

Abstract
α-Synuclein (α-Syn), one of the most abundant proteins in the CNS, is known to be a major player in the neurodegeneration observed in Parkinson's disease. We currently report that transient focal ischemia upregulates α-Syn protein expression and nuclear translocation in neurons of the adult rodent brain. We further show that knockdown or knock-out of α-Syn significantly decreases the infarction and promotes better neurological recovery in rodents subjected to focal ischemia. Furthermore, α-Syn knockdown significantly reduced postischemic induction of phospho-Drp1, 3-nitrotyrosine, cleaved caspase-3, and LC-3 II/I, indicating its role in modulating mitochondrial fragmentation, oxidative stress, apoptosis, and autophagy, which are known to mediate poststroke neuronal death. Transient focal ischemia also significantly upregulated serine-129 (S129) phosphorylation (pα-Syn) of α-Syn and nuclear translocation of pα-Syn. Furthermore, knock-out mice that lack PLK2 (the predominant kinase that mediates S129 phosphorylation) showed better functional recovery and smaller infarcts when subjected to transient focal ischemia, indicating a detrimental role of S129 phosphorylation of α-Syn. In conclusion, our studies indicate that α-Syn is a potential therapeutic target to minimize poststroke brain damage.
SIGNIFICANCE STATEMENT Abnormal aggregation of α-synuclein (α-Syn) has been known to cause Parkinson's disease and other chronic synucleinopathies. However, even though α-Syn is linked to pathophysiological mechanisms similar to those that produce acute neurodenegerative disorders, such as stroke, the role of α-Syn in such disorder is not clear. We presently studied whether α-Syn mediates poststroke brain damage and more importantly whether preventing α-Syn expression is neuroprotective and leads to better physiological and functional outcome after stroke. Our study indicates that α-Syn is a potential therapeutic target for stroke therapy.
Keywords: α-synuclein, brain, neuroprotection, stroke
Introduction
α-Synuclein (α-Syn) is a 14.5 kDa protein that is predominantly localized in the presynaptic terminals of the mammalian brain (Takeda et al., 2006). Physiological functions of α-Syn are not completely understood, but at elevated levels, it is known to participate in the neurodegeneration associated with Parkinson's disease (PD) and other synucleinopathies (Stoica et al., 2012; Lashuel et al., 2013). The α-Syn monomer has an amphiphatic N terminal, a hydrophobic central region (nonamyloid component), and an acidic C terminal (Oueslati et al., 2010). Under native conditions, α-Syn is known to exist as a helically folded tetramer that resists aggregation (Bartels et al., 2011). But inactivation of N and C terminals due to post-translational modifications disrupts stability of the tetrameric structure and leads to oligomerization and aggregation of α-Syn, which are thought to be responsible for its toxicity; however, increased levels of α-Syn monomers also promote neuronal death (Diógenes et al., 2012; Stoica et al., 2012).
Several mechanisms proposed for α-Syn-induced neuronal death in PD include inflammation, oxidative stress, mitochondrial fragmentation, and autophagy (Pacheco et al., 2012; Dias et al., 2013). Interestingly, all of these pathophysiologic mechanisms also mediate poststroke neuronal death (Kim and Vemuganti, 2015; Lopez et al., 2015; Nakka et al., 2016). A recent study showed that α-Syn transgenic mice are more susceptible to ischemic brain damage (Unal-Cevik et al., 2011). However, these mice already have high levels of α-Syn from birth and so might not shed light on the significance of poststroke induction of α-Syn. Hence, we presently evaluated the role of α-Syn in postischemic brain damage using siRNA-mediated knockdown and α-Syn knock-out mice (incapable of inducing α-Syn after stroke). We further evaluated effects of α-Syn on poststroke expression of proteins that control mitochondrial fragmentation, autophagy, apoptosis, and oxidative stress.
