

Abstract
Alterations in tissue oxygen pressure contribute to a number of diseases, including rheumatoid arthritis (RA). Low partial pressure of oxygen, a condition known as hypoxia, is a relevant feature in RA since it is involved in angiogenesis, inflammation, apoptosis, cartilage degradation, energy metabolism, and oxidative damage. Therefore, alterations in hypoxia-related signaling pathways are considered potential mechanisms of disease pathogenesis. The objective of this review is to highlight and update our current knowledge of the role of hypoxia in the pathogenesis of RA. We describe the experimental evidence that RA synovial tissue exists in a hypoxic state, as well as the origin and involvement of synovial hypoxia in different aspects of the pathogenic process.
Keywords: Hypoxia-inducible factor (HIF), Autoimmune disease, Inflammation
Background
Rheumatoid arthritis (RA) is a severe chronic autoimmune disease characterized by joint inflammation and destruction and the presence of autoantibodies (rheumatoid factor and anti-citrullinated protein antibodies). RA affects 1 % of the world’s population [1], and its prevalence among Mexicans is 1.6 % [2]. RA inflames the synovial membrane of diarthrodial joints and damages the articular tissues, leading to severe functional disarrangement of the entire joint. The initial stages of RA synovitis are characterized by proliferation of the microvasculature and secondary edema. Eventually, this process matures into a progressive infiltration of immune cells, including B cells, T cells, and monocytes, from the bloodstream. These immune cells are activated in the joint, differentiate, and acquire mature phenotypes. The influx of immune cells is also associated with phenotypic changes in synoviocytes, the typical resident cells. Both fibroblast- and monocyte-derived synoviocytes proliferate extensively and participate in the inflammatory process. In the latter stages of disease, the synovium becomes a mass of growing tissue, called pannus, which has the capacity to overlay, degrade, and invade the cartilage and resorb the bone [3]. In addition, RA patients develop systemic complications such as vasculitis and cardiovascular, pulmonary, skeletal, and psychological disorders. The etiology of RA remains elusive; however, there is evidence that both genetic and environmental factors are involved [4].
Unexpectedly, synovial hypoxia (defined as low oxygen partial pressure, pO2) is a constant feature of RA. Hypoxia can induce angiogenesis, inflammation, apoptosis, cartilage erosion, abnormal energy metabolism, and oxidative damage. Synovial hypoxia is thus considered a potential pathogenic factor in RA.
The aim of this review is to highlight and update our understanding of the potential roles of hypoxia in the pathogenesis of RA. We describe the evidence supporting the existence of hypoxia in the synovial tissue, its origin, and the potential connections between synovial hypoxia and the cells, soluble mediators, and processes currently considered relevant to the pathogenesis of RA.
Review
Hypoxia and signaling mediators
Atmospheric oxygen is extracted and transported to tissues via the blood. Under normal circumstances, the pO2 is not uniform across tissues but varies in the different body compartments. The pO2 can reach 100 mm Hg (16 % O2) in the pulmonary alveoli but decreases to ~40 mm Hg (6 % O2) in most tissues [5].
Tissue hypoxia occurs when there is an imbalance between the supply and demand for oxygen and can result in cell dysfunction and even death. Under hypoxic conditions, cells activate mechanisms to respond and adapt to this environment. One of the key regulators of the response is the transcription factor hypoxia-inducible factor (HIF) [6], which regulates genes related to angiogenesis, apoptosis, cell migration, vasomotor control, erythropoiesis, pH regulation, energy metabolism, and many other processes [7].
HIF was first detected in a nuclear extract from the human hepatoma cell line Hep3B [6] and was later described as a heterodimeric transcription factor composed of a HIF-1α subunit, which is regulated by oxygen levels, and a HIF-1β subunit, which is expressed constitutively [8]. Both subunits are members of a subfamily of factors with a basic helix-loop-helix (bHLH) domain and a Per, ARNT, and Sim (PAS) domain [9]. To date, three isoforms of the HIF-α subunit have been described—HIF-1α, HIF-2α, and HIF-3α—of which HIF-1α is the most studied. The three isoforms are characterized by the presence of bHLH, PAS, and oxygen-dependent degradation (ODD) domains. HIF-1α and HIF-2α share a number of structural and functional similarities, but HIF-1α is ubiquitously expressed, whereas HIF-2α is restricted to certain cell types and, in some cases, mediates different biological functions [10]. HIF-α protein levels are regulated at the stability and synthesis levels. Under normoxic conditions, HIF-α is modified by prolyl hydroxylases (PHD), which require ferrous iron (Fe2+) as an enzymatic cofactor and α-ketoglutarate and oxygen as substrates. The three forms of PHD (PHD-1, -2, and -3) differ in their affinities for the 1α, 2α, and 3α isoforms of HIF [11, 12]. PHD hydroxylates proline residues in the ODD domain of HIF-α [13]. The hydroxylated residues are subsequently recognized by the tumor suppressor von Hippel Lindau (vHL) protein, which recruits the Elongin-C–Elongin-B–Cullin 2–E3 ubiquitin ligase complex, leading to polyubiquitination and proteasomal degradation of HIF-α [14]. The activity of HIF-α is also regulated through hydroxylation of asparagine residues in its C-terminal transactivation domain by asparagyl β-hydroxylases (factor-inhibiting HIF), which are also dependent on oxygen, α-ketoglutarate and Fe2+. In this case, hydroxylation prevents HIF-α from interacting with its coactivator [15–17]. During hypoxia, these oxygen-dependent hydroxylation reactions are not performed, and HIF-α does not bind to vHL. HIF-α is thus stabilized and concentrates in the cytoplasm, subsequently translocating to the nucleus where it dimerizes with HIF-β. The HIF-αβ heterodimer binds to specific DNA sequences known as hypoxia response elements, and finally, recruitment of coactivators enables transcription of HIF-dependent genes (Fig. 1). Recent work has shown that PHD-2 is the major hydroxylase regulating HIF-α levels and the expression of angiogenic genes in fibroblasts derived from patients with RA [18].
