Beijing, China) for 1 hour at room temperature. The reaction was visualized by enhanced chemiluminescence (ECL), and fluorescence densitometry was performed (Bio-Rad, Hercules, CA, USA). The protein signals were normalized to the beta actin signal. Results were expressed as the ratio of the fluorescence intensity of LC3-II or cleaved caspase-3/beta-actin.
Immunofluorescence staining
Six rats from each group at each time point were anesthetized with 10% chloral hydrate (i.p., 400 mg/kg) and transcardially perfused with physiological saline, followed by 4% paraformaldehyde (Beijing Tiangen Biotechnology Co. Ltd., Beijing, China) at 1, 2, 3, 4, 5, 6, 12, 24, 48 and 72 hours after permanent MCAO. The brains were then removed and dehydrated in 20% sucrose solution. Subsequently, the brain tissues were cut into sections (20 μm in thickness) using a freezing microtome (SLEE, Mainz, Germany). The sections were washed with PBS three times, 5 min each, and permeabilized with 0.2% Triton X-100 in PBS for 15 minutes at room temperature. After washing, the sections were blocked with 10% normal goat serum for 1 hour. The sections were then incubated with rabbit antibody against rat LC3-II (Cell Signaling Technology; 1:400) or cleaved caspase-3 (Cell Signaling Technology; 1:300), or with PBS (negative control) overnight at 4°C. After washing, the sections were labeled with Alexa Fluor 594-conjugated anti-rabbit IgG (Invitrogen, Shanghai, China; 1:800) for 2 hours in the dark. Then, the sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in PBS (1:1,000) for 5 minutes. After a wash step, the fluorescence signals were detected with a fluorescence microscope (Nikon Instruments Co., Ltd., Tokyo, Japan). The results were expressed as the percentage of positive cells. At high power (× 200), the number of positive cells and the total number of cells were counted in 10 randomly selected fields for each section, and five sections were counted from each tissue sample. All counting was manually performed by an investigator blinded to treatment. The results were expressed as the percentage of LC3-II or cleaved caspase-3-positive cells/total cells.
Statistical analysis
All data were presented as the mean ± SEM. Statistical analyses were carried out using SPSS 13.0 software (SPSS China, Shanghai, China). Statistical differences were evaluated by one-way analysis of variance followed by Dunnett’s test. P < 0.05 was considered statistically significant.
Results
Neurological deficit
There was an obvious neurological deficit in the rats 2–5 hours following permanent MCAO. Between 5 and 12 hours, the neurological deficit worsened dramatically, with the neurological deficit score reaching a maximum by 12 hours and remaining elevated until 72 hours after permanent MCAO (Figure 1). None of rats in the control group exhibited neurological deficits.
Figure 1.

Effect of ischemic stroke on the neurological deficit score.
Two hours following MCAO, the rats displayed a significant neurological deficit. The neurological deficit score was notably increased 5 hours after ischemia. **P < 0.01, vs. control, 2-h, 3-h and 4-h groups; ##P < 0.01, vs. 5-h and 6-h groups. Data are expressed as the mean ± SEM (n = 6; one-way analysis of variance and Dunnett’s test). MCAO: Middle cerebral artery occlusion; h: hour(s).
Cerebral infarct volume
Figure 2 shows that the infarct is detectable at 2 hours, and that it slowly expands within 4 hours after MCAO. The ischemic core then rapidly expands 5–12 hours post-MCAO to occupy the territory of the penumbra, which is nearly undetectable at 12 hours, with the infarct volume peaking at this time.
Figure 2.

Effects of ischemic stroke on cerebral infarct volume.
(A) The infarct volume was evaluated by TTC staining at 1, 2, 3, 4, 5, 6, and 12 h following permanent ischemic stroke. White and red regions indicate infarcted and normal tissue, respectively. (B) The infarcted area expands progressively over time. At 12 h, the infarct volume reaches a maximum and remains at this high level between 24 and 72 h after MCAO (the data for the last three time points is not shown). *P < 0.05, vs. control, 1-h, 2-h and 3-h groups; **P < 0.01, vs. control, 1-h, 2-h, 3-h and 4-h groups; ##P < 0.01, vs. 5-h and 12-h groups. Data are expressed as the mean ± SEM (n = 6; one-way analysis of variance and Dunnett’s test). MCAO: Middle cerebral artery occlusion; TTC: triphenyltetrazolium chloride; h: hour(s).
