

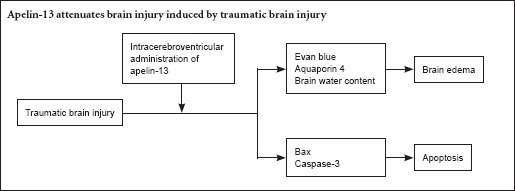
Keywords: nerve regeneration, apoptosis, apelin-13, traumatic brain injury, brain edema, blood-brain barrier, brain water content, aquaporin-4, caspase-3, neural regeneration
Abstract
The adipocytokine, apelin-13, is an abundantly expressed peptide in the nervous system. Apelin-13 protects the brain against ischemia/reperfusion injury and attenuates traumatic brain injury by suppressing autophagy. However, secondary apelin-13 effects on traumatic brain injury-induced neural cell death and blood-brain barrier integrity are still not clear. Here, we found that apelin-13 significantly decreases cerebral water content, mitigates blood-brain barrier destruction, reduces aquaporin-4 expression, diminishes caspase-3 and Bax expression in the cerebral cortex and hippocampus, and reduces apoptosis. These results show that apelin-13 attenuates secondary injury after traumatic brain injury and exerts a neuroprotective effect.
Introduction
Survival of traumatic brain injury (TBI) patients from the primary injury (e.g., development of brain edema or delayed axonal or neuronal degeneration) is mainly dependent on secondary insults (Plesnila et al., 2007). Brain edema is still one of the main causes of death in TBI (Unterberg et al., 2004; Khan et al., 2009). Edema is harmful because it changes metabolite concentration and alters cellular physiology, biochemistry, and function (Unterberg et al., 2004; Khan et al., 2009). Previous research, including our own, has shown that both brain edema and apoptosis participate in neuronal cell death and functional loss after TBI (Liu et al., 2008; Luo et al., 2010; Bao et al., 2012; Zhang et al., 2014).
The peptide, apelin, is an endogenous ligand for the apelin receptor (also known as the APJ receptor). Apelin was first isolated from bovine stomach (Tatemoto et al., 1998). The APJ receptor was first discovered by O‘Dowd et al. (1993), and is analogous to the angiotensin-I receptor. There are three biologically active forms of apelin, which are composed of 13, 17, or 36 amino acids. All the peptides are derived from a 77-amino acid prepropeptide precursor (O‘Carroll et al., 2013), although apelin-13 has much greater biological potency than apelin-36 (Tatemoto et al., 1998). Accordingly, apelin-13 is regarded as an important neuroprotective reagent in the nervous system (Khaksari et al., 2012; Jiao et al., 2013).
The central nervous system distribution pattern of apelinergic neurons suggests diverse roles of apelin, for example in circadian rhythms, controlling feeding behavior, body fluid homeostasis, and pituitary hormone release (Hosoya et al., 2000). Additionally, apelin and the APJ receptor are broadly expressed in neurons and oligodendrocytes, but less so in astrocytes (Choe et al., 2000). To the best of our knowledge, no experimental studies have addressed the role of apelin-13 on cerebral water content and neural cell death due to apoptosis after TBI. Thus, based on the above-mentioned findings, we investigated the hypothesis that apelin-13 may ameliorate brain edema and reduce cellular death in mice after TBI.
Materials and Methods
Ethics statement
The animal studies were approved by Institutional Animal Care and Use Committee of Xuzhou Medical College, China, and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Precautions were taken to minimize suffering and the number of animals used in each experiment.
Experimental animals
In total, 108 adult specific-pathogen-free male CD1 mice weighing 20–25 g and aged 8 weeks were purchased from the Experimental Animal Center of Xuzhou Medical College of China (lisence No. SCXK (Su) 2010-0003). Mice were equally and randomly assigned to sham, TBI (TBI only), and apelin-13 (TBI + apelin-13) groups.