Fig. 1.
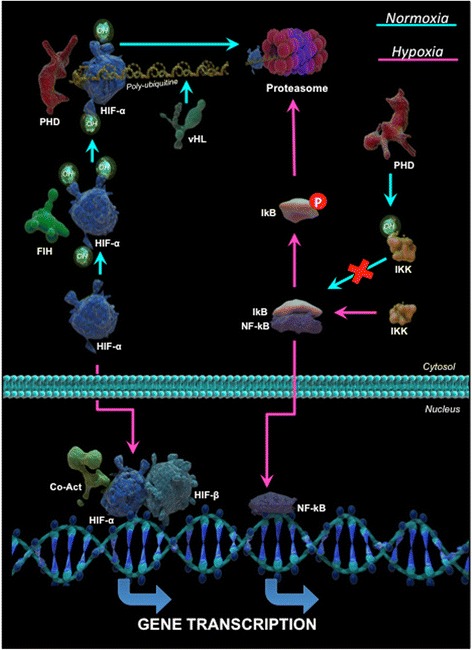
Hypoxia and signaling mediators. Representation of the signaling pathways mediated by NF-kB and HIF under normoxic and hypoxic conditions. Under normoxic conditions, HIF-α is hydroxylated by PHD and FIH enzymes to promote its ubiquitination and proteasomal degradation. During hypoxia, stabilized HIF-α relocates to the nucleus where it forms a dimer with HIF-β, recruits coactivators, and initiates transcription of hypoxia-regulated genes. NF-kB is inactive in the cytoplasm owing to its association with IkB. Activation of this pathway is regulated by IKK, which mediates the phosphorylation and degradation of IkB, allowing NF-kB to translocate to the nucleus where it activates gene transcription. PHD has been proposed to inhibit IKK under normoxic conditions. CoAct, coactivators; FIH, factor-inhibiting HIF (asparagyl β-hydroxylase); HIF, hypoxia-inducible factor; IkB, inhibitory protein; IKK, IkB kinase; NFkB, nuclear factor-kappa B; PHD, prolyl hydroxylase; vHL, von Hippel Lindau tumor suppressor
Although a role for HIF in RA pathogenesis has been inferred, the precise mechanisms by which this occurs remain to be defined. In the past, little was known about the contributions of the different HIF-α isoforms, but recent studies have shown that HIF-2α plays a fundamental role in RA independently of HIF-1α. HIF-2α is mainly overexpressed in fibroblasts, where it enhances their osteoclastogenic potential and regulates cell proliferation, expression of receptor activator of nuclear factor kB ligand, and induction of several catabolic factors [19]. The HIF-2α isoform is also involved in cartilage destruction. In a mouse arthritis model, HIF-2α–induced chemokine production by chondrocytes stimulated the migration and invasion of synovial fibroblasts, leading to cartilage erosion [20].
Hypoxia is not the only condition to activate and stabilize HIF-α. Physical factors such as heat and low pH, and biochemical factors such as hormones, cytokines, growth factors, and reactive oxygen species (ROS), may also induce and activate HIF-α. Bacterial lipopolysaccharides have been reported to induce HIF-1α activation in human monocytes and macrophages via nuclear factor-kappa B (NF-kB) and p44/42 mitogen-activated protein kinase (MAPK) pathways [21]. Under normoxic conditions, proinflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β can increase HIF expression at the mRNA and protein levels in synovial and gingival fibroblasts through the MAPK and phosphoinositide 3-kinase pathways [22, 23].
In addition to its effects on HIF-1α, hypoxia can activate signaling through members of the NF-kB family, which are considered the main proinflammatory transcription factors. NF-kB is overexpressed in RA synovial tissue [24] and regulates the expression of genes such as TNF-α, IL-6, IL-8, various chemokines, vascular endothelial growth factor (VEGF), and matrix metalloproteinase (MMP) 1, 3, and 13, which are associated with inflammation, angiogenesis, and tissue destruction [25]. NF-kB therefore acts in an additive and synergistic manner with HIF to contribute to the maintenance of the inflammatory response [26, 27]. In the classical pathway of NF-kB activation, NF-kB is retained in the cytoplasm by binding to the inhibitory protein IkB. Cell stimulation activates IkB kinase (IKK), which phosphorylates IkB and promotes its degradation. The release from IkB thus allows NF-kB to translocate to the nucleus where it binds to specific DNA sequences (kB sites) to activate transcription of genes appropriate to the activating stimulus. It has been proposed that PDHs hydroxylate and inhibit IKK activity under normoxic conditions [27], but under hypoxic conditions, the lack of hydroxylation permits IKK to phosphorylate and promote the degradation of IkB, leading to activation of NF-kB (Fig. 1).
Hypoxia in RA
Evidence of hypoxia in RA synovial tissue
Hypoxia is a feature of the synovial membrane in RA. This was first detected in 1970, when the oxygen tension in the synovial fluid of RA patients was found to be lower than that in healthy controls and osteoarthritis patients [28, 29]. Subsequently, tissue oximeters were used to confirm that hypoxia is a characteristic of RA synovial tissue [30, 31] and correlates with the intensity of the inflammatory process and cell migration [31].
Other studies have reported an inverse relationship between oxygen tension and synovial fluid volume [32] associated with acidosis, low glucose concentrations, high lactic acid concentrations [33, 34], and increased activity of some glycolytic enzymes, indicating that anaerobic metabolism is promoted as a survival mechanism within the hypoxic environment of the synovium [34].