α-Syn can undergo several post-translational modifications. Of those, phosphorylation of the serine-129 (S129) residue in the C terminus that is highly conserved is thought to be a defining pathological marker of PD and other synucleinopathies (Schmid et al., 2013). Under normal conditions, α-Syn predominantly exists in its unphosphorylated form, but oxidative stress and proteasomal dysfunction due to pathologies promote S129 phosphorylation, which is thought to modulate α-Syn oligomerization and aggregation leading to toxicity (Lee and Trojanowski, 2006; Lashuel et al., 2013). While many kinases can mediate phosphorylation of α-Syn, Polo-like kinases (PLK1, 2, and 3) are thought to be the major players (Tenreiro et al., 2014). Of those, PLK2 was shown to have an exclusive preference for the α-Syn S129 site. More importantly PLK2, but not other kinases, mediates quantitative conversion (>95%) of α-Syn to phosphorylated α-Syn (pα-Syn; Inglis et al., 2009; Salvi et al., 2012). PLK2 colocalizes with S129-pα-Syn in the cortical and hippocampal neurons of α-Syn transgenic mice (Mbefo et al., 2010), and mediates the phosphorylation of α-Syn (Waxman and Giasson, 2011). PLK2 directly binds to α-Syn, and PLK2-mediated S129 phosphorylation has been shown to regulate α-Syn turnover and autophagy-mediated disposal in an in vivo model of PD (Oueslati et al., 2010). Furthermore, plasmid-mediated overexpression of human α-Syn S129D form (phosphomimic) accelerates, while α-Syn S129A form (nonphosphorylatable) slows down, the dopaminergic neural fiber loss in the rat brain (Febbraro et al., 2013). Hence, to identify the functional role of S129 phosphorylation of α-Syn in poststroke brain damage, we evaluated effects of focal ischemia in PLK2 knock-out mice.
Materials and Methods
All experimental protocols using animals were approved by the University of Wisconsin Research Animal Resources and Care Committee. Care of animals followed the Guide for the Care and Use of Laboratory Animals, U.S. Department of Health and Human Services Publication Number 86-23 (revised). In all experiments, animals were randomly assigned to groups. Behavioral and histological analyses were performed by an investigator blinded to the study groups. Experiments were conducted in compliance with the “Animal Research: Reporting of In Vivo Experiments (ARRIVE)” guidelines.
In vitro ischemia.
Rat pheochromocytoma (PC12) cells (ATCC, CRL-1721.1) were grown in DMEM (Invitrogen) supplemented with 4.5 mg/ml glucose, 5% bovine calf serum, and 15% equine serum and then differentiated into neuronal phenotype by treating with NGF. On the day of the experiment, cells were washed twice with oxygen-glucose deprivation (OGD) buffer containing glucose-free isotonic salt solution (Invitrogen), and incubated in fresh OGD buffer for 6 h in an OGD chamber that was continuously infused with gas mixture containing 94% nitrogen, 5% carbon dioxide, and <1% oxygen at 37°C. The cells were then transferred to normoglycemic medium and incubated under normoxic conditions for 24 h at 37°C. Cells incubated under normoxic conditions in OGD buffer supplemented with 4.5 mg/ml glucose for 6 h followed by incubation in normal medium for 24 h at 37°C were used as control. In some experiments, PC12 cells (1 × 106 cells/well) were transfected with α-Syn siRNA or control siRNA (50 nm; Ambion) using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. The specificity of α-Syn siRNA was validated by the Silencer Select Algorithm (Ambion) and BLAST alignment. Each transfection was conducted in triplicate and each experiment was repeated four times. After 24 h of transfection, cells were either subjected to 6 h of OGD and 24 h of reoxygenation, or incubated under normoxic conditions with glucose (control). Cell viability was analyzed by trypan blue exclusion assay as per the manufacturer's instructions. Briefly, 100 μl of 0.4% trypan blue stock solution (Invitrogen) was added to 1 ml of cell suspension. Then the trypan blue-stained and total cells were counted using a hemocytometer (Hausser Scientific). The number of viable cells were calculated using the following formula: viable cell counts/quadrants counted × dilution factor × 10,000 (hemocytometer factor) × volume. Cell counting was performed by an investigator blinded to the study groups.
Animals.
Adult, male, spontaneous hypertensive rats (3 months old; 250–275 g) were obtained from Charles River. α-Syn−/− (C57BL/6N-Sncatm1Mjff/J) and PLK2−/− (129S.B6N-Plk2tm1Elan/J) mice (3 months old; 25–28 g) and the respective wild-type controls were obtained from Jackson Labs.
In vivo ischemia.