The autophagy process after stroke
Compared with the control group, LC3-II was prominently expressed in the ischemic penumbra 1 hour following permanent MCAO, and then levels gradually increased 2 to 5 hours after MCAO, peaking at 5 hours. Thereafter, levels rapidly declined between 6 and 72 hours after MCAO. Levels of this autophagy marker were low by 48 hours (Figure 3).
Figure 3.
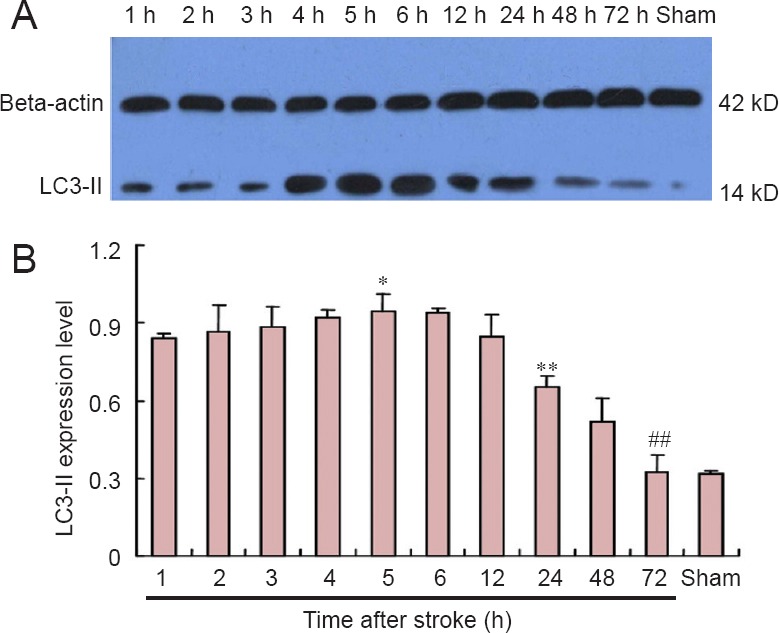
Effects of ischemic stroke on LC3-II protein levels in the rat ischemic penumbra (western blot assay).
(A) Representative western blot assay. (B) Densitometric quantification showing that LC3-II protein was significantly expressed 1 h following stroke compared with the sham control group. LC3-II protein levels were stable from 2 to 5 h after permanent MCAO. Levels rapidly declined starting at 6 h, and were similar to those in the control group at 72 h. *P < 0.05, vs. 1-h and 12-h groups; **P < 0.01, vs. 12-h and 48-h groups; ##P < 0.01, vs. 24-h and 48-h groups. Data are expressed as the mean ± SEM (n = 6; one-way analysis of variance and Dunnett’s test). h: Hour(s).
The apoptotic process after stroke
Similar to LC3-II, cleaved caspase-3 was robustly detected in the ischemic penumbra at 1 hour, and then gradually increased from 2 to 5 hours after permanent MCAO, reaching the highest level at 5 hours. Afterwards, levels declined from 6 to 72 hours after MCAO. Notably, the reduction in cleaved caspase-3 levels was milder than that for LC3-II. 72 hours after ischemic stroke, caspase-3 levels remained elevated, indicating that the apoptotic rate remained high 72 hours after stroke (Figure 4).
Figure 4.

Effects of ischemic stroke on cleaved caspase-3 levels in the ischemic penumbra (western blot assay).
(A) Representative western blot. (B) Densitometric quantification showing that cleaved caspase-3 protein levels gradually increased between 1 and 5 h, reaching the highest level at 5 h after MCAO. The levels declined from 6 to 72 h after MCAO, but were persistently high compared with the sham control, indicating that the apoptotic rate remained high 72 h after stroke. *P < 0.05, vs. 1-h, 2-h, 3-h and 4-h groups; **P < 0.01, vs. 5 h and 6 h groups; ##P < 0.01, vs. 12 h and sham control groups. Data are expressed as the mean ± SEM (n = 6; one-way analysis of variance and Dunnett’s test). h: Hour(s).