Establishment of the TBI model
The procedure for performing TBI in mice has been previously described (Bao et al., 2012). Briefly, mice were anesthetized with 0.4 mg/g chloral hydrate (4%). Then to induce TBI, a 40 g weight was dropped from 20 cm onto a 2-mm-diameter footplate on the left part of the brain, using a controlled depth of 1.0 mm. Mice in the sham group received craniotomy only, without impact injury. Injured cortical (including the impact site and surrounding area; 2 × 2 × 2 mm3 tissue block) and hippocampal (including the impact site and surrounding area of the entire ipsilateral hippocampus) tissue was dissected for use in several assays.
Apelin-13 administration
Apelin-13 (sc-351718; Santa Cruz Biotechnology, Santa Cruz, Northern CA, USA) was dissolved in normal saline. Apelin-13 was administered ipsilaterally intracerebroventricularly (1 mg/mL; 5 μL) immediately after TBI in the apelin-13 group. The sham group did not receive apelin-13 or normal saline, while the TBI group received vehicle (saline, 5 μL) by intracerebroventricular injection.
Measurement of cerebral water content
Adult male CD1 mice were anesthetized with 4% chloral hydrate (0.4 mg/g), and sacrificed at 24 or 48 hours after apelin-13 injection. The brain was removed and placed in a glass petri-dish. Cerebellar tissue was removed, and the left and right hemispheres divided along the anatomic midline. The wet weight of each hemisphere was weighed. The tissue was completely dried in an oven at 100°C for 5 days, and the dry weight of each hemisphere measured. Brain water content (% water) was calculated using the Elliott formula: brain water content = (wet weight – dry weight) / wet weight × 100% (Xi et al., 2006; Bierbach et al., 2008; Bao et al., 2012).
Evaluation of blood-brain barrier permeability
Evans Blue (4 mL/kg, 2% solution; Sigma-Aldrich, St. Louis, MO, USA) in saline was injected via the tail vein (6 mice in each group). The dye was allowed to circulate for 2 hours. Mice were then treated with 50 mL ice-cold phosphate buffered saline (PBS) by transcardiac perfusion. The brain was separated into right and left hemispheres and stored at –80°C. Samples were homogenized in 1.1 mL PBS, sonicated, and centrifuged for 30 minutes at 15,000 r/min, 4°C. Supernatants were collected in aliquots, and trichloroacetic acid (50%) added to 500 μL aliquots. The mixture was incubated at 4°C overnight, and then centrifuged for 30 minutes at 15,000 r/min, 4°C. Evans blue concentration was determined at 620 nm using a spectrophotometer (Thermo Spectronic Genesys 10 UV, Thermo Fisher Scientific Inc., Waltham, MA, USA). Data were quantified from a standard curve. The results are presented as: Evans Blue stain (μg)/tissue (g).
Terminal deoxyribonucleotidyl transferase (TDT)- mediated dUTP-digoxigenin nick end labeling (TUNEL) assay
Brain cell apoptosis was detected using a TUNEL assay kit, specifically, In Situ Cell Death Detection Kit (Roche, Basle, Switzerland; cat. no. 116848179). First, one of eight 12-μm serial sections was collected from the injured cortex of each animal (Bao et al., 2012). Next, paraffin sections were deparaffinized in xylene and rehydrated through a graded ethanol series. After washing with PBS, sections were digested in 20 mg/mL protease at 37°C for 15 minutes, and then washed again with PBS. Prepared sections were incubated in TUNEL reaction mixture at 37°C for 1 hour. After washing in PBS, sections were labeled with converter-POD for 30 minutes at 37°C, and stained with 3,3′-diaminobenzidine. TUNEL-positive cells were counted under a light microscope (BX53; Olympus, Tokyo, Japan) (Bao et al., 2012).