Under isoflurane anesthesia, the middle cerebral artery was occluded for 60 min (rats) or 90 min (mice) using silicone-coated nylon monofilaments (4-0 for rats and 6-0 for mice; Doccol) followed by various periods of reperfusion as described previously (Dharap et al., 2011, 2012). Sham-operated animals served as control. Regional cerebral blood flow and physiological parameters (pH, PaO2, PaCO2, hemoglobin, and blood glucose) were monitored, and rectal temperature was maintained at 37.0 ± 0.5°C during surgery. At 1 h of reperfusion, animals were tested with neurological severity scoring where a score of 0 suggests no neurological deficit (normal), 1 being mild neurological deficit (failure to fully extend right forepaw), 2 being moderate neurological deficit (circling to the right), 3 being severe neurological deficit (falling to the right), and 4 being very severe neurological deficit (no spontaneous movement). Rodents with no evidence of neurological deficit (score 0) were excluded. After death, rodents that showed hemorrhage were also excluded from analyses. Adult rats were injected intracerebrally with a mixture of three in vivo grade Silencer Select siRNAs targeting nonoverlapping regions of α-Syn mRNA and nontargeting negative control siRNAs (Life Technologies) at either 2 h before middle cerebral artery occlusion (MCAO) or at 30 min of reperfusion after MCAO, as described previously (Satriotomo et al., 2006; Pandi et al., 2013). In brief, siRNA mixture (7 nmol in 6 μl of buffer) was mixed with 2 μl of invivofectamine, incubated at room temperature for 15 min, and injected stereotaxically (1 μl/5 min) into the cerebral cortex (bregma: −0.2 mm posterior, 3 mm dorsoventral, 4.5 mm lateral; Paxinos and Watson, 1998).
Motor-function and infarct-volume determination.
Postischemic motor function was evaluated by the rotarod test (4 min on a cylinder rotating at 8 rpm), the beam-walk test (number of foot faults while crossing a tapered 120-cm-long beam for rats and a 5 mm × 60 cm beam for mice), and the adhesive-removal test (time taken to remove a small adhesive sticker placed on each forepaw) at days 1, 3, 5, and 7 of reperfusion as described previously (Schaar et al., 2010; Nakka et al., 2011). On day 7, animals were killed by transcardiac 4% phosphate-buffered paraformaldehyde (PFA) perfusion. Each brain was postfixed, cryoprotected, and sectioned (coronal; 30 μm thickness at an interval of 320 μm). Serial sections were stained with cresyl violet and scanned using the National Institutes of Health ImageJ software. Ischemic infarct volume was estimated by numeric integration of data from five serial coronal sections in respect to the sectional interval as described previously (Nakka et al., 2011; Mehta et al., 2015). Each infarct volume was corrected to account for edema and differential shrinkage during tissue processing using the Swanson formula (Swanson et al., 1990).
Western blotting.
Cells or brain tissue were homogenized in ice-cold 25 mm Tris-HCl buffer, pH 7.4, containing 2 mm EDTA and a protease inhibitor mixture (Sigma Chemical). Homogenates were centrifuged at 1500 × g for 10 min at 4°C. In some experiments, subcellular fractionation was performed using the NE-PER Nuclear and Cytoplasmic Extraction Kit (ThermoFisher Scientific) as per manufacturer's instructions. Proteins were solubilized by adding Laemmli electrophoresis sample buffer (5% SDS, 20% glycerol, 10% 2-mercaptoethanol, 125 mm Tris-HCl, pH 6.8, and 0.004% bromophenol blue; Sigma-Aldrich) and denatured by heating at 94°C for 5 min. Samples (40 μg of protein equivalent) were electrophoresed, transferred to PVDF membranes and probed with antibodies against α-Syn (1:1500; monoclonal; Cell Signaling Technology), S129 pα-Syn (1:1000; polyclonal; GeneTex), dynamin-related protein-1 (Drp1; 1:1000; monoclonal; Cell Signaling Technology), phosphorylated Drp1 (pDrp1; 1:1000; monoclonal; Cell Signaling Technology), cleaved caspase-3 (1:1000; monoclonal; Cell Signaling Technology), 3-nitrotyrosine (3-NT; 1:1400; monoclonal; Abcam), and LC-3 (1:1000; monoclonal; Cell Signaling Technology), followed by HRP-conjugated anti-rabbit or anti-mouse IgG (1:5000; Cell Signaling Technology). Blots were stripped and reprobed with antibodies against β-actin (1:3000; monoclonal; Cell Signaling Technology) or GAPDH (1:1000; monoclonal; Santa Cruz Biotechnology). Blots were developed using enhanced chemiluminescence and quantified with Image Studio software (LI-COR Biotechnology). For identifying α-Syn oligomers, in vivo crosslinking was used as described previously with slight modifications (Dettmer et al., 2013). Briefly, homogenates were incubated with 1 mm disuccinimidyl glutarate in DMSO for 30 min at 37°C with rotation. The reaction was quenched with the addition of 1 m Tris, pH 7.6, for 15 min at room temperature. Samples were briefly sonicated and incubated on ice for 10 min with addition of 1% Triton X-100 (v/v) followed by centrifugation for 1 h at 108,800 × g at 4°C. Supernatants were mixed with the NuPage LDS sample buffer (Invitrogen) without any reducing agent, electrophoresed, and blotted onto PVDF membranes, which were treated with 0.4% PFA (30 min at room temperature), before blocking with 5% BSA in TBS-T and probed with an α-Syn antibody (1: 1500; monoclonal; Cell Signaling Technology).