Origins of hypoxia in RA
Two main theories have been proposed to explain hypoxia in RA synovial tissue. The first suggests that increased cell proliferation generates a high metabolic demand and increases the distance between proliferating cells and nearby blood vessels, which cannot meet the tissue oxygen requirements [35]. The second posits that the increased intraarticular pressure resulting from hyperplasia, synovial fluid effusion, and joint movements within the rigid joint capsule intermittently collapse the network of capillaries and therefore decrease the blood flow in the synovial tissue [36].
Another potential explanation for synovial hypoxia could be the enhanced expression of angiotensin converting enzyme (ACE), a membrane metalloprotease that catalyzes the formation of angiotensin II from its inactive precursor, angiotensin I. Angiotensin II is a potent vasoconstrictor and ACE is overexpressed in stromal cells in RA synovial tissue, generating local vasoconstriction and enhancing hypoxia [37].
Hypoxia might play a role not only in perpetuating synovial inflammation in RA but also in inducing it, as suggested by work from Jeon and colleagues [38] using a murine model of collagen-induced arthritis (CIA). Hypoxia levels were evaluated from the early stages of the disease and related to clinical aspects of the inflammatory process. Interestingly, in this model, hypoxia was present from the pre-arthritic stages. From the perspective of these findings, in which synovial hypoxia is detected before clinical onset of arthritis, the two theories mentioned earlier become insufficient to explain the origin of hypoxia in RA. It is likely that cell proliferation and intraarticular high pressure explain only the maintenance of the hypoxic state once the inflammatory process is established in the tissue. This highlights the importance of exploring the pre-arthritic phases in relation to the onset of the hypoxic state, for which signaling mediators have already been demonstrated in the arthritic phase.
Hypoxia, inflammation, and angiogenesis in RA
The first evidence that HIF-1α participates in the inflammatory process came from Cramer et al. [39], who showed that deletion of HIF-1α in macrophages reduces disease severity in different models of acute and chronic inflammation (including a passively induced arthritis model). Moreover, the bactericidal capacity of these macrophages was also decreased by HIF-1α deletion [39, 40].
HIF-1α is upregulated in RA fibroblasts [41], CD3+ T cells [42], and CD68+ synovial macrophages [43]. The latter cells have been shown to express several hypoxia-regulated genes in RA, including stromal cell-derived factor-1 (SDF-1), IL-8, VEGF, IL-1β, and TNF-α [44]. In addition, HIF-2α nuclear and cytoplasmic expression has also been demonstrated in the synovial lining and stromal cells of patients with RA and osteoarthritis [45].
SDF-1 is a potent angiogenic and chemotactic factor that is upregulated in synovial fibroblasts and promotes the recruitment of CXCR4+ ly mphocytes to the joints in RA. Expression of SDF-1 and VEGF in RA synovial tissue is hypoxia dependent [41], and CXCR4 expression can be induced by hypoxia. Celastrol, a triterpine compound with antioxidant and anti-inflammatory activity, inhibits hypoxia-induced migration and invasion of synovial fibroblasts via suppression of HIF-1α-mediated CXCR4 expression [46]. The increased survival of T cells under hypoxic conditions is another factor that contributes to their accumulation in the synovium [42, 47]. Additionally, HIF-1α overexpression is involved in the regulation of apoptosis in neutrophils [48, 49].
Hypoxia and IL-17 have a synergistic effect on the migration and invasion of synovial fibroblasts by increasing the expression of MMP2 and MMP9 via the NF-kB–HIF-1α pathway [50]. HIF-1α also promotes the activation of signaling pathways and controls IL-33 production by fibroblasts, which in turn induces expression of HIF-1α and generates a regulatory cycle that perpetuates inflammation in RA [51]. A recent study found evidence for a functional interaction during HIF-1α, Notch-3, and STAT-1 to regulate proinflammatory mechanisms in RA synovial fibroblasts during hypoxia [52].
Hypoxia is also associated with the differentiation of some synovial cells. Differentiation of Th0 lymphocytes towards Th17 cells, which are important for the development of autoimmune diseases such as RA, is associated with HIF-1α–regulated expression of the glucose transporter GLUT-1 [53]. In contrast, hypoxia inhibits the expression of several differentiation and maturation markers in dendritic cells, and reduces their ability to stimulate T cells and upregulate the production of proinflammatory cytokines such as TNF and IL-1β [54].
Toll-like receptors (TLRs) sense highly conserved structural motifs found in many molecules of bacterial and viral origin. Recognition of these molecules (e.g., bacterial lipoproteins, double-stranded RNA, and lipopolysaccharides) by TLRs upregulates costimulatory molecules and induces the production of proinflammatory and tissue-destructive mediators. Many cells within the joint express TLRs, and they can be activated by a variety of endogenous TLR ligands present within the inflamed RA joints. Current evidence suggests that TLR activation may contribute to the persistent expression of proinflammatory cytokines in RA through activation of NF-kB [55]. Stimulation of synovial fibroblast TLRs leads to increased expression of inflammatory cytokines, MMPs, and adhesion molecules. Moreover, induction of HIF-1α by hypoxia synergizes with TLR signaling to produce inflammatory cytokines (IL-6, IL-8, TNF-α), MMP-1, -3, -9, and VEGF, thereby exacerbating RA [56]. NFkB links TLR signaling to the hypoxic response through transcriptional regulation of HIF-1α, providing a likely explanation for these observations.
High-mobility group box 1 (HMGB-1), a non-histone nuclear protein, is an endogenous TLR ligand and has been detected in the synovial fibroblasts of RA patients. HMGB-1 stimulates the expression of VEGF, and this is attenuated by inhibition of HIF-1α. Similarly, treatment of CIA mice with a neutralizing anti-HMGB-1 prevents the formation of blood vessels, which is associated with a decrease in HIF-1α expression in the synovial tissue. This suggests that HMBG-1 participates in the process of HIF-1-mediated angiogenesis [57].