Western blot assay
Injured cortical and hippocampal samples were homogenized using western blot analysis buffer that included 10 mM Tris-HCl (pH 7.4), Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 0.1% sodium dodecyl sulphate, 1 mM phenylmethyl sulfonylfluoride, 5 mM ethylenediamine tetraacetic acid, 0.28 kU/L aprotinin, 1 mM benzamidine, 50 mg/L leupeptin, and 7 mg/L pepstain A (all chemicals were from Sigma-Aldrich). Homogenates were centrifuged for 10 minutes at 12,000 r/min, 4°C. Supernatants were stored at –80°C for later use. Protein concentration was determined using a bicinchoninic acid kit (Pierce, Appleton, WI, USA). Protein (30 mg) extracted from each sample underwent sodium dodecyl sulfate polyacrylamide gel electrophoresis using 10% electropheresis gels. Proteins were then transferred to polyvinylidene fluoride membranes using a semidry electrotransferring unit (Bio-Rad, Hercules, CA, USA). Membranes were incubated in antibodies against aquaporin 4 (AQP4) (1:600; rabbit polyclonal IgG; Santa Cruz Biotechnology), pro-caspase-3/caspase-3 (1:1,000; rabbit monoclonal IgG; Cell Signaling Technology, Danfoss, MA, USA), BCL2-associated X protein (Bax) (1:1,000; rabbit monoclonal IgG; Cell Signaling Technology), and β-actin (1:2,000; rabbit monoclonal IgG; Bioworld Technology, Minneapolis, MN, USA), in Tris-buffered saline containing 0.1% Tween-20 and 5% nonfat dry milk overnight at 4°C. After overnight incubation with primary antibodies, membranes were washed and incubated for 2 hours in horseradish peroxidase conjugated goat anti-rabbit IgG (1:2,000; Bioworld Technology) in Tris-buffered saline containing 0.1% Tween-20. PageRulerTM Prestained Protein Ladder (5 μL; Thermo Fisher Scientific Inc.) was used according to the manufacturer’s instructions. Immunoreactivity was tested by enhanced chemiluminescence autoradiography (Amersham Life Science, Chicago, IL, USA), according to the manufacturer’s instructions. After stripping, membranes were reprobed using β-actin. The signal intensity for binding of each primary antibody was quantitatively analyzed from optical density values using Quantity One (Bio-Rad). Results were normalized to β-actin.
Statistical analysis
Data are presented as the mean ± SEM, and were analyzed using SPSS.13.0 software (SPSS, Chicago, IL, USA). The TUNEL-positive cell count was analyzed using the rank-sum test. Brain water content, Evans Blue stain, and western blot data were analyzed by one-way analysis of variance with Dunnett’s t-test. Values of P < 0.05 were considered statistically significant.
Results
Apelin-13 reduced brain edema in TBI mice
Compared with the sham group, brain water content was higher (P < 0.01) and Evans blue leakage reduced (P < 0.01) in the TBI group. Additionally, apelin-13 significantly reduced brain water content in the ipsilateral hemisphere (P < 0.01) and increased Evans blue leakage in the injured hemisphere at 24 and 48 hours after TBI (P < 0.01; Figures 1, 2).
Figure 1.
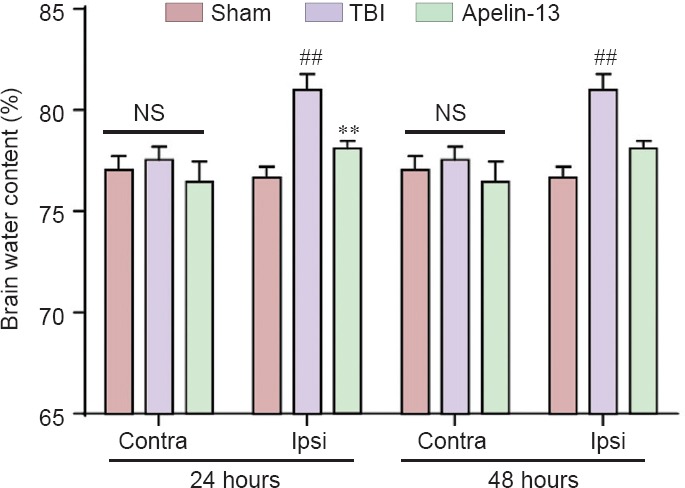
Apelin-13 ameliorates TBI-induced brain edema.
