

Abstract
Introduction
The approval of blinatumomab signals the long awaited arrival of immunotherapy for acute lymphoblastic leukemia (ALL). Previous options for relapsed or refractory disease were restricted to combination cytotoxic chemotherapy with limited efficacy and significant toxicity. Through an innovative mechanism of action, blinatumomab stimulates a polyclonal antitumor T cell response, yielding unprecedented single agent efficacy in the relapsed/refractory setting. Success comes at the cost of immunological toxicities rarely encountered with previous therapies and challenging administration logistics requiring clinical expertise.
Areas covered
All published clinical and preclinical studies using blinatumomab were reviewed in addition to all registered ongoing clinical trials and data published in abstract form. The search was limited to the English language. The pharmacology, clinical efficacy, toxicity profile, and logistical considerations for drug administration are discussed.
Expert Opinion
Blinatumomab is an exciting addition to the treatment armamentarium for relapsed/refractory ALL, yet several questions remain regarding optimal implementation into the current treatment paradigm. A unique toxicity profile should be weighed against promising benefits in a poor prognosis population. Other emerging therapies, such as chimeric antigen receptor-modified T cells and inotuzumab ozogamicin, with different side effect profiles and administration schedules, may prove to be more beneficial for specific patient populations.
1. Introduction
Management of lymphoid malignancies was revolutionized with the introduction of the first immunotherapeutic agent, rituximab, in the late 1990's. Since that time, numerous immunotherapeutic agents have been approved but have largely bypassed acute lymphoblastic leukemia (ALL), with most leukemic clones lacking expression of a consistent drug target. Moreover, patients with relapsed or refractory (RR) ALL, particularly those who relapse following allogeneic stem cell transplantation (alloSCT), have dismal outcomes [1-3]. Blinatumomab (B lineage-specific anti-tumor mouse monoclonal antibody) (drug summary box) is the first therapeutically useful bispecific single-chain antibody construct [4] and was recently approved by the United States Food and Drug Administration (FDA) for treatment of RR-ALL. This unique molecule harnesses the power of antitumor immune surveillance to display impressive single agent efficacy in patients with RR B cell malignancies, including B cell ALL and non-Hodgkin lymphoma (NHL). This review will provide a detailed overview of the immunopharmacology of blinatumomab and summarize currently available clinical data for its use in both ALL and NHL. Information on toxicity management and the role of blinatumomab in the current treatment paradigm of lymphoid malignancies is also discussed.
2. Pharmacology, Pharmacokinetics, and Immunologic Response to Blinatumomab
Blinatumomab is a non-immunogenic, single chain (sc) protein consisting of an anti-CD19 variable fragment (Fv) and an anti-CD3 Fv, joined by a flexible 5 amino acid glycine-serine linker. Blinatumomab is produced in Chinese hamster ovary cells containing a cDNA vector that constitutively expresses the blinatumomab protein [4,5]. The construct has a molecular mass of only 55 kilodaltons (kDa); approximately one-third the size of most therapeutic monoclonal antibodies (mAbs) [4]. Small size theoretically improves tumor penetration and permits close target-effector cell interaction [5]. Blinatumomab is the first of a class of antibodies known as bi-specific T-cell engagers (BiTEs). Many previously approved cytotoxic mAbs rely on antibody-dependent cellular cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC) for cell kill and involve immune cells expressing Fc receptors, such as natural killer cells and macrophages. Such mAbs are incapable of directly activating T cells, the most potent anti-tumor cells, as evidenced by the variety of T cell immune escape mechanisms employed by various cancers. BiTEs have the direct capacity to link cancer cells and T cells, inducing a polyclonal T-cell anti-tumor response independent of the processing and presentation of any specific single peptide antigen to the effector cell [6].
Conventionally, T cell activation depends on the formation of the immunologic synapse, involving the T cell receptor (TCR), CD3 signaling complex, costimulatory molecules (CSMs), and cell adhesion molecules (CAMs) along with the major histocompatibility complex (MHC) on the antigen presenting cell [4,6]. Tumor cell MHC expression is often downregulated as an escape mechanism from T cell surveillance. BiTEs bypass this complex cascade, limiting the T cell activation signal to a single protein interaction between CD19 and CD3 but otherwise forming an identical immunologic synapse [7]. Blinatumomab, unlike prior BiTEs, does not require CSMs (such as interleukin (IL)-2 or anti-CD28 mAb) to induce T cell response due to close proximity of the target and effector cells brought together by the drug [8,9]. Even in the presence of blinatumomab concentrations exceeding the median effective dose (ED50) by several thousand-fold, T cell activation only occurs in the dual presence of effector (CD3+) and target (CD19+) cells together in an immune synapse [5,6]. Cytotoxicity is CD19-dependent, as treatment with an anti-CD22 mAb does not impair efficacy and no cytotoxicity is observed against CD19-negative B cell lineages or in the presence of a competing anti-CD19 mAb [5].
Blinatumomab functions to induce cancer cell death via 4 distinct phases [10,11]. In the first phase, drug preferentially binds the B cell given the greater affinity for CD19 [equilibrium dissociation constant (KD)of 10-9 M] compared with CD3 (KD 10-7 M), resulting in B cells displaying a matrix of membrane-bound blinatumomab molecules awaiting CD3 binding [11]. The second phase is closing of the gap between the T and B cells to elicit T cell-mediated cytotoxicity. Third is the synergistic phase, involving the formation of the immunologic synapse. In the fourth phase, the T cell eliminates the target cell through perforin and granzyme (P/G) release (Figure 1). Perforins form cell membrane channels to permit granzyme entry into the target cell, which activate caspases, resulting in target cell apoptosis. Cytotoxicity is inhibited by calcium chelation, the perforin inhibitor concanamycin, and caspase 3, 8, and 9 inhibitors, highlighting the importance of the calcium-dependent P/G system in triggering apoptosis [5,8,12]. Fas (CD95) upregulation follows blinatumomab treatment; however, an agonistic anti-CD95 mAb failed to induce apoptosis, suggesting that Fas activation alone is insufficient for apoptosis [8,12]. Following apoptosis, the T cell dissociates from the blinatumomab-bound B cell, allowing the T cell to find another target cell.
Figure 1.
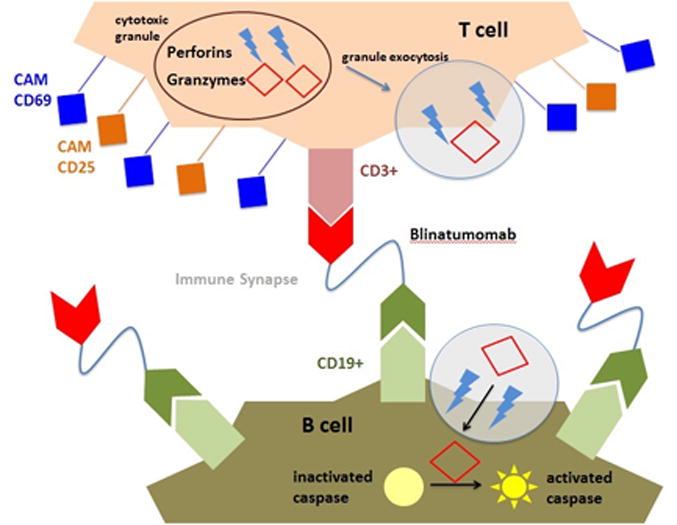
Unlike previous BiTEs, blinatumomab requires low effector to target (E:T) ratios for activity [10,11]. At 10:1, 50% target cell lysis occurred after 20 minutes and plateaued near 100% at 100 minutes. Lysis was similar at 1:5, but substantially lessened at 1:10 [11]. When B cells greatly outnumber T cells, serial killing occurs, with a single T cell eliminating up to 6 B cells in 9 hours, such that the limiting factor for apoptosis is duration of drug exposure. At higher E:T ratios, T cells have immediate access to B cells, limiting cell-to-cell movement and serial killing. Since cytotoxicity is partially dependent on T cell numbers, depletion with chemotherapy or radiotherapy may impair drug efficacy. In chronic lymphocytic leukemia (CLL) samples, a 50% response was observed even in samples from patients receiving prior chemotherapy (including fludarabine) or radiotherapy between 2 days and 2 or more years prior to blinatumomab. IL-2 costimulation augmented response in some non-responders. Lack of response in remaining samples was presumed to be related to low E:T ratios (less than 1:100 in 2/3 samples) or very recent chemotherapy in the remaining sample [8].