Stimulation of angiogenesis in hypoxic conditions is part of the cellular effort to restore oxygen delivery to the tissue. HIF-1α and HIF-2α isoforms are expressed in the RA synovium at levels related to the magnitude of the angiogenic response [58].
VEGF is one of the most potent and well-studied angiogenic stimuli, and VEGF mRNA levels are increased by HIF-1α during hypoxia. VEGF contributes to the initial vasodilation through induction of nitric oxide and increased endothelial cell permeability. In addition, VEGF prevents apoptosis of endothelial cells and stimulates their proliferation and migration [59]. Hypoxia also induces the expression of other angiogenic factors such as IL-8 [60], CCL20 [61], and SDF1 [41, 62].
NADPH oxidase enzyme-2 (NOX2) is overexpressed in the joints of RA patients and those of rats in the CIA model of arthritis. This enzyme generates superoxide by transferring electrons from NADPH inside the cell across the plasma membrane to molecular oxygen to produce superoxide anion, a reactive free radical. NOX-2 is overexpressed in RA joints in inverse proportion to the pO2. NOX-2 activators and 3 % hypoxia promote human microvascular endothelial cell migration, tubule formation, and secretion of proangiogenic mediators, suggesting that oxidative stress derived from the hypoxia-induced increase in NOX-2 activity is an initiating factor in angiogenesis [63]. The placental growth factors PlGF-1 and PlGF-2 are also hypoxia-inducible angiogenic growth factors and promote the proliferation, survival, migration, and invasion of RA synovial fibroblasts [64]. In a rat arthritis model, synovial angiogenesis was associated with overexpression of VEGF, CD34, and HIF-1α, and the levels of HIF-1α were positively correlated with the arthritis index [65] (Fig. 2).
Fig. 2.
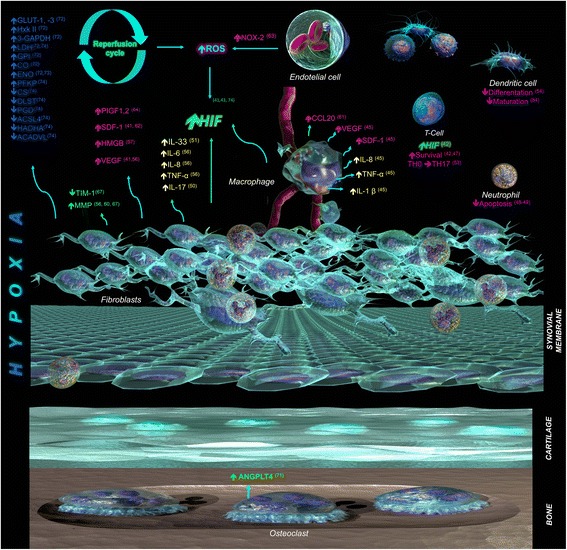
Direct and indirect effects of hypoxia and/or HIF on the different cell types contributing to RA pathogenesis. References relevant to the indicated phenomena are provided in the scheme. The colors of the molecules name indicate if the molecule has effect on: inflammation (yellow), oxidative stress (cyan), energy metabolism (blue), angiogenesis (magenta), destruction (green). ACADVL very long-chain acyl-CoA dehydrogenase; ACSL, long-chain fatty acid-CoA ligase; ANGPLT, angiopoietin-like; CCL20, chemokine (C-C motif) ligand 20; CO, mitochondrial cytochrome; CS, citrate synthase; DLST, oxoglutarate dehydrogenase complex component E2; ENO, enolase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GLUT, glucose transporter; GPI, glucose phosphate isomerase; HADHA, hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase; HIF, hypoxia-inducible factor; HMGB, high-mobility group protein B; Hxk, hexokinase; IL, interleukin; LDH, lactate dehydrogenase; MMP, matrix metalloproteinase; NOX, NADPH oxidase; PFKP phosphofructokinase; PGD, phosphogluconate dehydrogenase; PLGF, placental growth factor; ROS, reactive oxygen species; SDF, stromal cell-derived factor; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor
Although angiogenesis is increased in RA, the formation of new blood vessels fails to keep pace with the rapid thickening caused by synovial tissue hyperplasia. Moreover, the presence of immature blood vessels in the synovial tissue may explain the increased density of blood vessels and persistent hypoxia in the RA synovium [66].
Hypoxia and cartilage destruction in RA
The destruction of articular cartilage in RA is associated with increased activity of MMPs. These enzymes degrade extracellular matrix proteins, and their activity is regulated by endogenous tissue inhibitors of MMPs (TIMPs). MMP expression is elevated during repair and remodeling of damaged tissues. RA synovial fibroblasts exposed to hypoxia express increased levels of MMP-1 and MMP-3 and decreased levels of TIMP-1 at both the mRNA and protein levels [60, 67].
The MMPs promote remodeling of the extracellular matrix during angiogenesis, and since their expression is mainly induced by Ets-1, this transcription factor is considered to be involved in the invasion and destruction of cartilage and bone in RA [68]. Ets-1 is inducible by hypoxia [69] and co-localizes with HIF-1α in the synovial inflammatory infiltrate in an adjuvant-induced arthritis model in rats [70].
Angiopoietin-like 4 (ANGPTL4) stimulates bone resorption mediated by osteoclasts, which are the cells responsible for bone loss in RA. ANGPLT4 is overexpressed in RA osteoclasts in a HIF-1α–dependent manner. Other cell types in RA synovial tissue also overexpress ANGPLT4, suggesting that there are several sources of ANGPLT4 to promote bone resorption [71] (Fig. 2).