Dynamic changes in autophagy and apoptosis after stroke
Because ischemia activated apoptosis and autophagy in the ischemic penumbra (Zhang et al., 2013; Yu et al., 2015), we investigated the dynamic changes in autophagy and apoptosis after stroke. The levels of cleaved caspase-3 were higher than that of LC3-II at each time point. The levels of LC3-II were about half that of cleaved caspase-3, with a slight change 1 day after stroke. After that, the LC3-II/cleaved caspase-3 ratio rapidly declined (Figure 5). At 72 hours after stroke, more neurons were apoptotic, without autophagy.
Figure 5.

Dynamic changes in autophagy and apoptosis after stroke.
Within 5 h of permanent ischemic stroke, the LC3-II/cleaved caspase-3 ratio displayed a slight decreasing trend. From 12 to 72 h following ischemia, the ratio declined, showing that autophagy was reduced during this stage. At 72 h after stroke, more neurons underwent apoptosis, but not autophagy. *P < 0.05, vs. 1-h, 2-h and 12-h groups; **P < 0.01, vs. 24-h and 72-h groups. Data are expressed as the mean ± SEM (n = 6; one-way analysis of variance and Dunnett’s test). h: Hour(s).
Immunofluorescence staining
Similar to the western blot assay results, both the percentage of LC3-II and cleaved caspase-3-positive cells in the ischemic penumbra gradually increased within 5 hours following permanent ischemic stroke, and reached a maximum at 5 hours (Figure 6). After that, the ratio of LC3-II-positive cells declined by 12 hours after stroke. In comparison, the percentage of cleaved caspase-3-positive cells showed only a slight reduction.
Figure 6.

Effects of ischemic stroke on the immunoreactivities for LC3-II and cleaved caspase-3 in the ischemic penumbra (immunofluorescence staining, × 200).
(A, B) Representative fluorescence microscopic images of immunofluorescence staining for LC3-II and cleaved caspase-3. Abundant positive cells (including LC3-II and cleaved caspase-3-positive cells) were found in the penumbra 1–12 h following ischemic stroke; however, a few positive cells were detected in the corresponding area in the control group (C, D). Both the percentage of LC3-II-positive cells (C, **P < 0.01, vs. control, 3-h and 6-h groups; ##P < 0.01, vs. 6-h and 72-h groups) and cleaved caspase-3-positive cells (D, *P < 0.05, vs. 1-h group; **P < 0.01, vs. control group; ##P < 0.01, vs. 6-h and 72-h groups) reached the highest level 5 h after permanent ischemic stroke. Thereafter, the ratio of LC3-II-positive cells gradually declined, and remained at a very low level 72 h after MCAO. Comparatively, the percentage of cleaved caspase-3-positive cells only slightly decreased, and remained high between 12 and 72 h after MCAO. The arrow indicates LC3-II or cleaved caspase-3-positive cells (red-colored). Data are expressed as the mean ± SEM (n = 6; one-way analysis of variance and Dunnett’s test). MCAO: Middle cerebral artery occlusion; h: hour(s).
Discussion
Both researchers and clinicians have given much attention to the ischemic penumbra in the study of cerebral injury caused by ischemic stroke, because cellular energy metabolism is preserved in the penumbra, and there is the potential for recovery when the blood supply is restored. Notably, cell damage in this area appears reversible (Lo, 2008). Comparatively, the type of cell death in the ischemic core is necrosis, which is impossible to survive after ischemia. Autophagy and apoptosis in the ischemic penumbra are activated after stroke (Pamenter et al., 2012), showing that they may play important roles in the pathophysiology of ischemic stroke.