Linear, dose-dependent apoptosis occurs following blinatumomab. Minimally, 10-2 or 10-1 ng/mL is necessary for cell lysis with a plateau noted between 1 and 10 ng/mL [10,13]. The cytotoxic effects of blinatumomab and rituximab were compared in vitro against CD19+CD20+ lymphoma cell lines. At 24 hours, apoptosis rates at E:T ratios of 1:1 and 1:10 were 50% and 25%, respectively. On the contrary, rituximab ADCC was 25% at 1:1 and undetectable at 1:10 [13]. Rituximab's cytotoxic efficacy peaked at 100 mcg/mL with a resultant 60% target cell lysis, and a calculated ED50 of 10 mcg/ml, while blinatumomab exhibited 100,000-fold greater potency, with an ED50 of 30 pg/ml and 100% tumor cell lysis at 1 ng/ml. Such difference in potency in spite of similar KD values suggests a difference in cytotoxic efficiency between T cell activation with blinatumomab and ADCC following rituximab [10]. Interestingly, the combination of rituximab and blinatumomab has an additive apoptotic effect, likely due to non-overlapping recruitment of T cells from blinatumomab in addition to Fc receptor bearing effector cells by rituximab. Upon addition of serum (to enable CDC), additive apoptotic effect persisted, suggesting possible further benefit from this mechanism [13]. Combination therapy taking advantage of these additive non-cross resistant mechanisms may be a potential area for future research.
CD19 is not a new drug target. This 5 kDa transmembrane glycoprotein is a CSM expressed on malignant and non-malignant B cells throughout maturation until terminal plasma cell differentiation where expression is lost [14]. As determined by flow cytometry in 42 [15] and 451 [16] cases with B cell ALL, the incidence of CD19 expression ranges from 93 to 100%, respectively, making it an attractive therapeutic target. As with CD20 drug targeting, CD19 inhibition causes non-malignant B cell aplasia. This is expected to be transient following blinatumomab, given the lack of CD19 expression on stem cells [17].
Following blinatumomab, 50% B cell depletion occurs at 1 hour, 90% depletion is noted at 4 hours, and by day 2 the B-cell count drops to less than 1 cell/mcL. B cell aplasia continues throughout the cycle without recovery during the 2-week treatment-free intervals [9,10]. CD8+ memory T cells are responsible for this rapid cytotoxicity, with naïve CD8+ and CD4+ T cell contribution observed at 16 hours [10]. T cell redistribution occurs following blinatumomab initiation, where T cell counts drop within 8 hours, then recover to 50% of baseline by day 3, with full recovery around day 9. T cell count continues to climb to an average of 2 times the baseline value, peaking around day 17 following initiation [9]. Redistribution occurs with each cycle, with more rapid recovery observed in later cycles. Dose-dependent cytokine release from activated T cells rapidly follows blinatumomab initiation, the profile of which shows significant interpatient variability. From greatest to least, IL-10, IL-6, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and IL-2 are elevated, yet half of patients show minimal elevations [9,18]. Peak TNF-α, IL-2, and IL-4 levels are observed between 8-12 hours following initiation, with peaks of IFN-γ, IL-6, and IL-10 within 24 hours [19]. Despite continuous drug infusion, cytokines declined by day 2 and were hardly detectable at infusion completion with no elevation observed during subsequent cycles. Cytokine release does not correlate with response but may be responsible for numerous toxicities during blinatumomab infusion. Pre-incubation with dexamethasone for up to 24 hours reduces cytokine release (most potently TNF-α, IL-4, and IL-2) without inhibiting CAM upregulation [19]. Concentrations of 1 to 3 × 10-7 M (approximately 8 mg of prednisolone or 1 mg of dexamethasone used clinically) produced maximal cytokine reduction. Importantly, no change in cytotoxicity was observed in the presence of dexamethasone. Still, use of higher doses of dexamethasone may have direct lympholytic effects on T cells, with the potential to impair blinatumomab cytotoxicity.
The half-life of blinatumomab was determined to be only 2.11 hours, possibly due to its small molecular mass and lack of Fc domain [20]. Administration via continuous intravenous infusion (CIVI) is required for maintaining therapeutic levels while also granting tight control of drug exposure, allowing quick drug elimination by infusion discontinuation in the case of toxicity [9,17]. Steady state is obtained within 1 day of initiation and maintained throughout each cycle, suggesting that cell binding does not significantly alter serum concentration [9]. The effective concentration 90% (EC90) is 470 pg/mL for ALL, which can be obtained using either flat or body surface area (BSA) based dosing in adults [21]. Pharmacokinetics were linear when 5 to 90 mcg/m2/day (d), about 9-162 mcg/d in adults, was administered via CIVI [20]. Although drug metabolism and elimination routes have not been fully characterized, only minimal urinary excretion has been reported [21]. Extent of penetration into the CNS is unknown. The small size of the molecule and its unique neurological side effect profile support the potential entry of blinatumomab into the cerebrospinal fluid. However, given the presence of limited effector cells in the CNS, significant activity is not expected, suggesting potential benefit of administrating prophylactic intrathecal chemotherapy together with blinatumomab.
3.1 Clinical Data: Non-Hodgkin's Lymphoma
The first clinical trial of blinatumomab was in RR-NHL patients, the majority of whom had follicular lymphoma (FL) or mantle cell lymphoma (MCL) [22]. Patients had received a median of 3 prior chemotherapy regimens and 87% received prior rituximab. In this phase 1 dose-escalation trial, patients received blinatumomab 0.5 mcg/m2/d to 60 mcg/m2/d via CIVI for 4-8 weeks. The overall response rate (ORR) was 29% (11/38), including 10% with complete remission (CR). A dose response relationship was identified. All responders received at least 15 mcg/m2/d (ORR 21% for 15-30 mcg/m2/d, with rates similar between 15 and 30 mcg/m2/d) and all patients receiving 60 mcg/m2/d (7/7) had either a partial or complete response. Of 11 patients with bone marrow disease, 6 had complete and 3 had partial clearing of disease from the marrow. In addition, hepatic and splenic involvement responded to doses of 15 mcg/m2/d or greater.
The phase 1 trial was then expanded to include more patients with diffuse large B-cell lymphoma (DLBCL) and MCL. In a 2011 abstract including 76 patients, an ORR of 69% (37% CR) was reported across all NHL subtypes with a median duration of response of 404 days [23,24]. By subtype, the ORRs for patients with FL, MCL, and DLBCL were 80% (12/15), 71% (5/7), and 55% (6/11), respectively. Specific to DLBCL, 4/6 responders had CR, despite poor prognostic factors such as extra-nodal disease and receipt of prior alloSCT [4,25]. Interestingly, a correlation between a low B to T cell (B:T) ratio (<1:8) and CNS adverse drug reactions (ADRs) was discovered. Implementation of dexamethasone prophylaxis and treatment for CNS ADRs and “double-step” dose escalation (in 1 week increments from 5 to 15 to 60 mcg/m2/d) evolved to limit CNS ADRs [4,24,26]. In addition, dexamethasone 20 mg administered either 12 hours or 1 hour prior to infusion start and dose escalations, followed by 8 mg 3 times daily for 2 additional days yielded better outcomes than prednisolone prophylaxis, with no CNS ADRs warranting treatment discontinuation in the dexamethasone group [4,25]. All 7 patients who received dexamethasone for treatment of CNS ADRs and continued blinatumomab treatment had objective responses [4].
At the time of this review, the latest trial results assessing blinatumomab in DLBCL were from a phase 2 study of 25 patients, with a median age of 66 years. Patients had received a median of 3 prior therapies and 28% had prior autologous SCT (autoSCT). Patients received either a “double-step” fixed-dose escalation of 9 mcg/d for week 1, 28 mcg/d for week 2, then 112 mcg/d for the remaining 6 weeks or a flat dose of 112 mcg/d for 8 weeks [27]. Of 21 evaluable patients, the ORR was 43% lasting a median of 11.6 months, with 19% having a CR. Stepwise dosing was determined to be the safest, most effective approach.
Across NHL trials, higher blinatumomab doses seem to be necessary to produce maximal cell kill compared to ALL studies. BSA-based dosing in the NHL trials used a stepwise approach to reach 60 mcg/m2/d [26] or a flat dose of 112 mcg/d was used [27], while impressive outcomes were obtained with lower doses of 15 mcg/m2/d [28] or 28 mcg/d [29] in ALL. In addition, many NHL trials extended treatment to 8 weeks of continuous blinatumomab exposure [27] while ALL trials have used a 4 week on, 2 week off strategy. The reason for differences in drug sensitivity is not entirely clear, but may be related to drug penetration into malignant nodal tissue compared to elimination of malignant peripheral blood lymphocytes [18]. The extent and rate of tumor reduction in NHL is dose dependent, as evidenced by the lack of response with doses less than 15 mcg/m2/d [18,22].