Hypoxia and energy metabolism in RA
Energy metabolism has recently become an important area of study in RA, and hypoxia is known to induce substantial metabolic changes in RA synovial tissue. Under aerobic conditions, the oxidation of one molecule of glucose to carbon dioxide (CO2) produces ~36 molecules of adenosine triphosphate (ATP), and this is the most efficient route of energy generation. However, anaerobic glycolysis is much less efficient and produces only two molecules of ATP during conversion of one molecule of glucose to lactate. Anaerobic glycolysis is relevant to conditions associated with a restricted oxygen supply, such as the hypoxia observed in RA joints. Indeed, the increased activity of some glycolytic enzymes, diminished glucose concentrations, and elevated lactate concentrations in RA synovial tissue indicate that anaerobic glycolysis is favored in this hypoxic environment.
Under hypoxic conditions, HIF-1α increases the expression of the glucose transporters GLUT1 and GLUT3 to enhance glucose uptake, and it regulates the expression of hexokinase II, glyceraldehyde 3-phosphate dehydrogenase, lactate dehydrogenase (LDH), and mitochondrial cytochrome oxidase to enhance glycolysis in the RA synovial tissue. HIF-1α also upregulates glucose phosphate isomerase, which together with enolase, aldolase, and triose phosphate isomerase, act as autoantigens [72]. Therefore, during its attempt to increase energy production for cell survival, HIF also generates antigenic targets and promotes autoimmunity. Moreover, upregulation of α-enolase in RA fibroblasts during hypoxia is associated with hyperproliferation and overexpression of anti-apoptotic proteins, such as Bcl-2, survivin, and cyclin B1 [73].
Interestingly, a recent study confirmed that concentrations of glucose and lactate are decreased and increased, respectively, in the synovial fluid of RA patients [74]. Levels of glucose-1-phosphate and D-mannose (glycolytic donors) were also decreased. In these patients, enzymes involved in anaerobic catabolism were upregulated (6-phosphofructokinase type C [PFKP] and LDH A), and enzymes involved in aerobic oxidation and fatty acid oxidation were downregulated (citrate synthase [CS], 2-oxoglutarate dehydrogenase complex component E2 [DLST], 6-phosphogluconate dehydrogenase [PGD], long-chain fatty acid-CoA ligase 4 [ACSL4], very long-chain acyl-CoA dehydrogenase [ACADVL], and hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase [HADHA]). CS and DLST are the rate-limiting enzymes of the tricarboxylic acid cycle; therefore, the significantly reduced expression of these enzymes together with the decreased levels of citric acid suggest that ATP production through aerobic oxidation is decreased in RA synovial fluid. Furthermore, expression of HIF-1α, PFKP, and LDHA is decreased and expression of CS, DLST, PGD, ACSL4, HADHA, and ACADVL is increased in synovial fibroblasts after HIF-1α knockdown. Collectively, these results suggest that HIF plays a significant role in regulating the metabolic flux (Fig. 2).
Protein citrullination may also be associated with hypoxia. α-Enolase and peptidylarginine deiminase type II, which are upregulated in RA fibroblasts under hypoxic conditions, have known links to citrullinated antigens. This link between hypoxia and citrullination has been described in astrocytes, suggesting that it might also exist in the RA synovium [75].
Hypoxia and reactive oxygen species in RA
Synovial perfusion is directly affected by the high intraarticular pressure prevailing in the synovial tissue of RA. This pressure may be further increased by movement, which contributes to local tissue hypoxia followed by reoxygenation when the joint is unloaded. These repeated cycles of hypoxia–reoxygenation lead to increased levels ROS and consequently to NF-kB activation [76]. Experimental evidence suggests that ROS produced by the NOX system maintains HIF-1α in an inactive state. Under hypoxic conditions, insufficient amounts of ROS are formed and HIF-1α is activated [77]. In contrast, ROS produced by mitochondria during hypoxia have a stabilizing effect on HIF-1α [78, 79]. Although ROS have several physiological functions in the cell, an imbalance between oxidant and antioxidant activity can lead to a state of oxidative stress. Such stress affects many cellular components, including DNA, lipids, and proteins, causing changes in their structures and functions, as is observed in RA (Fig. 2).
Conclusions
The pathogenesis of RA is incompletely understood. Although inflammatory and autoimmune phenomena are believed to be the central abnormalities, the precise etiology remains elusive. Synovial hypoxia is a consistent finding and can be linked to several pathogenic processes through direct and indirect effects on angiogenesis, cartilage damage, bone resorption, oxidative damage, and inflammation. It should be noted, however, that hypoxia is not the only promoter of inflammation in RA, and it can be induced and perpetuated by diverse stimuli. Nevertheless, the induction of an anaerobic glycolytic phenotype in the synovium is specifically induced by hypoxia. Once anaerobic glycolysis is established in the synovium, several of the enzymes induced by this metabolic shift may become antigenic. Hypoxia modifies the metabolic environment in the synovium, but in the context of cellular stress, presentation of the upregulated antigenic enzymes could initiate an autoimmune response. For example, glucose phosphate isomerase, a glycolytic enzyme induced by hypoxia, is autoantigenic. The possible involvement of hypoxia in the citrullination process has also been proposed.
Overall, hypoxia is an interesting condition with effects that reflect a complex mechanism of action in RA. This requires further study, especially in the pre-arthritic stages when hypoxia is already present, according to the results of Jeon and colleagues. These findings provide new perspectives to understand the pathogenesis of RA and to identify new therapeutic targets.
Acknowledgments
The authors express their gratitude to Consejo Nacional de Ciencia y Tecnología (CONACYT) for financial support to CMQF, student enrolled in a PhD program during the preparation of this review.
Funding
Not applicable.
Availability of data and material
Not applicable.