Brain water content in the ipsilateral (injured) and contralateral hemisphere were measured at 24 and 48 hours after TBI. Data are expressed as the mean ± SEM, and were analyzed by one-way analysis of variance followed by Dunnett’s t-test (6 mice in each group). **P < 0.01, vs. TBI group; ##P < 0.01, vs. sham group. Contra: Contralateral; Ipsi: ipsilateral; NS: not significant; TBI: traumatic brain injury.
Figure 2.
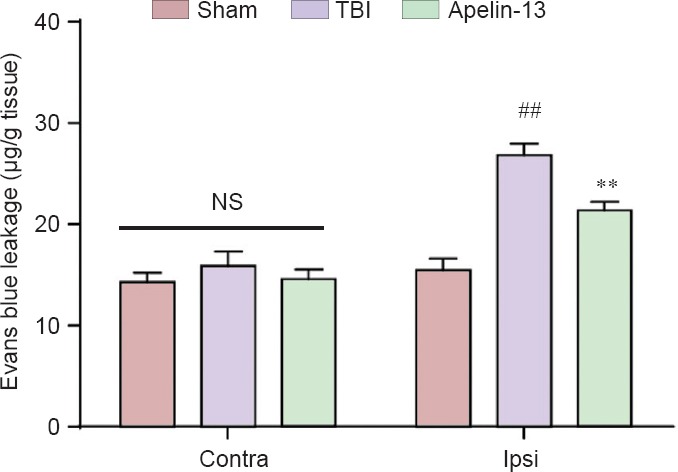
Reduction of blood-brain barrier permeability is affected by apelin-13 at 48 hours after TBI.
Data are expressed as the mean ± SEM, and were analyzed by one-way analysis of variance followed by Dunnett’s t-test (6 mice in each group). **P < 0.01, vs. TBI group; ##P < 0.01, vs. sham group. Contra: Contralateral; Ipsi: ipsilateral; NS: not significant; TBI: traumatic brain injury.
To further investigate the mechanism of apelin-13 attenuating brain edema after TBI, AQP4 protein levels were also measured. Apelin-13 significantly diminished AQP4 protein expression in the injured cortex and hippocampus (P < 0.05 or P < 0.01; Figures 3).
Figure 3.

Apelin-13 acutely reduces AQP4 protein expression in the cortex and hippocampus at 24 and 48 hours post-TBI.
(A, C) Representative western blots of AQP4 protein the cortex (A) and hippocampus (C) were detected by western blot assay. (B, D) Quantitative analysis of AQP4 protein expression in the cortex (B) and hippocampus (D). Optical density of the respective protein bands were analyzed by Quantity One (Bio-Rad) and normalized to β-actin. Data are expressed as the mean ± SEM (6 mice in each group). Statistical comparisons were performed by analysis of variance followed by Dunnett’s t-test. *P < 0.05, **P < 0.01, vs. TBI group; ##P < 0.01, vs. sham group. AQP4: Aquaporin-4; TBI: traumatic brain injury; h: hours.
Apelin-13 decreased TBI-induced cell apoptosis
Compared with the TBI group, apelin-13 led to a striking reduction of TUNEL-positive cells 48 hours after TBI (P < 0.01; Figure 4).
Figure 4.

Apelin-13 reduces TBI-induced cell apoptosis in the brain 48 hours after TBI.
Arrows show normal cells and arrowheads point to TUNEL-positive cells in the sham (A), TBI (B), and apelin-13 (C) groups. Scale bars: 50 μm. (D) Quantification of TUNEL-positive cells in the injured cortex (in 400-fold fields). Data are presented as the mean ± SEM (6 mice in each group), and were analyzed by the rank-sum test. **P < 0.01, vs. TBI group; ##P < 0.01, vs. sham group. TBI: Traumatic brain injury; TUNEL: terminal deoxyribonucleotidyl transferase (TDT)-mediated dUTP-digoxigenin nick end labeling.