3.2 Clinical Data: Acute Lymphoblastic Leukemia
Following the success of blinatumomab against lymphoma marrow infiltration at 15 mcg/m2/d [22], phase 2 investigation began in the treatment of precursor B cell ALL (Pre-B-ALL) in morphologic remission but with minimal residual disease (MRD).MRD positivity was defined as the presence of at least 1 × 10-4 leukemic cells following induction and consolidation chemotherapy, detected by real time quantitative polymerase chain reaction for immunoglobulin or TCR gene rearrangements [30]. Despite low disease burden, most MRD-positive patients are destined for clinical relapse [31-33], many within 8 months of morphologic CR in the absence of alloSCT [34]. As such, treatment with blinatumomab was hypothesized to prevent future hematologic relapse in high-risk patients. Patients received blinatumomab 15 mcg/m2/d 4 weeks on, 2 weeks off, for a maximum of 4 cycles. Twenty patients were evaluable, including 5 with Philadelphia chromosome positive (Ph+) disease. The primary endpoint, MRD negativity, was met in 80% (16/20), including 3 of 5 Ph+ patients, with all responses following only 1 cycle. Median RFS (mRFS) at 13 months of median follow up (mFU) was 78% [30], while at 33 months mFU, a 61% rate of RFS was reported [35]. Blinatumomab served as an effective bridge to alloSCT for 45% (9/20). At 33 months of mFU, 67% of transplanted patients remained in CR with 2 CD19+ relapses occurring post alloSCT. Four patients not receiving alloSCT relapsed, all within 7 months of blinatumomab completion. Two patients relapsed with CD19-negative marrow disease, the other 2 with extramedullary relapse (1 testicular, 1 CNS), perhaps an early indicator of the limited efficacy of blinatumomab in these sanctuary sites [35]. One hundred-sixteen patients with MRD-positive ALL (defined as >1 × 10-3 in this study) in morphologic remission, received the previously studied blinatumomab dosing scheme [29,30] in a follow up phase 2 study, which further substantiated the efficacy of blinatumomab in eliminating remaining MRD after at least 3 chemotherapy courses. As the primary endpoint, 80% achieved MRD negativity, 98% of which occurred after only 1 cycle. Response did not correlate with MRD burden, age, sex, or the number of prior therapies.
Subsequent trials were expanded to patients with greater disease burden in the setting of overt morphologic relapse. In a phase 2 study of 36 patients with RR-ALL, blinatumomab was administered at 5 mcg/m2/d for 1 week followed by 15 mcg/m2/d for the remaining 3 weeks of the first treatment cycle and all subsequent cycles [28]. Enrolled patients had a median age of 32 years, at least 5% marrow blasts, and had received at least 1 prior induction and consolidation with standard chemotherapy. Nearly half had undergone prior alloSCT and 2 patients had Ph+ ALL. Twenty-five (69%) achieved CR, 10 of which were CR with partial hematologic recovery (CRh). Eighty-eight percent of responders (22/25) achieved a MRD response, 18 of which occurred after 1 cycle. The timing of relapse appeared to correlate with response. Of the 21 patients treated after first relapse, 20 (95%) achieved CR, whereas 6 of 15 (40%) who relapsed more than once prior to enrollment reached CR. With a 12-month mFU, the median overall survival (mOS) for all patients was 9.8 months and that of responders was 14.1 months, with a mRFS of 7.6 months. Thirty-six percent of patients received alloSCT following blinatumomab. A later follow up revealed a mOS of 12.9 months and a mRFS of 8.8 months [36]. Of the 12 responders who did not receive subsequent alloSCT, 8 (67%) relapsed, 4 during active blinatumomab treatment. Response rates were lower for those who relapsed after prior alloSCT (8 of 15; 53%) versus patients without prior alloSCT in first or subsequent relapse [11/11 (100%) and 6/10 (60%), respectively]. One of 2 enrolled Ph+ patients had a CR. Although limited by a small size, this study suggested the efficacy of blinatumomab even in patients with greater disease burden than only MRD-positive status with morphologic remission. Despite a high number of MRD-negative responses, relapses were common [28]. In comparison with the MRD studies, patients with morphologic relapse displayed more intense cytokine release syndrome (CRS), likely associated with greater disease burden. There were 2 cases of grade 4 CRS, both with high tumor burden (88% and 90% marrow blasts). To mitigate this, the two-step dose escalation pattern was adopted, in addition to permitting dexamethasone and cyclophosphamide debulking prior to blinatumomab, resulting in no further cases of grade 3 or higher CRS.
A follow up phase 2 trial secured FDA approval of blinatumomab for RR-ALL. The trial evaluated 189 heavily pretreated patients with very poor prognosis RR-ALL and a median age of 39 years [37]. Thirty-four percent (64/189) had prior alloSCT, 39% (74/189) had at least 2 prior salvage regimens, and 56% (105/189) had at least 75% marrow blasts. The dosing scheme and inclusion criteria were similar to the smaller phase 2 trial [28], with the exception of requiring at least 10% marrow blasts and limiting inclusion only to patients with early first relapse (within 12 months of initial remission) [37]. Within 2 cycles, 33% (63/189) achieved CR and 10% (18/189) achieved CRh, with 80% of responses occurring within the first cycle. Response rates are lower than the previous trial [28], possibly due to inclusion of patients with more aggressive disease (minimum of 10% marrow blasts versus 5% blasts and excluding late relapses) [37]. A potential predictor of response was identified, in that patients with less than 50% marrow blasts at the start of therapy had a CR rate of 73% as compared to 29% in patients with 50% or greater blasts. The CR rate did not vary between those with (45%) and without (42%) prior alloSCT [38]. Among responders with evaluable MRD data, 82% had a MRD response, which was not influenced by prior alloSCT. MRD responders had a mRFS of 6.9 months and mOS of 11.5 months, compared to a 2.3 month mRFS and 6.7 month mOS for MRD non-responders. Previously published data clearly suggest the detrimental effect of MRD following first line ALL therapy [31-33]; however, this trial suggests MRD may be an important prognostic factor in RR-ALL as well and demonstrates the importance of the depth of response following blinatumomab. Forty percent of enrolled patients proceeded to alloSCT following blinatumomab, including second alloSCTs in 17% of previously transplanted patients. Further details regarding the 32 transplanted patients have been reported, suggesting the feasibility of alloSCT following blinatumomab, with a 100-day mortality rate of 11% [38].
To limit CRS, neurotoxicity, and other ADRs potentially related to higher tumor burden, this larger phase 2 trial required a debulking pre-phase with dexamethasone 10-24 mg/m2/d for up to 5 days, completed 3 days prior to the start of blinatumomab. Fifty-one percent of patients received pre-phase treatment, which did not influence ORR to blinatumomab. Only 1.6% (3/189) of patients experienced grade 3/4 CRS or grade 4 neurotoxicity, all of which resolved with treatment discontinuation.
Limited pediatric RR-ALL data with blinatumomab are available. The FDA approval information does not specifically list dosing information for pediatric patients but acknowledges that available pediatric trials use BSA-based dosing. The adult fixed doses of 9 mcg/d and 28 mcg/d may be used as long as the patient weighs at least 45 kg. Initial experience in pediatrics comes from compassionate use data, in which 3 patients with RR-ALL, who had relapsed after alloSCT, received blinatumomab 15 mcg/m2/d for 4 to 6 weeks [39]. All 3 patients achieved MRD-negative CR, and 1 of 3 was able to proceed to alloSCT. A follow up report of 9 pediatric patients from compassionate use data (of which 2 were previously reported [39]) was published [40]. Six of these patients had 1 prior alloSCT and 3 had received 2 prior alloSCTs. The median age was 10.4 years. Patients received 5 or 15 mcg/m2/d for 4 weeks, followed by 1 to 2 weeks off. In the case of no response, patients treated at 5 mcg/m2/d were increased to 15 mcg/m2/d after receiving a course of chemotherapy or stem cell boost to control progressive disease. Six of 9 patients achieved CR, 4 after a single cycle. Two responders required treatment with chemotherapy following the failure of the first cycle of blinatumomab, with response occurring after the second blinatumomab cycle. These demonstrate some of the very limited available clinical data on potential synergy between tumor debulking chemotherapy followed by blinatumomab, in congruence with adult data suggesting patients with lower marrow blast counts are more likely to respond to blinatumomab [37]. With a 4 year mFU, the mOS was 33% (44% for responders). Of the 6 responders, 4 went on to receive haploidentical alloSCT.