Authors’ contributions
CMQF prepared the manuscript, collected the information, prepared the figures, and provided overall supervision. SAGC prepared the manuscript and collected the information. CPT prepared the figures and provided overall supervision. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ACADVL
Very long acyl-CoA dehydrogenase
- ACE
Angiotensin converting enzyme
- ACSL
Long chain fatty acid-CoA ligase
- ANGPTL
Angiopoietin-like
- ATP
Adenosine triphosphate
- bHLH
Basic helix-loop-helix
- CIA
Collagen-induced arthritis
- CO
Mitochondrial cytochrome
- CO2
Carbon dioxide
- CS
Citrate synthase
- DLST
Oxoglutarate dehydrogenase complex component E2
- ENO
Enolase
- Fe2+
Ferrous iron
- FIH
Factor-inhibiting HIF
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GLUT
Glucose transporter
- GPI
Glucose phosphate isomerase
- HADHA
Hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase
- HIF
Hypoxia inducible factor
- HMGB
High-mobility group box
- Hxk
Hexokinase
- IKK
IkB kinase
- IL
Interleukin
- LDH
Lactate dehydrogenase
- MAPK
Mitogen-activated protein kinase
- MMP
Metalloproteinase
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NF-kB
Nuclear factor-kappa B
- NOX
NADPH oxidase enzyme
- ODD
Oxygen-dependent degradation
- PAS
Per, ARNT and Sim
- PFKP
Phosphofructokinase
- PGD
Phosphugluconate dehydrogenase
- PHD
Prolyl hydroxylases
- PIGF
Placental growth factor
- pO2
Oxygen partial pressure
- RA
Rheumatoid Arthritis
- ROS
Reactive oxygen species
- SDF
Stromal cell-derived factor
- TIM
Tissue inhibitors of metalloproteinases
- TLR
Toll-like receptors
- TNF
Tumor necrosis factor
- VEGF
Vascular endotelial growth factor
- vHL
von Hippel Lindau
Contributor Information
Celia María Quiñonez-Flores, Email: celiamaria07@hotmail.com.
Susana Aideé González-Chávez, Email: susanaglezch@gmail.com.
César Pacheco-Tena, Email: dr.cesarpacheco@gmail.com.
References
- 1.Gibofsky A. Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis. Am J Manag Care. 2012;18:S295–302. [PubMed] [Google Scholar]
- 2.Moreno-Montoya J, Alvarez-Nemegyei J, Sanin LH, Pérez-Barbosa L, Trejo-Valdivia B, Santana N, et al. Association of regional and cultural factors with the prevalence of rheumatoid arthritis in the mexican population: a multilevel analysis. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. 2015;21:57–62. doi: 10.1097/RHU.0000000000000223. [DOI] [PubMed] [Google Scholar]
- 3.Izquierdo E, Pablos JL. Fibroblastos sinoviales. Semin Fund Esp Reumatol. 2013;14:121–8. doi: 10.1016/j.semreu.2013.06.001. [DOI] [Google Scholar]
- 4.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/MCB.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19:12–6. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 9.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 2008;15:628–34. doi: 10.1038/cdd.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–90. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Appelhoff RJ, Tian Y-M, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–65. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 13.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 14.Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 18.Muz B, Larsen H, Madden L, Kiriakidis S, Paleolog EM. Prolyl hydroxylase domain enzyme 2 is the major player in regulating hypoxic responses in rheumatoid arthritis. Arthritis Rheum. 2012;64:2856–67. doi: 10.1002/art.34479. [DOI] [PubMed] [Google Scholar]
- 19.Ryu J-H, Chae C-S, Kwak J-S, Oh H, Shin Y, Huh YH, et al. Hypoxia-inducible factor-2α is an essential catabolic regulator of inflammatory rheumatoid arthritis. PLoS Biol. 2014;12:e1001881. doi: 10.1371/journal.pbio.1001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huh YH, Lee G, Lee K-B, Koh J-T, Chun J-S, Ryu J-H. HIF-2α-induced chemokines stimulate motility of fibroblast-like synoviocytes and chondrocytes into the cartilage-pannus interface in experimental rheumatoid arthritis mouse models. Arthritis Res Ther. 2015;17:302. doi: 10.1186/s13075-015-0816-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006;396:517–27. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westra J, Brouwer E, Bos R, Posthumus MD, Doornbos-van der Meer B, Kallenberg CGM, et al. Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann N Y Acad Sci. 2007;1108:340–8. doi: 10.1196/annals.1422.035. [DOI] [PubMed] [Google Scholar]
- 23.Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E. Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J. 2000;350(Pt 1):307–12. doi: 10.1042/bj3500307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benito MJ, Murphy E, Murphy EP, van den Berg WB, FitzGerald O, Bresnihan B. Increased synovial tissue NF-kappa B1 expression at sites adjacent to the cartilage-pannus junction in rheumatoid arthritis. Arthritis Rheum. 2004;50:1781–7. doi: 10.1002/art.20260. [DOI] [PubMed] [Google Scholar]
- 25.Simmonds RE, Foxwell BM. Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatol Oxf Engl. 2008;47:584–90. doi: 10.1093/rheumatology/kem298. [DOI] [PubMed] [Google Scholar]
- 26.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–9. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliver KM, Taylor CT, Cummins EP. Hypoxia. Regulation of NFkB signalling during inflammation: the role of hydroxylases. Arthritis Res Ther. 2009;11:215. doi: 10.1186/ar2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund-Olesen K. Oxygen tension in synovial fluids. Arthritis Rheum. 1970;13:769–76. doi: 10.1002/art.1780130606. [DOI] [PubMed] [Google Scholar]