Autophagy is a cell survival process that allows cells to recycle essential nutrients and cytoplasmic constituents, and which helps retain ATP (Glick et al., 2010). In ischemic stroke, autophagy is up-regulated for cell survival by stressors such as ischemia, nutrient deprivation and reactive oxygen species (ROS). The role that autophagy plays in stroke has been studied in animal models, but the results have been inconclusive and even contradictory. In particular, it is unclear whether increasing or inhibiting autophagy is protective after ischemic stroke (Su et al., 2014; Zheng et al., 2014). Differences in study design and the use of inhibitors and activators, as well as differences in animal models may contribute to the discrepancies. The appropriate animal model is critical for reliable outcome. In the use of animal models, the severity of the reduction in cerebral blood flow (CBF), as well as its restoration, should be considered to better mimic the pathophysiological mechanisms of human stroke (Hossmann et al., 2012). The severity of the reduction in CBF can be easily controlled, but the dynamics of CBF restoration are frequently not addressed. Rapid complete reperfusion is commonly used in animal models by most researchers. However, in clinical experience, less than 5% of all patients achieve complete reperfusion with timely tPA and thrombectomy treatments, and most patients seldom achieve recanalization (Kahle et al., 2012). Therefore, permanent ischemic stroke models more effectively mimic human stroke damage. Hence, an animal model of permanent focal ischemia was used in the present study.
Apoptosis is a self-killing function, and cells undergoing apoptosis are disposed of in a controlled way that minimizes damage and disruption to neighboring cells (Chen et al., 2014; Ji et al., 2014). Under certain conditions, autophagy is a stress adaptation that suppresses apoptosis (Maiuri et al., 2007). In ischemic stroke, both autophagy and apoptosis are simultaneously activated. To clarify the functional relationship between these processes, we investigated the dynamics of autophagy and apoptosis following ischemia.
We found that both autophagy and apoptosis were increased in the penumbra 1 hour after ischemic stroke, compared with the control group. Then, levels of LC3-II (an indicator of autophagy; Kabeya et al., 2004) and cleaved caspase-3 (a marker of apoptosis; Moskowitz et al., 2010) steadily increased 2–5 hours following ischemic stroke, reaching a maximum at 5 hours. During this stage, infarct volume and the severity of the neurological deficit increased slowly, suggesting that activation of autophagy and apoptosis might have a neuroprotective function, by promoting neuronal survival and inhibiting the infarct core from expanding into the penumbra. However, our results are not consistent with the findings of previous studies (Hossmann et al., 2008), which showed that the penumbra disappears and the ischemic core reaches its maximum volume between 3 and 6 hours after MCAO.
At 12–72 hours post MCAO, LC3-II levels in the penumbra rapidly decreased, suggesting that autophagy was reduced 48 to 72 hours after ischemic stroke. The LC3-II/cleaved caspase-3 ratio was reduced to a very low level 24 hours after MCAO, showing that the shift from autophagy to apoptosis occurs 24 hours following ischemic stroke. After that, more neurons underwent apoptosis, but not autophagy. TTC staining showed that the infarct volume reached a maximum at 12 hours, indicating that the infarction progressively enlarged 5–12 hours post-ischemic stroke. Interestingly, LC3-II and cleaved caspase-3 levels were still detectable in this area 12 to 72 hours after stroke, suggesting that there are significant numbers of autophagic and apoptotic neurons that can potentially be rescued. It should be mentioned that although TTC staining is an indicator of tissue dehydrogenase and mitochondrial dysfunction, and is commonly used to differentiate viable tissues from nonviable tissues (Benedek et al., 2006), it cannot be used to observe the penumbra. Thus, although infarct volume had reached a maximum, autophagy and apoptosis might continue to play important roles, and LC3-II and cleaved caspase-3 might still be detectable in the ischemic core.
In the present study, we found differences in temporal dynamics between autophagy and apoptosis following ischemia. Studies show that both autophagy and apoptosis are involved in neuroprotection and recovery after stroke (Pamenter et al., 2012). Suppressing apoptosis after stroke with caspase inhibitors, overexpressing anti-apoptotic proteins and exercise rescues injured neurons in the penumbra and promotes functional recovery (Zhao et al., 2004; Singh et al., 2010; Zhang et al., 2013). Previous studies have focused on apoptosis during the early phase after stroke, but there is a lack of studies on temporal changes after the early phase. Although suppressing apoptosis is regarded as a neuroprotective strategy, autophagy has dual effects in ischemia. Activation of autophagy by rapamycin before ischemia and in ischemic preconditioning protects neurons from death (Sheng et al., 2010; Yan et al., 2011). However, increased autophagy exerts a different effect after ischemia (Ravikumar et al., 2010). Some reports showed that activation of autophagy protects neurons from death (Carloni et al., 2008; Buckley et al., 2014), whereas others indicate that it increases neuronal death or abolishes the neuroprotective effects of ischemic postconditioning (Koike et al., 2008; Gao et al. 2012). Autophagy also significantly reduces the lesion volume in the acute phase (4 hours after the beginning of ischemia) (Puyal et al., 2009). Interestingly, in an in vitro ischemia model, inhibiting autophagy 24 hours prior to reperfusion markedly increases neuronal death, while it significantly protects neurons from death 48–72 hours after reperfusion (Shi et al., 2012). These observations indicate that autophagy plays different roles based on the extent of autophagy and the time window in ischemia. Thus, a comprehensive comparison of the effects of different time points of intervention on functional recovery is necessary before the autophagy pathway can be used as a therapeutic target for stroke.