Results of a phase 1 trial of 41 pediatric patients with primary-refractory B-ALL, second or greater relapse, or relapse after alloSCT were recently reported [41]. Patients initially received 5, 15, or 30 mcg/m2/d on a 4 week on, 2 week off treatment cycle, later changed to 5 mcg/m2/d on days 1-7 then 15mcg/m2/d thereafter following 2 cases of severe CRS at 30 mcg/m2/d. Sixty-three percent of patients enrolled had relapsed following prior alloSCT. Despite very poor prognosis, 13/41 (32%) achieved CR, 77% of which were MRD-negative. With a 12.4 month mFU, the mOS was 5.7 months for all patients with a mRFS of 8.3 months. Nine patients went on to receive alloSCT.
To date, the most advanced report of blinatumomab in pediatric RR-ALL is a phase 2 study of 39 patients; 18 of which were enrolled in the aforementioned phase 1 trial [42]. The median age was 9 years with 41% receiving 2 or more prior salvage regimens and 64% receiving prior alloSCT. Sixty-nine percent of patients had at least 50% marrow blasts. Patients received 5 mcg/m2/d on days 1-7 and 15 mcg/m2/d thereafter, on a 4 week on, 2 week off treatment cycle. Only about half of patients completed cycle 1 and only 10% completed 2 cycles. Thirty-one percent (12/39) achieved CR, 5 of whom were MRD responses. With 6 month mFU, the mOS of all patients was 4.3 months and mRFS was 5.6 months. Half of patients in CR went on to receive alloSCT and 5 non-responders received alloSCT after blinatumomab was stopped. In summary, the available data on the use of blinatumomab in pediatric RR-ALL is promising and the results of the ongoing phase 2 trial are eagerly awaited.
3.3. Clinical Data: Use of Blinatumomab Beyond NHL and ALL
One of the earliest reports assessing blinatumomab was an ex vivo analysis demonstrating cytotoxicity against chronic lymphocytic leukemia (CLL), described above [8]. A recent in vitro study investigated blinatumomab in CLL and demonstrated similar cytotoxicity in treatment naïve and RR-CLL cell lines [43]. A third publication cites the feasibility of utilizing blinatumomab for in vitro T cell expansion, called blinatumomab-expanded T cells (BET), to be used as an immune reconstitution method after lymphocyte depleting therapy in CLL [44]. BET cells could additionally be used in combination with blinatumomab to provide a means of eliminating MRD after primary therapy. The only available published clinical data to date in CLL is the inclusion of 2 patients with CLL and 1 patient with small lymphocytic lymphoma in the initial phase 1 NHL study [22]. These patients received 0.5-15 mcg/m2/d for variable durations, with no CRs reported. Within the same study, 2 patients with marginal zone lymphoma (MZL) were treated with 15-30 mcg/m2/d for a short period and were not evaluable for response. Lack of response in this small analysis can be attributed to suboptimal dosing and duration of therapy. Additionally, CD4+ and CD8+ regulatory T cells are positively correlated with disease progression in CLL and likely function to suppress T cell expansion induced by blinatumomab, thereby constituting a potential mechanism of resistance to therapy [45]. We are unaware of any currently available blinatumomab trials enrolling patients with CLL or MZL.
In theory, acute myeloid leukemia (AML) may represent another potential therapeutic target for blinatumomab as CD19 expression has been described in association with the presence of t(8;21) and monocytic lineages [46,47] with variable prognostic effect [48,49]. Although there is no published clinical data assessing blinatumomab for AML, novel BiTE constructs against different myeloid and stem cell antigens expressed by AML cells, such as CD33 [50-52] and CD123 [53,54] are under active development.
4. Adverse Drug Reactions and Management
With a unique mechanism of action comes distinct ADRs, many of which substantially differ from combination chemotherapy that patients would otherwise receive in the RR setting. Arguably the most significant ADRs include CRS and neurotoxicities, including encephalopathy, convulsions, tremor, speech disturbances, confusion, and ataxia [20]. While CRS is less common (all grades: 11%, grade 3 or higher: 1%), neurological toxicities remain a common issue (all grades: roughly 50%, grade 3 or higher: 15%).
Supraphysiologic cytokine release following initial exposure to blinatumomab may result in CRS. Symptoms of CRS range from flu-like symptoms to hypotension, capillary leak syndrome, and multi-organ failure. CRS exclusively occurs during the first cycle of therapy with clinical sequelae coinciding with peak T cell expansion and cytokine release within the first few days of blinatumomab administration [9]. CRS incidence and intensity correlates with disease burden, as demonstrated by the lack of significant CRS in MRD-positive patients in morphologic CR [30] compared to 2% grade 3/4 CRS in RR-ALL with dexamethasone prephase treatment [37] and 6% grade 3/4 CRS without pre-phase treatment [28]. Interestingly, the degree of CRS severity does not correlate with treatment response in the initial MRD studies [9]. The two-step dose escalation scheme in addition to pre-treatment with dexamethasone and/or chemotherapy reduces the severity of CRS [28,37]. Grade 3/4 CRS requires infusion interruption (Table 1), while less severe cases can be managed with dexamethasone. The optimal dexamethasone regimen for CRS treatment remains unclear. A tapered regimen of 24 mg intravenous (IV) divided every 8 hours on day 1, 16 mg divided every 12 hours on day 2, followed by 8 mg daily on days 3 and 4 has been used with success.
Table 1. Toxicity Management.
| Toxicity | Grade | Action |
|---|---|---|
| Cytokine Release Syndrome | 3 | Withhold infusion until resolved, then restart at 9 mcg/d* |
| 4 | Permanently discontinue | |
|
| ||
| Neurologic Toxicity | Seizure | If more than one seizure occurs, permanently discontinue blinatumomab |
| Grade 3 | Withhold the infusion until resolved to ≤ Grade 1 for at least 3 days, then restart at 9 mcg/d*. If the toxicity does not recur after 7 days, escalate to 28 mcg/d*. If the toxicity recurs or if it takes more than 7 days to resolve, permanently discontinue blinatumomab. |
|
| Grade 4 | Permanently discontinue | |
|
| ||
| Other Clinically Relevant Adverse Reactions | Grade 3 | Withhold the infusion until resolved to ≤ Grade 1 for at least 3 days, then restart at 9 mcg/d*. If the toxicity does not recur after 7 days, escalate to 28 mcg/d*. If the toxicity takes more than 14 days to resolve, permanently discontinue. |
| Grade 4 | Consider permanent discontinuation. | |
Dexamethasone 20 mg should be administered 1 hour prior to the reinitation of infusion (If blinatumomab was held for more than 4 hours) and prior to dose increases.
Secondary macrophage activation syndrome (MAS) correlating with CRS is rare but serious and should be considered in patients with hyperferritinemia and pathological evidence of hemophagocytosis [55]. Pathogenesis is partially dependent on IL-6. Treatment with the anti-IL-6 receptor mAb tocilizumab has been used for the treatment of CRS following chimeric antigen receptor modified T cells (CAR T) infusion and may be considered in critical situations. However, it is unclear if such data should be extrapolated to blinatumomab treated patients [56]. Although IL-6 blockade is not expected to impair blinatumomab cytotoxicity, given the limited clinical experience in this setting, its use should be limited to patients with life-threatening MAS.
Neurotoxicities following blinatumomab may be caused by irritation of the neuroendothelium by neurotoxic cytokines and chemokines following T cell activation [4]. The only identified risk factor at this time is a low B:T cell ratio of <1:8 [26]. The median time to onset of any neurotoxicity is 7 days and given the presumed inflammatory pathology, is often successfully managed with dexamethasone and/or infusion interruption (Table 1) [37]. Dexamethasone is the preferred corticosteroid given its known CNS penetration and published clinical experience. In the entire phase 2 RR-ALL trial, 86% of patients required dexamethasone within 30 days of initiation for ADR management, which did not seem to impair outcomes. Infusion interruption did not seem to impair outcomes, as roughly half of patients requiring interruption obtained CR/CRh [37]. If a grade 3 or higher seizure occurs, dose interruption is recommended along with administration of anti-epileptic therapy. Blinatumomab may be restarted upon resolution to grade 1 or less. Most seizure episodes have been treated with levetiracetam; minimal data exist using alternative drugs. Given the rarity of seizures, routine prophylaxis is not warranted due to a high number needed to treat, but may be considered in patients with pre-existing CNS pathology. As patients with CNS leukemia or other CNS pathology were excluded from trials, drug tolerability in these populations is unknown.