- 29.Falchuk KH, Goetzl EJ, Kulka JP. Respiratory gases of synovial fluids. An approach to synovial tissue circulatory-metabolic imbalance in rheumatoid arthritis. Am J Med. 1970;49:223–31. doi: 10.1016/S0002-9343(70)80078-X. [DOI] [PubMed] [Google Scholar]
- 30.Sivakumar B, Akhavani MA, Winlove CP, Taylor PC, Paleolog EM, Kang N. Synovial hypoxia as a cause of tendon rupture in rheumatoid arthritis. J Hand Surg. 2008;33:49–58. doi: 10.1016/j.jhsa.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Ng CT, Biniecka M, Kennedy A, McCormick J, Fitzgerald O, Bresnihan B, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheum Dis. 2010;69:1389–95. doi: 10.1136/ard.2009.119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richman AI, Su EY, Ho G. Reciprocal relationship of synovial fluid volume and oxygen tension. Arthritis Rheum. 1981;24:701–5. doi: 10.1002/art.1780240512. [DOI] [PubMed] [Google Scholar]
- 33.Treuhaft PS, MCCarty DJ. Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis Rheum. 1971;14:475–84. doi: 10.1002/art.1780140407. [DOI] [PubMed] [Google Scholar]
- 34.Naughton D, Whelan M, Smith EC, Williams R, Blake DR, Grootveld M. An investigation of the abnormal metabolic status of synovial fluid from patients with rheumatoid arthritis by high field proton nuclear magnetic resonance spectroscopy. FEBS Lett. 1993;317:135–8. doi: 10.1016/0014-5793(93)81508-W. [DOI] [PubMed] [Google Scholar]
- 35.Larsen H, Akhavani M, Raatz Y, Paleolog E. Gene expression studies to investigate disease mechanisms in rheumatoid arthritis: does angiogenesis play a role? Curr Rheumatol Rev. 2007;3:243–51. doi: 10.2174/157339707782408991. [DOI] [Google Scholar]
- 36.Jawed S, Gaffney K, Blake DR. Intra-articular pressure profile of the knee joint in a spectrum of inflammatory arthropathies. Ann Rheum Dis. 1997;56:686–9. doi: 10.1136/ard.56.11.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh DA, Catravas J, Wharton J. Angiotensin converting enzyme in human synovium: increased stromal [(125)I]351A binding in rheumatoid arthritis. Ann Rheum Dis. 2000;59:125–31. doi: 10.1136/ard.59.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeon CH, Ahn J-K, Chai J-Y, Kim HJ, Bae E-K, Park SH, et al. Hypoxia appears at pre-arthritic stage and shows co-localization with early synovial inflammation in collagen induced arthritis. Clin Exp Rheumatol. 2008;26:646–8. [PubMed] [Google Scholar]
- 39.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–15. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46:2587–97. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 42.Makino Y, Nakamura H, Ikeda E, Ohnuma K, Yamauchi K, Yabe Y, et al. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol Baltim Md 1950. 2003;171:6534–40. doi: 10.4049/jimmunol.171.12.6534. [DOI] [PubMed] [Google Scholar]
- 43.Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001;44:1540–4. doi: 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 44.Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol Baltim Md 1950. 2005;175:6257–63. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- 45.Giatromanolaki A, Sivridis E, Maltezos E, Athanassou N, Papazoglou D, Gatter KC, et al. Upregulated hypoxia inducible factor-1alpha and -2alpha pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2003;5:R193–201. doi: 10.1186/ar756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li G, Liu D, Zhang Y, Qian Y, Zhu Y, Guo S, et al. Anti-invasive effects of celastrol in hypoxia-induced fibroblast-like synoviocyte through suppressing of HIF-1α/CXCR4 signaling pathway. Int Immunopharmacol. 2013;17:1028–36. doi: 10.1016/j.intimp.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, et al. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol Baltim Md 1950. 2005;174:7592–9. doi: 10.4049/jimmunol.174.12.7592. [DOI] [PubMed] [Google Scholar]
- 48.Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, et al. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 2002;100:3008–16. doi: 10.1182/blood-2002-02-0454. [DOI] [PubMed] [Google Scholar]
- 49.Vissers MCM, Wilkie RP. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1alpha. J Leukoc Biol. 2007;81:1236–44. doi: 10.1189/jlb.0806541. [DOI] [PubMed] [Google Scholar]
- 50.Li G, Zhang Y, Qian Y, Zhang H, Guo S, Sunagawa M, et al. Interleukin-17A promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing MMP2 and MMP9 expression through NF-kB/HIF-1α pathway. Mol Immunol. 2013;53:227–36. doi: 10.1016/j.molimm.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 51.Hu F, Shi L, Mu R, Zhu J, Li Y, Ma X, et al. Hypoxia-inducible factor-1α and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PLoS One. 2013;8:e72650. doi: 10.1371/journal.pone.0072650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis. 2014;74:1275-83 [DOI] [PubMed]
- 53.Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen I-H, et al. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008;112:3723–34. doi: 10.1182/blood-2008-02-142091. [DOI] [PubMed] [Google Scholar]
- 55.Huang Q-Q, Pope RM. The role of toll-like receptors in rheumatoid arthritis. Curr Rheumatol Rep. 2009;11:357–64. doi: 10.1007/s11926-009-0051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu F, Mu R, Zhu J, Shi L, Li Y, Liu X, et al. Hypoxia and hypoxia-inducible factor-1α provoke toll-like receptor signalling-induced inflammation in rheumatoid arthritis. Ann Rheum Dis. 2014;73:928–36. doi: 10.1136/annrheumdis-2012-202444. [DOI] [PubMed] [Google Scholar]