In this study, we found that both LC3-II and cleaved caspase-3 levels in the penumbra gradually increased from 1 to 5 hours post permanent ischemic stroke. Thereafter, autophagy and apoptosis declined, but the reduction in autophagy was more marked from 24 to 72 hours after MCAO, suggesting that within 12 hours after ischemic stroke, treatment should target both autophagy and apoptosis. Cleaved caspase-3 continued to be found at high levels, suggesting that treatment should focus on apoptosis 12 hours after the insult.
There are several issues that need to be addressed by future studies, including (1) the interrelationships and the dynamics of autophagy, apoptosis and necrosis in ischemic stroke; (2) the mechanisms of neuroprotection of autophagy and apoptosis; (3) the use of different animal models, including permanent ischemic stroke, gradually reversed transient ischemia and rapid complete reperfusion, to clarify the crosstalk between autophagy, apoptosis and necrosis; and (4) the development of effective methods for improving neuronal rescue in the penumbra.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 81460351; the Doctoral Foundation of Kunming University of Science and Technology of China, No. KKSY201360112; the Scientific Research Foundation of Yunnan Provincial Department of Education of China, No. 2014Y070.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Copyedited by Patel B, Norman C, Wang J, Li JY, Li CH, Song LP, Zhao M
References
- 1.Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med. 2012;25:30–34. doi: 10.3109/14767058.2012.663176. [DOI] [PubMed] [Google Scholar]
- 2.Benedek A, Móricz K, Jurányi Z, Gigler G, Lévay G, Hársing LG, Jr, Mátyus P, Szénási G, Albert M. Use of TTC staining for the evaluation of tissue injury in the early phases of reperfusion after focal cerebral ischemia in rats. Brain Res. 2006;1116:159–165. doi: 10.1016/j.brainres.2006.07.123. [DOI] [PubMed] [Google Scholar]
- 3.Buckley KM, Hess DL, Sazonova IY, Periyasamy-Thandavan S, Barrett JR, Kirks R, Grace H, Kondrikova G, Johnson MH, Hess DC, Schoenlein PV, Hoda MN, Hill WD. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6:8. doi: 10.1186/2040-7378-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Chen WQ, Sun YY, Liu KY, Sun XJ. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res. 2014;9:1210–1216. doi: 10.4103/1673-5374.135329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 7.Descloux C, Ginet V, Clarke PG, Puyal J, Truttmann AC. Neuronal death after perinatal cerebral hypoxia-ischemia: Focus on autophagy-mediated cell death. Int J Neurosci. 2011;45:75–85. doi: 10.1016/j.ijdevneu.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- 11.Hossmann KA. Cerebral ischemia: models, methods and outcomes. Neuropharmacology. 2008;55:257–270. doi: 10.1016/j.neuropharm.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Hossmann KA. The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J Cereb Blood Flow Metab. 2012;32:1310–1316. doi: 10.1038/jcbfm.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji XY, Zhang LN, Liu R, Liu YZ, Song JF, Dong H, Jia YF, Zhou ZG. Potential targets for protecting against hippocampal cell apoptosis after transient cerebral ischemia-reperfusion injury in aged rats. Neural Regen Res. 2014;9:1122–1128. doi: 10.4103/1673-5374.135314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Z, Chen CH, Chen YY, Han JY, Riley J, Zhou CM. Autophagic effect of programmed cell death 5 (PDCD5) after focal cerebral ischemic reperfusion injury in rats. Neurosci Lett. 2014;566:298–303. doi: 10.1016/j.neulet.2014.02.066. [DOI] [PubMed] [Google Scholar]