Almost 100% of blinatumomab-treated patients will experience an ADR, with roughly 30% having a grade 3/4 ADR [37]. Febrile neutropenia, infection, and myelosuppression are common and expected given the treated population and mechanism of action of the drug. Pyrexia is the single most common ADR, and of all grades occurs in 62% of patients with 7% experiencing grade 3 or higher febrile episodes. Treatment with acetaminophen can be considered, with the caution that it may mask neutropenic fever. Gastrointestinal toxicity, transaminitis, and electrolyte disturbances are reported but rarely severe [20]. Despite impressive anti-tumor effect, tumor lysis syndrome following blinatumomab remains very rare. After the first cycle, frequency and intensity of ADRs typically lessen, presumably due to less cytokine release than in cycle 1. Immunoglobulin deficiencies follow blinatumomab treatment; however, a correlation with increased risk of infection remains to be determined [57]. Despite widespread T cell activation, autoimmune phenomena and increased risk of graft-versus-host disease in previously transplanted patients have not been reported. Less than 1% of treated patients develop anti-blinatumomab antibodies [20].
5. Administration Logistics
Hospitalization is recommended for the first 9 days of the first cycle and the first 2 days of cycle 2 to monitor for CRS and neurotoxicity [20]. Patients should receive dexamethasone 20 mg intravenously 1 hour prior to infusion start, 1 hour prior to dose escalation on day 8 of the 1st cycle, and 1 hour prior to restarting after any treatment interruptions lasting 4 hours or more. Blinatumomab is administered via CIVI over 28 days and therefore arrangements for continuous intravenous home administration using an infusion pump must be made prior to hospital discharge. Insurance coverage and coordination with homecare agencies for drug and pump procurement well in advance of discharge is essential. Reconstituted IV bags are stable for 8 days while refrigerated and 48 hours at room temperature, permitting home delivery in advance. At our center, a single cycle of blinatumomab currently costs roughly $89,890, which obstructs access to this medication for some patients.
6. Future Directions
Clinical trials with blinatumomab which are currently enrolling patients in the U.S. and registered on www.clinicaltrials.gov are presented (Table 2). Although the available data clearly suggest single agent benefit in RR-ALL and RR-NHL, the use of this agent in combination with other drugs or for the treatment of other CD19+ cancers (including CLL, other B cell lymphomas and t(8;21) AML) is still an uncharted territory. The current FDA approval is also limited to Ph- RR-ALL, likely due to the limited number of Ph+ patients studied thus far, although the National Comprehensive Cancer Network does include blinatumomab as an option for tyrosine kinase inhibitor-refractory Ph+ ALL [58]. Other trials comparing the efficacy of blinatumomab vs. multi-agent chemotherapy or assessing the feasibility of combining blinatumomab with chemotherapy, in both adult and pediatric RR and newly diagnosed (ND) ALL, are ongoing.
Table 2. Ongoing clinical trials.
| Trial Identifier | Phase | Official Title | Status | Condition |
|---|---|---|---|---|
| NCT020000427 | Phase II | A Phase 2 Single Arm, Multicenter Trial to Evaluate the Efficacy of the BiTE Antibody Blinatumomab in Adult Subjects With Relapsed/Refractory Philadelphia Positive B-precursor Acute Lymphoblastic Leukemia (Alcantara Study) | Recruiting | Adult Ph+ pre-B, RR-ALL |
| NCT0210853 | Phase III | Risk-Stratified Randomized Phase III Testing of Blinatumomab (NSC#765986) in First Relapse of Childhood B-Lymphoblastic Leukemia (B-ALL) | Recruiting | Childhood relapsed ALL |
| NCT02143414 | Phase II | A Phase II Study of Blinatumomab (NSC-765986) and POMP (Prednisone, Vincristine, Methotrexate, 6-Mercaptopurine) for Patients ≥ 65 Years of Age With Newly Diagnosed Philadelphia-Chromosome Negative (Ph-) Acute Lymphoblastic Leukemia (ALL) and of Dasatinib (NSC-732517), Prednisone and Blinatumomab for Patients ≥ 65 Years of Age With Newly Diagnosed Philadelphia-Chromosome Positive (Ph+) ALL | Recruiting | Adult ND ALL |
| NCT02013167 | Phase III | A Phase 3, Randomized, Open Label Study Investigating the Efficacy of the BiTE Antibody Blinatumomab Versus Standard of Care Chemotherapy in Adult Subjects With Relapsed/Refractory B-precursor Acute Lymphoblastic Leukemia (ALL) (TOWER Study) | Recruiting | Adult RR-ALL |
| NCT02003222 | Phase III | A Phase III Randomized Trial of Blinatumomab for Newly Diagnosed BCR-ABL-Negative B Lineage Acute Lymphoblastic Leukemia in Adults | Recruiting | Ph-negative ND Adult ALL |
| NCT02187354 | Expanded Access | An Open-Label, Multi-center, Expanded Access Protocol of Blinatumomab for the Treatment of Pediatric and Adolescent Subjects With Relapsed and/or Refractory B-precursor Acute Lymphoblastic Leukemia (ALL) | Available | Pediatric and adolescent B-precursor RR-ALL |
7. Conclusion
As the first approved BiTE, blinatumomab brings a long-awaited promise of effective immunotherapy to patients with RR-ALL with the potential to change the treatment paradigm and open the door for future BiTE constructs. The unique design of this agent permits formation of a pharmacologic immune synapse occurring independent of antigen processing and MHC presentation to trigger a polyclonal antitumor cytotoxic T cell response and rapid, sustained B cell apoptosis. T cell activation occurs at the cost of supraphysiologic cytokine release, resulting in a unique side effect profile requiring clinical expertise for appropriate management and monitoring.
Blinatumomab has been most extensively studied in RR-ALL after initial success in RR-NHL, where experience remains limited to early phase data with limited patient numbers. Nonetheless, efficacy in NHL looks promising, particularly in RR-DLBCL, with response rates around 40% [27]. Blinatumomab shows impressive efficacy in RR-ALL in morphologic relapse, with an approximate 40% CR rate [37] in patients failing multiple lines of prior therapy including alloSCT. Its promising ability to eliminate MRD and thus potentially prevent future clinical relapse may question the need for alloSCT to prolong survival in such patients. Future trials to assess the efficacy of blinatumomab combined with cytotoxic chemotherapy for both ND and RR lymphoid neoplasms, as well as a comparative phase 3 trial randomizing patients to blinatumomab versus chemotherapy, are ongoing. Furthermore, expansion of treatment with this agent to other CD19+ malignancies may broaden the number of patients able to benefit from this innovative immunotherapeutic agent.
8. Expert Opinion
B cell neoplasms were the first malignancies to reap the benefit of immunotherapy with the introduction of rituximab in the late 1990's. Only recently has immunotherapy begun to be applied for the treatment of other hematological neoplasms, including ALL. In the future, ND and heavily pretreated ALL patients are likely to have several immunotherapy options in addition to combination chemotherapy. Here we have presented currently available clinical data supporting the use of blinatumomab for RR-NHL and RR-ALL. However, given the drug's current approval and the rapidly growing competition in this sector, we will limit our discussion here to RR-ALL.
During our review, we discovered several unanswered questions regarding this agent. Would outcomes in RR-ALL be improved by higher doses of drug, similar to those initially used in NHL studies? Is there a difference in maximum tolerated dose between these diseases and if so, why? What is the role of alloSCT following blinatumomab? Many studied patients proceeded to alloSCT without an obvious increase in transplant related mortality; however the necessity of alloSCT following CR with blinatumomab is unclear, especially in the setting of MRD negativity. At present, the available data are inadequate to address whether the use of single agent blinatumomab obviates the need for alloSCT in RR-ALL.
An important unanswered question is which patient or disease characteristics predict response? Prior studies have suggested that increased marrow disease burden (>50% blasts) [37], multiple previous relapses [28], or relapsed disease after prior alloSCT [36] are all associated with worse response rates to blinatumomab. In addition, higher regulatory T cell counts and elevated lactate dehydrogenase may also predict poor response [59]. To date, analyses of T cell numbers and cytokine kinetics show no correlation to response rate [9]. Other markers remain to be identified, including a clear correlation between the occurrence of CRS and therapeutic efficacy.