- 57.Park SY, Lee SW, Kim HY, Lee WS, Hong KW, Kim CD. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1α activation. Eur J Immunol. 2014 [DOI] [PubMed]
- 58.Brouwer E, Gouw ASH, Posthumus MD, van Leeuwen MA, Boerboom AL, Bijzet J, et al. Hypoxia inducible factor-1-alpha (HIF-1alpha) is related to both angiogenesis and inflammation in rheumatoid arthritis. Clin Exp Rheumatol. 2009;27:945–51. [PubMed] [Google Scholar]
- 59.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahn JK, Koh E-M, Cha H-S, Lee YS, Kim J, Bae E-K, et al. Role of hypoxia-inducible factor-1alpha in hypoxia-induced expressions of IL-8, MMP-1 and MMP-3 in rheumatoid fibroblast-like synoviocytes. Rheumatol Oxf Engl. 2008;47:834–9. doi: 10.1093/rheumatology/ken086. [DOI] [PubMed] [Google Scholar]
- 61.Bosco MC, Delfino S, Ferlito F, Battaglia F, Puppo M, Gregorio A, et al. Hypoxic synovial environment and expression of macrophage inflammatory protein 3gamma/CCL20 in juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1833–8. doi: 10.1002/art.23516. [DOI] [PubMed] [Google Scholar]
- 62.Santiago B, Calonge E, Del Rey MJ, Gutierrez-Cañas I, Izquierdo E, Usategui A, et al. CXCL12 gene expression is upregulated by hypoxia and growth arrest but not by inflammatory cytokines in rheumatoid synovial fibroblasts. Cytokine. 2011;53:184–90. doi: 10.1016/j.cyto.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Biniecka M, Connolly M, Gao W, Ng CT, Balogh E, Gogarty M, et al. Redox-mediated angiogenesis in the hypoxic joint of inflammatory arthritis. Arthritis Rheumatol Hoboken NJ. 2014;66:3300–10. doi: 10.1002/art.38822. [DOI] [PubMed] [Google Scholar]
- 64.Yoo S-A, Park J-H, Hwang S-H, Oh S-M, Lee S, Cicatiello V, et al. Placental growth factor-1 and -2 induce hyperplasia and invasiveness of primary rheumatoid synoviocytes. J Immunol Baltim Md 1950. 2015;194:2513–21. doi: 10.4049/jimmunol.1402900. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X, Liu J, Wan L, Sun Y, Wang F, Qi Y, et al. [Up-regulated expressions of HIF-1α, VEGF and CD34 promote synovial angiogenesis in rats with adjuvant arthritis]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi Chin. J Cell Mol Immunol. 2015;31:1053–6. [PubMed] [Google Scholar]
- 66.Kennedy A, Ng CT, Biniecka M, Saber T, Taylor C, O’Sullivan J, et al. Angiogenesis and blood vessel stability in inflammatory arthritis. Arthritis Rheum. 2010;62:711–21. doi: 10.1002/art.27287. [DOI] [PubMed] [Google Scholar]
- 67.Cha HS, Ahn KS, Jeon CH, Kim J, Song YW, Koh EM. Influence of hypoxia on the expression of matrix metalloproteinase-1, -3 and tissue inhibitor of metalloproteinase-1 in rheumatoid synovial fibroblasts. Clin Exp Rheumatol. 2003;21:593–8. [PubMed] [Google Scholar]
- 68.Behrens P, Mathiak M, Mangold E, Kirdorf S, Wellmann A, Fogt F, et al. Stromal expression of invasion-promoting, matrix-degrading proteases MMP-1 and -9 and the Ets 1 transcription factor in HNPCC carcinomas and sporadic colorectal cancers. Int J Cancer. 2003;107:183–8. doi: 10.1002/ijc.11336. [DOI] [PubMed] [Google Scholar]
- 69.Oikawa M, Abe M, Kurosawa H, Hida W, Shirato K, Sato Y. Hypoxia induces transcription factor ETS-1 via the activity of hypoxia-inducible factor-1. Biochem Biophys Res Commun. 2001;289:39–43. doi: 10.1006/bbrc.2001.5927. [DOI] [PubMed] [Google Scholar]
- 70.Peters CL, Morris CJ, Mapp PI, Blake DR, Lewis CE, Winrow VR. The transcription factors hypoxia-inducible factor 1alpha and Ets-1 colocalize in the hypoxic synovium of inflamed joints in adjuvant-induced arthritis. Arthritis Rheum. 2004;50:291–6. doi: 10.1002/art.11473. [DOI] [PubMed] [Google Scholar]
- 71.Swales C, Athanasou NA, Knowles HJ. Angiopoietin-Like 4 Is Over-Expressed in Rheumatoid Arthritis Patients: Association with Pathological Bone Resorption. PLoS One. 2014;9:e109524. doi: 10.1371/journal.pone.0109524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang X, Wei C. Glycolysis and rheumatoid arthritis. Int J Rheum Dis. 2011;14:217–22. doi: 10.1111/j.1756-185X.2011.01598.x. [DOI] [PubMed] [Google Scholar]
- 73.Fan S, Zong M, Zhang H, Lu Y, Lu T, Fan L. Decreased expression of alpha-enolase inhibits the proliferation of hypoxia-induced rheumatoid arthritis fibroblasts-like synoviocytes. Mod Rheumatol Jpn Rheum Assoc. 2015;25:701–7. [DOI] [PubMed]
- 74.Yang XY, Zheng KD, Lin K, Zheng G, Zou H, Wang JM, et al. Energy metabolism disorder as a contributing factor of rheumatoid arthritis: a comparative proteomic and metabolomic study. PLoS One. 2015;10:e0132695. doi: 10.1371/journal.pone.0132695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pacheco-Tena C, González-Chávez SA. The danger model approach to the pathogenesis of the rheumatic diseases. J Immunol Res. 2015;2015:506089. doi: 10.1155/2015/506089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bonizzi G, Piette J, Merville MP, Bours V. Cell type-specific role for reactive oxygen species in nuclear factor-kappaB activation by interleukin-1. Biochem Pharmacol. 2000;59:7–11. doi: 10.1016/S0006-2952(99)00290-7. [DOI] [PubMed] [Google Scholar]
- 77.Huang LE, Willmore WG, Gu J, Goldberg MA, Bunn HF. Inhibition of hypoxia-inducible factor 1 activation by carbon monoxide and nitric oxide. Implications for oxygen sensing and signaling. J Biol Chem. 1999;274:9038–44. doi: 10.1074/jbc.274.13.9038. [DOI] [PubMed] [Google Scholar]
- 78.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 79.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]