To further investigate the role of apelin-13 TBI-induced apoptosis, caspase-3 and Bax protein levels were examined in the injured cortex and hippocampus. Lower caspase-3 protein levels were found in the apelin-13 group compared with the TBI group (P < 0.01; Figure 5). Additionally, Bax is a key protein in regulation of apoptosis (Plesnila et al., 2007), and its protein levels were significantly lower in the apelin-13 group compared with the TBI group at 24 and 48 hours post-TBI (Figure 6).
Figure 5.

Inhibition of TBI-induced caspase-3 expression in the cortex and hippocampus by apelin-13 at 24 and 48 hours post TBI.
(A, C) Representative western blots of caspase-3 protein in the cortex (A) and hippocampus (C) were detected by western blot assay. (B, D) Quantitative analysis of caspase-3 protein expresssion in the cortex (B) and hippocampus (D). Data are expressed as the mean ± SEM (6 mice in each group). Statistical comparisons were performed by analysis of variance followed by Dunnett’s t-test. **P < 0.01, vs. TBI group; ##P < 0.01, vs. sham group. TBI: Traumatic brain injury; h: hours.
Figure 6.

Inhibition of TBI-induced Bax expression in the cortex and hippocampus by apelin-13 at 24 and 48 hours post TBI.
(A, C) Representative western blots of Bax protein in the cortex (A) and hippocampus (C) were detected by western blot assay. (B, D) Quantitative analysis of Bax protein expresssion in the cortex (B) and hippocampus (D). Data are expressed as the mean ± SEM (6 mice in each group). Statistical comparisons were perfomred by analysis of variance followed by Dunnett’s t-test. **P < 0.01, vs. TBI group; ##P < 0.01, vs. sham group. TBI: Traumatic brain injury.
Discussion
We have investigated the effect of apelin-13 on secondary events, specifically, brain edema and apoptosis after TBI. To our knowledge, these are the first observations showing that apelin-13 restores the blood-brain barrier, attenuates brain edema, regulates AQP4 expression, and suppresses apoptosis in an in vivo TBI model.
Brain edema and subsequent increased intracranial pressure are severe complications that increase mortality and long-term disability in patients (Rangel-Castillo et al., 2008; Eghwrudjakpor and Allison, 2010). The potential therapeutic benefits of apelin-13 for ischemia/reperfusion injury, and its neuroprotective effects, have previously been recognized (Aboutaleb et al., 2013; Xin et al., 2015; Yan et al., 2015). However, whether apelin-13 can alleviate brain edema after TBI remains unclear.
Blood-brain barrier disruption occurs at a high incidence after TBI and contributes to pathological changes such as brain edema, inflammation, and loss of neuronal viability or function after brain injury (Khan et al., 2009). Here, we found that apelin-13 prevents blood-brain barrier disruption following TBI and alleviates brain edema.
Previous studies have shown that brain water content reaches a peak at 24 to 48 hours after TBI. Interestingly, intracranial pressure also peaks at this time (Bao et al., 2012; Zhang et al., 2014). Accordingly, we determined water content in the injured hemisphere at 24 and 48 hours after TBI. Our results show that brain water content is markedly decreased in the ipsilateral hemisphere of the apelin-13 group 24 and 48 hours after TBI, compared with the TBI group, which is consistent with the Evans blue leakage outcome.