What is the efficacy of blinatumomab against extramedullary disease and disease in tumor sanctuary sites? Several reports describe extramedullary, CNS, or testicular relapse after blinatumomab treatment; the exact significance of these findings is unclear [28,30,40]. Because patients with CNS pathology or CNS leukemia were excluded from trials, it is unknown if these patients can safely be treated with additional measures for screening and managing neurotoxicity. Patients with hepatitis B and C infection were also excluded, presumably due to fear of reactivation, yet it is unclear if blinatumomab carries the same viral reactivation risk as other anti-B cell mAbs (e.g., rituximab). Finally, is there a dose threshold after which dexamethasone interferes with cytotoxicity? Mechanistically, high dose dexamethasone resulting in T cell lysis may suppress blinatumomab cytotoxicity. Available data does not suggest an obvious impairment in efficacy with dexamethasone [19,37]; however, until a randomized trial is completed to directly assess this point, judicious use of dexamethasone may be warranted.
Prior to blinatumomab, the only FDA approved single agent treatment for RR B-ALL was liposomal vincristine, producing a dismal CR/CRh rate of 20% [60]. In addition to blinatumomab, 2 exciting new immunotherapeutic agents have emerged for treatment of RR-ALL: CAR T cells and inotuzumab ozogamicin (IO). Groups at the University of Pennsylvania (UPenn)/Children's Hospital of Philadelphia (CHOP), Memorial Sloan Kettering Cancer Center (MSKCC), and the National Cancer Institute (NCI) published their initial experiences with differently engineered CAR T cells in adult and pediatric patients with RR-ALL. The UPenn/CHOP group developed CTL019 autologous T cells expressing an anti-CD19 scFv with CD3ζ and CD133 (4-1BB) [61]. Cells are collected via leukapheresis, transduced via a lentiviral vector, expanded ex vivo with anti-CD3/anti-CD28 beads, and then transfused back into the patient. In a recent long-term update of their results, the group reported a 90% CR rate in 25 pediatric and 5 adult RR-ALL with 85% MRD-negative responses [61]. Ten patients who achieved a CR at 1 month have relapsed, 50% with CD19-negative disease. Of note, CTL019 cells cross the blood brain barrier, potentially preventing CNS relapse. All responders developed CRS with 1/3 requiring tocilizumab [61,62]. In adult patients with RR-ALL treated by the same group, 92% (11/12) developed severe CRS, 6 required tocilizumab, and 3 died with documented infections [63]. CRS correlated with disease burden.
Researchers at MSKCC have engineered CAR T cells expressing an anti-CD19 scFv linked to CD28 and CD3ζ signaling domains (19-28z) via a retroviral vector [64]. They have reported a 91% CR rate in 22 evaluable adults with RR-ALL, with 90% being MRD-negative responses [65]. Seventy-seven percent of eligible patients proceeded to alloSCT and 5 have relapsed, 1 with CD19-negative disease. Sixty-nine percent (9/13) with overt morphologic marrow disease prior to CAR T cell therapy developed CRS requiring tocilizumab or corticosteroids. Among ALL patients treated at the NCI with CAR T cells expressing the CD-19 specific CD3ζ and CD28 domains, 70% (14/20) achieved CR and 60% (12/20) attained MRD negativity [66]. Of note, all 6 patients who had primary refractory disease demonstrated responses. Ten of the patients with MRD negative remission subsequently received alloSCT, with the 2 remaining patients later relapsing with CD19-negative disease. Grade 3 or 4 CRS occurred in 26% of patients treated at the MTD of 1×106 CAR T cells.
Compared with blinatumomab, CAR T cell infusion results in a higher incidence and severity of CRS (often with concurrent MAS) that frequently requires tocilizumab. Like blinatumomab, the incidence and severity of CRS correlates with disease burden. However, despite this increased toxicity, response rates are clearly higher following CAR T cell therapy than with blinatumomab, albeit in limited numbers of patients. No comparative trials exist between CAR T and blinatumomab therapies and data regarding treatment with one modality after failure of the other is limited. Currently, the selection of the appropriate therapeutic approach for RR-ALL is primarily dictated by tolerability, treatment availability, and potential logistical issues (i.e. financial, insurance coverage, and accessibility to a clinical cancer center with available accruing CAR T cell clinical trials). CAR T cells appear to be most beneficial in fit patients able to tolerate the risk of severe CRS or those with low disease burden, due to the lower risk of CRS. In addition, although CIVI administration of blinatumomab is inconvenient, CAR T cell therapy is only available at select institutions and can be cumbersome as well, due to the need for autologous T cell harvest, transduction, and interim disease control while genetically altered T cells are being generated. Perhaps the most compelling reason to use blinatumomab over CAR T cells is the short half-life of blinatumomab, which grants strict control of severe ADRs, like CRS. CRS following blinatumomab can be rapidly reversed with infusion interruption and dexamethasone, which does not seem to impair efficacy. On the other hand, CRS following CAR T may be long lived and require management with tocilizumab, intensive care monitoring, pressor support, and dexamethasone; the latter of which can impair the efficacy of CAR T therapy [64]. In addition, many patients receiving CAR T received leukoreducing cytotoxic chemotherapy a short time prior to CAR T infusion to minimize CRS risk and to control rapidly proliferating disease until T cell infusion. In contrast, administration of cytotoxic chemotherapy prior to blinatumomab is not standard. Lastly, relapsed disease with CD19-negative B cells remain an issue for both CD19-targeted modalities, presumably due to variable expression of CD19 across clones expressing similar aberrant cytogenetics [67], suggesting the multiclonal nature of acute leukemia. Further research on how to optimally manage CD19-negative relapses following CD19-directed therapies is needed [4,28,30].
IO is an immunoconjugate comprised of an anti-CD22 mAb linked to the toxin, calicheamicin. CD22 is expressed in 90% of B-ALL cases. Binding of IO to tumor cells results in drug internalization and cleavage of calicheamicin, allowing it to bind DNA and cause double strand breaks with subsequent cell death. Single center phase II studies from the MD Anderson Cancer Center [68,69] have reported 90 RR-ALL patients treated with IO as a single dose per cycle (every 3-4 weeks) or as weekly doses IV over 1 hour. Enrolled patients were heavily pretreated, with almost 70% receiving IO as second salvage therapy, but only 11% having prior alloSCT, a smaller percentage than blinatumomab trials [37]. Close to 20% of patients had Ph+ RR-ALL, which is a higher percentage than patients in blinatumomab trials. With a median of 2 cycles, CR/CRh was achieved in 58% of patients, with 72% of assessable patients having a MRD-negative response. However, responses were not durable, as the median remission duration was 7 months. Forty percent of IO-treated patients received subsequent alloSCT. Significant ADRs included pyrexia, hypotension, and drug-related hepatotoxicity, all of which were less common with weekly versus single dose regimens. Comparative data are unavailable between blinatumomab and IO, and differences in disease characteristics make it difficult to determine which agent is superior in the relapsed setting. Ongoing trials with IO combined with multi-agent chemotherapy for RR and ND ALL will likely yield exciting results in the near future. Unlike blinatumomab, the mechanism of action of IO does not involve T cell activation and therefore combination with other lymphotoxic chemotherapy or corticosteroids is not expected to impair efficacy or result in immunological toxicities.
Both IO and blinatumomab have demonstrated therapeutic efficacy in heavily pre-treated RR-ALL patients, with manageable and unique side effect profiles. Although IO inarguably has a more convenient administration schedule when compared to that of blinatumomab, both clearly have potential roles for the treatment of ND patients for the elimination of MRD. Trials investigating the role of blinatumomab in this setting are currently ongoing. The United States Intergroup Trial, E1910, is evaluating blinatumomab plus chemotherapy as compared with chemotherapy alone in adult patients between 35 to 70 years of age with ND B-ALL (see Table 2) [NCT02003222]. A second trial is examining the efficacy of blinatumomab in older patients with ND B-ALL in combination with chemotherapy or dasatinib and prednisone, depending on Philadelphia chromosome mutation status [NCT02143414]. CAR T cell therapy results in the highest response rates of the 3 modalities but may be more appropriate as a salvage therapy due to the high incidence of severe CRS and the limited number of clinical centers offering this therapy. Ongoing trials will hopefully shed light on the appropriate role of each of these therapies for the treatment of ALL.