- 15.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3 GABARAP and GATE16 localize to autophagosomal membrane depending on form-ii formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 16.Kahle MP, Bix GJ. Successfully Climbing the “STAIRs”: surmounting failed translation of experimental ischemic stroke treatments. Stroke Res Treat 2012. 2012:374098. doi: 10.1155/2012/374098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;44:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 19.Klowka SP, Wintermark M, Engelhorn T, Fiebach JB. Acute stroke magnetic resonance imaging: current status and future perspective. Neuroradiology. 2010;52:189–201. doi: 10.1007/s00234-009-0637-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele B, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight A, Piacentini M, Nagata S, Melino G. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12:1528–1534. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 21.Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- 22.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 23.Moskowitz MA, Lo EH, ladecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–98. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen TS, Larsen B, Herning M, Skriver EB, Lassen NA. Blood flow and vascular reactivity in collaterally perfused brain tissue. Evidence of an ischemic penumbra in patients with acutestroke. Stroke. 14:332–341. doi: 10.1161/01.str.14.3.332. [DOI] [PubMed] [Google Scholar]
- 25.Pamenter ME, Perkins GA, McGinness AK, Gu XQ, Ellisman MH, Haddad GG. Autophagy and appoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS One. 2012;7:e51469. doi: 10.1371/journal.pone.0051469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 27.Rami A, Kogel D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: two sides of the same coin? Autophagy. 2008;4:422–426. doi: 10.4161/auto.5778. [DOI] [PubMed] [Google Scholar]
- 28.Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 30.Rogers D, Campbell CA, Stretton JL, Mackay KB. Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke. 1997;28:2060–2066. doi: 10.1161/01.str.28.10.2060. [DOI] [PubMed] [Google Scholar]
- 31.Shao ZQ, Liu ZJ. Neuroinflammation and neuronal autophagic death were suppressed via Rosiglitazone treatment: New evidence on neuroprotection in a rat model of global cerebral ischemia. J Neurol Sci. 2015;349:65–71. doi: 10.1016/j.jns.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 32.Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- 33.Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 2012;18:250–260. doi: 10.1111/j.1755-5949.2012.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh MH, Brooke SM, Zemlyak I, Sapolsky RM. Evidence for caspase effects onrelease of cytochrome c and aif in a model of ischemia in cortical neurons. Neurosci Lett. 2010;469:179–183. doi: 10.1016/j.neulet.2009.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su J, Zhang T, Wang K, Zhu T, Li X. Autophagy activation contributes to the neuroprotction of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem Res. 2014;39:2068–2077. doi: 10.1007/s11064-014-1396-x. [DOI] [PubMed] [Google Scholar]
- 36.Yan W, Zhang H, Bai X, Lu Y, Dong H, Xiong L. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 2011;1402:109–121. doi: 10.1016/j.brainres.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimori T. Autophagy: paying Charon’s toll. Cell. 2007;128:833–836. doi: 10.1016/j.cell.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 38.Yu J, Bao C, Dong Y, Liu X. Activation of autophagy in rat brain cells following focal cerebral ischemia reperfusion through enhanced expression of Atg1/pULK and LC3. Mol Med Rep. 2015;12:3339–3344. doi: 10.3892/mmr.2015.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang P, Zhang Y, Zhang J, Wu Y, Jia J, Wu J, Hu Y. Early exercise protects against cerebral ischemic injury through inhibiting neuron apoptosis in cortex in rats. Int J Mol Sci. 2013;14:6074–6089. doi: 10.3390/ijms14036074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H, Yenari MA, Cheng D, Barreto-Chang OL, Sapolsky RM, Steinberg GK. Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J Cereb Blood Flow Metab. 2004;24:681–692. doi: 10.1097/01.WCB.0000127161.89708.A5. [DOI] [PubMed] [Google Scholar]
- 41.Zheng Y, Hou J, Liu J, Yao M, Li L, Zhang B, Zhu H, Wang Z. Inhibition of autophagy contributes to melatonin-mediated neuroprotection against transient focal cerebral ischemia in rats. J Pharmacol Sci. 2014;124:354–364. doi: 10.1254/jphs.13220fp. [DOI] [PubMed] [Google Scholar]