Aquaporins are involved in intracranial edema and permit selective bidirectional water movement (Verkman and Mitra, 2000; Marmarou, 2007; He and Lu, 2015). The predominant brain isoform, AQP4, is found within the perivascular end feet of astrocytes and can regulate fast water transport (Nicchia et al., 2004). We found that TBI up-regulates AQP4 protein expression at 24 and 48 hours post-TBI. These results are consistent with our profile of TBI-induced brain water content, which indicates that AQP4 up-regulation may exacerbate brain edema. In contrast, AQP4 protein levels are down-regulated in the apelin-13 group at 24 and 48 hours post-TBI. Despite the controversial role of AQP4 in TBI-induced brain edema (Ke et al., 2001; Saadoun et al., 2003; Sun et al., 2003), our results suggest that an apelin-13 effect on brain edema may be involved in AQP4 regulation.
Further, we counted TUNEL-positive cells to confirm apoptosis after TBI affected by apelin-13. Additionally, we also measured apoptosis-associated protein levels of caspase-3 and Bax. Our previous research has shown that apelin-13 results in a marked increase of Bcl-2 protein levels (Bao et al., 2014). However, Bcl-2 was also the first identified oncogene protein involved in blocking apoptotic cell death. Caspase-3 would be cleaved in both of the two pathways (Zhang et al., 2005). We found that cell apoptosis was inhibited, and caspase-3 and Bax down-regulated by apelin-13 after TBI. These results show that apoptosis may be involved in the protective function of apelin-13.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81302612; the Natural Science Foundation of Jiangsu Province of China, No. 12KJB310018; a grant from the Student Innovation Training Program of China, No. 201410313023Z.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Copyedited by James R, de Souza M, Yu J, Qiu Y, Li CH, Song LP, Zhao M
References
- 1.Aboutaleb N, Kalalianmoghaddam H, Eftekhari S, Shahbazi A, Abbaspour H, Khaksari M. Apelin-13 inhibits apoptosis of cortical neurons following brain ischemic reperfusion injury in a transient model of focal cerebral ischemia. Int J Pept Res Ther. 2013;20:127–132. [Google Scholar]
- 2.Bao HJ, Wang T, Zhang MY, Liu R, Dai DK, Wang YQ, Wang L, Zhang L, Gao YZ, Qin ZH, Chen XP, Tao LY. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem Res. 2012;37:2856–2867. doi: 10.1007/s11064-012-0880-4. [DOI] [PubMed] [Google Scholar]
- 3.Bao Hu, Zhang L, Han WC, Dai DK. Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem Res. 2014;40:89–97. doi: 10.1007/s11064-014-1469-x. [DOI] [PubMed] [Google Scholar]
- 4.Bierbach B, Meier M, Kasper-König W, Heimann A, Alessandri B, Horstick G, Oelert H, Kempski O. Emboli formation rather than inflammatory mediators are responsible for increased cerebral water content after conventional and assisted beating-heart myocardial revascularization in a porcine model. Stroke. 2008;39:213–219. doi: 10.1161/STROKEAHA.107.496620. [DOI] [PubMed] [Google Scholar]
- 5.Choe W, Albright A, Sulcove J, Jaffer S, Hesselgesser J, Lavi E, Crino P, Kolson DL. Functional expression of the seven-transmembrane HIV-1 co-receptor APJ in neural cells. J Neurovirol. 2000;6(Suppl 1):S61–69. [PubMed] [Google Scholar]
- 6.Eghwrudjakpor PO, Allison AB. Decompressive craniectomy following brain injury: factors important to patient outcome. Libyan J Med. 2010 doi: 10.4176/091104. doi: 104176/091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He ZP, Lu H. Aquaporin-4 gene silencing protects injured neurons after early cerebral infarction. Neural Regen Res. 2015;10:1082–1087. doi: 10.4103/1673-5374.160099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, Kitada C, Honda S, Kurokawa T, Onda H, Nishimura O, Fujino M. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem. 275:21061–21067. doi: 10.1074/jbc.M908417199. [DOI] [PubMed] [Google Scholar]