Acknowledgments
Dr. Meir Wetzler contributed significantly in the preparation of this manuscript prior to his unfortunate passing. It is with great reverence that we acknowledge the selfless dedication he had to his practice, research, patients, and the mentorship he provided the authors of this manuscript.
This study was supported in part by grants from the National Cancer Institute (grant number CA16056) to BK, CWF, EPO, EAG, ESW and MW, the Szefel Foundation, Roswell Park Cancer Institute, the Leonard S LuVullo Endowment for Leukemia Research, the Nancy C Cully Endowmend for Leukemia Research, the Babcock Family Endowment and the Heidi Leukemia Research Fund, Buffalo, NY. EW is a consultant and principal investigator for Juno and is also supported by a Cancer Clinical Investigator Team Leadership Award (CCITLA) awarded by National Cancer Institute through a supplement to P30CA016056. MW was a consultant for Amgen and Juno.
Footnotes
Financial competing interests disclosure: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Bibliography
- 1.Fielding AK, Richards SM, Chopra R, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944–50. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian HM, Thomas D, Ravandi F, et al. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer. 2010;116:5568–74. doi: 10.1002/cncr.25354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gökbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032–41. doi: 10.1182/blood-2011-12-399287. [DOI] [PubMed] [Google Scholar]
- 4**.Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: A historical perspective. Pharmacol Ther. 2012;136(3):334–42. doi: 10.1016/j.pharmthera.2012.07.013. A thorough review of the development of blinatumomab, immunopharmacologic responses to treatment, adverse events, pharmacokinetics, and additional clinical data available at that time. [DOI] [PubMed] [Google Scholar]
- 5*.Löffler A, Kufer P, Lutterbuse R, et al. A recombinant bispecific single-chain antibody, CD19xCD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95(6):2098–103. First description of blinatumomab pharmacodynamics, in vitro efficacy, immunopharmacologic response, and description of differences between blinatumomab and previous BiTE constructs. [PubMed] [Google Scholar]
- 6*.Wolf E, Hofmeister R, Kufer P, et al. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005;10(18):1237–44. doi: 10.1016/S1359-6446(05)03554-3. In depth review of general BiTE design and pharmacologic principles and immunopharmacologic responses following BiTE treatment. [DOI] [PubMed] [Google Scholar]
- 7.Offner S, Hofmeister R, Romaniuk A, et al. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43:763–71. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Löffler A, Gruen M, Wuchter C, et al. Efficient elimination of chronic lymphocytic leukemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia. 2003;17:900–9. doi: 10.1038/sj.leu.2402890. [DOI] [PubMed] [Google Scholar]
- 9**.Klinger M, Brandl C, Zugmaier G, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119(26):6226–33. doi: 10.1182/blood-2012-01-400515. Ex vivo analysis of clinical trial samples describing the immunopharmacologic response to blinatumomab therapy as well as pharmacokinetics and describes potential markers predictive of response. [DOI] [PubMed] [Google Scholar]
- 10*.Dreier T, Lorenczerski G, Brandl C, et al. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002;100:690–97. doi: 10.1002/ijc.10557. In vitro analysis of the pharmacokinetics and pharmacodynamics of blinatumomab. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman P, Hofmeister R, Brischwein K, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer. 2005;115:98–104. doi: 10.1002/ijc.20908. [DOI] [PubMed] [Google Scholar]
- 12.Gruen M, Bommert K, Bargou RC. T-cell-mediated lysis of B cells induced by a CD19xCD3 bispecific single-chain antibody is perforin depdendent and death receptor independent. Cancer Immunol Immunother. 2004;53:625–32. doi: 10.1007/s00262-003-0496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.d'Argouges S, Wissing S, Brandl C, et al. Combination of rituximab with blinatumomab (MT103/MEDI-538), a T cell-engaging CD19-/CD3-bispecific antibody, for highly efficient lysis of human B lymphoma cells. Leuk Res. 2009;33(3):465–73. doi: 10.1016/j.leukres.2008.08.025. [DOI] [PubMed] [Google Scholar]
- 14.Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1:36. doi: 10.1186/2162-3619-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piccaluga PP, Arpinati M, Candoni A, et al. Surface antigens analysis reveals significant expression of candidate targets for immunotherapy in adult acute lymphoid leukemia. Leuk Lymphoma. 2011;52(2):325–327. doi: 10.3109/10428194.2010.529206. [DOI] [PubMed] [Google Scholar]
- 16.Raponi S, De Propris MS, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52(6):1098–107. doi: 10.3109/10428194.2011.559668. [DOI] [PubMed] [Google Scholar]
- 17.Nagorsen D, Bargou R, Ruttinger D, et al. Immunotherapy of lymphoma and leukemia with T-cell engaging BiTE antibody blinatumomab. Leuk Lymphoma. 2009;50(6):886–91. doi: 10.1080/10428190902943077. [DOI] [PubMed] [Google Scholar]
- 18.Hijazi Y, Klinger M, Schub A, et al. Blinatumomab exposure and pharmacodynamic response in patients with non-Hodgkin lymphoma (NHL) J Clin Oncol. 2013;31(suppl; abstr 3051) [Google Scholar]
- 19*.Brandl C, Haas C, d'Argouges S, et al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol Immunother. 2007;56:1551–1563. doi: 10.1007/s00262-007-0298-z. Describes the in vitro effect of dexamethasone on blinatumomab cytotoxicity and inflammatory markers following drug exposure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blincyto ® [package insert] Thousand Oaks, CA: Amgen Inc; 2014. [Google Scholar]
- 21.Wu B, Hijazi Y, Wolf A, et al. Pharmacokinetics (PK) of blinatumomab and its clinical implications. J Clin Oncol. 2013;31(suppl; abstr 3048) [Google Scholar]
- 22.Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 23.Goebeler M, Viardot A, Noppeney R, et al. Blinatumomab (CD3/CD19 BiTE® Antibody) results in high response rate in patients with relapsed non-Hodgkin's lymphoma (NHL) including MCL and DLBCL. Ann Oncol. 2011;22(4):Abstr 068. [Google Scholar]
- 24.Goebeler ME, Viardot A, Kufer P, et al. Final results from a phase 1 study of blinatumomab in patients with relapsed/refractory Non-Hodgkin's lymphoma. Hematol Oncol. 2013;31(Suppl. 1):Abstr 302. [Google Scholar]
- 25.Viardot A, Goebeler M, Nopponey R, et al. Blinatumomab Monotherapy Shows Efficacy in Patients with Relapsed Diffuse Large B Cell Lymphoma. Blood (ASH Annual Meeting Abstracts) 2011:Abstr 1637. [Google Scholar]
- 26.Viardot A, Goebeler M, Scheele J, et al. Treatment of Patients with Non-Hodgkin Lymphoma (NHL) with CD19/CD3 Bispecific Antibody Blinatumomab (MT103): Double-Step Dose Increase to Continuous Infusion of 60 μg/m2/d Is Tolerable and Highly Effective. Blood (ASH Annual Meeting Abstracts) 2010:Abstr 2880. [Google Scholar]
- 27.Viardot A, Goebeler M, Hess G, et al. Treatment of Relapsed/Refractory Diffuse Large B-Cell Lymphoma with the Bispecific T-Cell Engager (BiTE®) Antibody Construct Blinatumomab: Primary Analysis Results from an Open-Label, Phase 2 Study. Blood. 2014:Abstr 4460. [Google Scholar]
- 28*.Topp MS, Gökbuget N, Zugmaier G, et al. Phase II Trial of the Anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–40. doi: 10.1200/JCO.2014.56.3247. First clinical trial to assess the efficacy of blinatumomab in patients with relapsed/refractory ALL. [DOI] [PubMed] [Google Scholar]
- 29.Goekbuget N, Dombret H, Bonifacio M, et al. BLAST: A Confirmatory, Single-Arm, Phase 2 Study of Blinatumomab, a Bispecific T-Cell Engager (BiTE®) Antibody Construct, in Patients with Minimal Residual Disease B-Precursor Acute Lymphoblastic Leukemia (ALL) Blood. 2014:Abstr 379. [Google Scholar]
- 30*.Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–8. doi: 10.1200/JCO.2010.32.7270. First clinical trial to assess the efficacy of blinatumomab in ALL, specifically in elimination of MRD in patients in hematologic CR. [DOI] [PubMed] [Google Scholar]
- 31.Brüggemann M, Raff T, Flohr T, et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107(3):1116–23. doi: 10.1182/blood-2005-07-2708. [DOI] [PubMed] [Google Scholar]
- 32.Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL) Blood. 2009;113(18):4153–62. doi: 10.1182/blood-2008-11-185132. [DOI] [PubMed] [Google Scholar]
- 33.Raff T, Gökbuget N, Lüschen S, et al. Molecular relapse in adult standard-risk ALL patients detected by prospective MRD monitoring during and after maintenance treatment: data from the GMALL 06/99 and 07/03 trials. Blood. 2007;109(3):910–5. doi: 10.1182/blood-2006-07-037093. [DOI] [PubMed] [Google Scholar]
- 34.Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868–76. doi: 10.1182/blood-2011-09-377713. [DOI] [PubMed] [Google Scholar]
- 35.Topp MS, Gökbuget N, Zugmaier, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120(26):5185–7. doi: 10.1182/blood-2012-07-441030. [DOI] [PubMed] [Google Scholar]
- 36.Zugmaier G, Gökbuget N, Viardot A, et al. Long-term survival in adult patients with relapsed/refractory B-precursor acute lymphoblastic leukemia (ALL) who achieved minimal residual disease (MRD) response following anti-CD19 BiTE® blinatumomab. Blood. 2014:Abstr 2287. [Google Scholar]
- 37***.Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. Largest trial to date in relapsed/refractory ALL demonstrating the efficacy of blinatumomab. This trial is the basis for the FDA approval of blinatuomomab. [DOI] [PubMed] [Google Scholar]
- 38.Stein A, Topp M, Gökbuget N, et al. Allogeneic Hematopoietic Stem Cell Transplantation Following Anti-CD19 BiTE® Blinatumomab in Adult Patients with Relapsed/Refractory B-Precursor Acute Lymphoblastic Leukemia (ALL) Blood. 2014:Abstr 965. [Google Scholar]
- 39.Handgretinger R, Zugmaier G, Henze G, et al. Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leukemia. 2011;25:181–4. doi: 10.1038/leu.2010.239. [DOI] [PubMed] [Google Scholar]
- 40.Schlegel P, Lang P, Zugmaier G, et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica. 2014;99(7):1212–19. doi: 10.3324/haematol.2013.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase 1/2 Study in Pediatric Patients with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) Receiving Blinatumomab Treatment. Blood. 2014:Abstr 2292. [Google Scholar]
- 42.Gore L, Locatelli F, Zugmaier G, et al. Initial Results from a Phase 2 Study of Blinatumomab in Pediatric Patients with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Blood. 2014:Abstr 3703. [Google Scholar]
- 43.Wong R, Pepper C, Brennan P, et al. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica. 2013;98(12):1930–8. doi: 10.3324/haematol.2012.082248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Golay J, D'Amico A, Borleri G, et al. A novel method using blinatumomab for efficient, clinical-grade expansion of polyclonal T cells for adoptive immunotherapy. J Immunol. 2014;193:4739–47. doi: 10.4049/jimmunol.1401550. [DOI] [PubMed] [Google Scholar]
- 45.Jadidi-Niaragh F, Yousefi M, Memarian A, et al. Increased frequency of CD8+ and CD4+ regulatory T cells in chronic lymphocytic leukemia: Association with disease progression. Cancer Invest. 2013;31:121–31. doi: 10.3109/07357907.2012.756110. [DOI] [PubMed] [Google Scholar]
- 46.Kita K, Nakase K, Miwa H, et al. Phenotypical characteristics of acute myelocytic leukemia associated with the t(8;21)(q22;q22) chromosomal abnormality: frequent expression of immature B-cell antigen CD19 together with stem cell antigen CD34. Blood. 1992;80(2):470–7. [PubMed] [Google Scholar]
- 47.Brandt JT, Tisone JA, Bohman JE, Theil KS. Aberrant expression of CD19 as a marker of monocytic lineage in acute myelogenous leukemia. Am J Clin Pathol. 1997;107(3):283–91. doi: 10.1093/ajcp/107.3.283. [DOI] [PubMed] [Google Scholar]
- 48.Ball ED, Davis RB, Griffin JD, et al. Prognostic value of lymphocyte surface markers in acute myeloid leukemia. Blood. 1991;77(10):2242–50. [PubMed] [Google Scholar]
- 49.Solary E, Casasnovas RO, Campos L, et al. Surface markers in adult acute myeloblastic leukemia: correlation of CD19+, CD34+ and CD14+/DR--phenotypes with shorter survival. Leukemia. 1992;6(5):393–9. [PubMed] [Google Scholar]
- 50.Laszlo GS, Gudgeon CJ, Harrington KH, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554–61. doi: 10.1182/blood-2013-09-527044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krupka C, Kufer P, Kischel R, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014;123(3):356–65. doi: 10.1182/blood-2013-08-523548. [DOI] [PubMed] [Google Scholar]
- 52.Aigner M, Feulner J, Schaffer S, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27(5):1107–15. doi: 10.1038/leu.2012.341. [DOI] [PubMed] [Google Scholar]
- 53.Bonifant CL, Torres D, Velasquez MP, et al. CD123-Engager T Cells As a Novel Immunotherapeutic for AML. Blood. 2014:Abstr 3762. doi: 10.1038/mt.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith BD, Roboz GJ, Walter RB, et al. First-in Man, Phase 1 Study of CSL362 (Anti-IL3Rα / Anti-CD123 Monoclonal Antibody) in Patients with CD123+ Acute Myeloid Leukemia (AML) in CR at High Risk for Early Relapse. Blood. 2014:Abstr 120. [Google Scholar]
- 55.Teachey DT, Rheingold SR, Maude SL, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–7. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee DW, Gardner R, Porter DL, et al. Current concepts in diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zugmaier G, Topp MS, Alekar S, et al. Long-term follow-up of serum immunoglobulin levels in blinatumomab-treated patients with minimal residual disease-positive B-precursor acute lymphoblastic leukemia. Blood Cancer J. 2014;4:244. doi: 10.1038/bcj.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.National Comprehensive Cancer Network. [Accessed Feb 8, 2015];Acute Lymphoblastic Leukemia [Version 2.2014] doi: 10.6004/jnccn.2015.0153. [DOI] [PubMed] [Google Scholar]
- 59.Duell J, Dittrich M, Bedke T, et al. Crucial Role of Regulatory T Cells in Predicting the Outcome of the T Cell Engaging Antibody Blinatumomab in Relapsed and Refractory B Precursor ALL Patients. Blood. 2014:Abstr 2291. [Google Scholar]
- 60.O'Brien S, Schiller G, Lister J, et al. High-dose vincristine sulfate liposome injection for advanced, relapsed, and refractory adult Philadelphia chromosome-negative acute lymphoblastic leukemia. J Clin Oncol. 2013;31(6):676–83. doi: 10.1200/JCO.2012.46.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grupp S, Maude SL, Shaw P, et al. T Cells Engineered with a Chimeric Antigen Receptor (CAR) Targeting CD19 (CTL019) Have Long Term Persistence and Induce Durable Remissions in Children with Relapsed, Refractory ALL. Blood. 2014:Abstr 380. [Google Scholar]
- 62.Maude SL, Frey N, Shaw PA, et al. Chimeric Antigen Receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frey NV, Levine BL, Lacey SF, et al. Refractory cytokine release syndrome in recipients of Chimeric Antigen Receptor (CAR) T cells. Blood. 2014:Abstr 2296. [Google Scholar]
- 64.Brentjens R, Davila M, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park JH, Riviere I, Wang X, et al. CD19-Targeted 19-28z CAR Modified Autologous T Cells Induce High Rates of Complete Remission and Durable Responses in Adult Patients with Relapsed, Refractory B-Cell ALL. Blood. 2014:Abstr 382. [Google Scholar]
- 66.Lee DW, Stetler-Stevenson M, Sabatino M, et al. Intent-to-treat results of a phase I trial of CD19 Chimeric Antigen Receptor T cells using a consistent treatment regimen reveals a 67% complete response rate in relapsed, refractory acute lymphoblastic leukemia. Blood. 2014;124(21):Abstr 381. [Google Scholar]
- 67.Francis J, Dharmadhikari AV, Sait SN, et al. CD19 expression in acute leukemia is not restricted to the cytogenetically aberrant populations. Leuk Lymphoma. 2013;57(7):1517–20. doi: 10.3109/10428194.2012.754096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kantarjian H, Thomas D, Jorgensen J, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13(4):403–11. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- 69.Kantarjian H, Thomas D, Jorgensen J, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119:2728–36. doi: 10.1002/cncr.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]