- 9.Jiao HU, Zhang ZH, Ma Q, Fu WE, Liu Z. Mechanism underlying the inhibitory effect of Apelin-13 on glucose deprivation-induced autophagy in rat cardiomyocytes. Exp Ther Med. 2013;5:797–802. doi: 10.3892/etm.2013.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ke C, Poon WS, Ng HK, Pang JC, Chan Y. Heterogeneous responses of aquaporin-4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett. 2001;301:21–24. doi: 10.1016/s0304-3940(01)01589-0. [DOI] [PubMed] [Google Scholar]
- 11.Khaksari M, Aboutaleb N, Nasirinezhad F, Vakili A, Madjd Z. Apelin-13 protects the brain against ischemic reperfusion injury and cerebral edema in a transient model of focal cerebral ischemia. J Mol Neurosci. 2012;48:201–208. doi: 10.1007/s12031-012-9808-3. [DOI] [PubMed] [Google Scholar]
- 12.Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, Singh I. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, Cao QQ, Ni H, Qin ZH, Tao LY. Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res. 2010;88:2847–2858. doi: 10.1002/jnr.22453. [DOI] [PubMed] [Google Scholar]
- 15.Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:E1. doi: 10.3171/foc.2007.22.5.2. [DOI] [PubMed] [Google Scholar]
- 16.Nicchia GP, Nico B, Camassa LM, Mola MG, Loh N, Dermietzel R, Spray DC, Svelto M, Frigeri A. The role of aquaporin-4 in the blood-brain barrier development and integrity: studies in animal and cell culture models. Neuroscience. 2004;129:935–944. doi: 10.1016/j.neuroscience.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 17.O‘Carroll AM, Lolait SJ, Harris LE, Pope GR. The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol. 2013;219:R13–35. doi: 10.1530/JOE-13-0227. [DOI] [PubMed] [Google Scholar]
- 18.O‘Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–360. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- 19.Plesnila N, von Baumgarten L, Retiounskaia M, Engel D, Ardeshiri A, Zimmermann R, Hoffmann F, Landshamer S, Wagner E, Culmsee C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007;14:1529–1541. doi: 10.1038/sj.cdd.4402159. [DOI] [PubMed] [Google Scholar]
- 20.Rangel-Castillo L, Gopinath S, Robertson CS. Management of intracranial hypertension. Neurol Clin. 2008;26:521–541, x. doi: 10.1016/j.ncl.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saadoun S, Papadopoulos MC, Krishna S. Water transport becomes uncoupled from K+ siphoning in brain contusion, bacterial meningitis, and brain tumours: immunohistochemical case review. J Clin Pathol. 2003;56:972–975. doi: 10.1136/jcp.56.12.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun MC, Honey CR, Berk C, Wong NL, Tsui JK. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg. 2003;98:565–569. doi: 10.3171/jns.2003.98.3.0565. [DOI] [PubMed] [Google Scholar]
- 23.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 24.Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 25.Verkman AS, Mitra AK. Structure and function of aquaporin water channels. Am J Physiol Renal Physiol. 2000;278:F13–28. doi: 10.1152/ajprenal.2000.278.1.F13. [DOI] [PubMed] [Google Scholar]
- 26.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. The Lancet Neurology. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 27.Xin Q, Cheng B, Pan Y, Liu H, yang C, Chen J, Bai B. Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides. 2015;63:55–62. doi: 10.1016/j.peptides.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 28.Yan XG, Cheng BH, Wang X, Ding LC, Liu HQ, Chen J, Bai B. Lateral intracerebroventricular injection of Apelin-13 inhibits apoptosis after cerebral ischemia/reperfusion injury. Neural Regen Res. 2015;10:766–771. doi: 10.4103/1673-5374.157243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M, Shan H, Chang P, Wang T, Dong W, Chen X, Tao L. Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PLoS One. 2014;9:e87241. doi: 10.1371/journal.pone.0087241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RS. Bench-to-bedside review: apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care. 2005;9:66–75. doi: 10.1186/cc2950. [DOI] [PMC free article] [PubMed] [Google Scholar]