Immunostaining.
PC12 cells were fixed with 4% PFA, permeabilized, blocked with 0.3% Triton X-100, and immunostained with an antibody against α-Syn (1:50; polyclonal; Cell Signaling Technology). The fluorescence intensity was measured as described previously (McCloy et al., 2014). Briefly, using the ImageJ software, an outline around each cell (10 randomly selected cells/well × 4 wells) was drawn and the area, as well as integrated density, along with five random adjacent background readings (mean fluorescence), were measured. Then, the fluorescence intensity was calculated using a following formula: integrated density − (area of selected cell × mean fluorescence of background readings). Brain sections from rats subjected to transient MCAO or sham surgery were immunostained with antibodies against α-Syn (1:200; monoclonal; Cell Signaling Technology), NeuN (1:300; monoclonal; Millipore), pDrp-1 (1:1000; monoclonal; Cell Signaling Technology), 3-NT (1:500; monoclonal; Abcam), cleaved caspase-3 (1:400; monoclonal; Cell Signaling Technology), and LC-3 (1:100; monoclonal; Cell Signaling Technology), as described previously (Yan et al., 2007). To ensure that the homologous areas of injury were samples between animals, sections between the coordinates +1 to +1.5 mm from bregma were used in all cases. To evaluate long-term effects of ischemia on α-Syn pathology, α-Syn+/+ and α-Syn−/− mice were subjected to 90 min transient MCAO and killed at 4 months of reperfusion. Brain sections (30 μm) from these mice were pretreated with proteinase K (Promega; 50 μg/ml) in buffer containing 50 mm Tris-HCl, pH 8.0, and 10 mm CaCl2 at 37°C for 5 min, and then stained with an antibody against α-Syn (1:200; monoclonal; Cell Signaling Technology). To confirm the colocalization of NeuN and α-Syn, human brain sections were stained with antibodies against α-Syn (1:50; polyclonal; Cell Signaling Technology) and NeuN (1:300; monoclonal; Millipore). The human stroke brain sections were obtained with informed consent from a male patient aged 48 years with ischemic stroke on the right hemisphere at biopsy performed for diagnostic purposes. The patient did not show any comorbidities. Brain specimens from a subject with matched age and without clinical or postmortem evidence of neurological disease were obtained at autopsy and served as control. The study was in accordance with protocols approved by the Institutional Research Review Board at Huashan Hospital of Fudan University, China (Jin et al., 2006). To quantify the number of NeuN+ cells colocalized with α-Syn immunoreactivity, the total number of NeuN+ cells as well as the number of NeuN+ cells colocalized with α-Syn present within a 0.1 mm × 0.1 mm square box in the penumbral area three times per section (n = 4) were counted. The number of cells was expressed as a percentage of the average number of colocalized cells/0.01 mm2.
Real-time PCR.
Real-time PCR was performed as described previously for rat α-Syn (NM_019169) with SYBR-green method using 18s rRNA as an internal control (Dharap et al., 2009).
Results
In vitro ischemia induced α-Syn expression and its prevention protected the cells
When PC12 cells were exposed to 6 h of OGD and 24 h of reoxygenation, there was significant induction of α-Syn mRNA (by ∼1.6-fold; p < 0.05) and protein (by ∼2-fold; p < 0.05) expression compared with normoxic control cells (Fig. 1A–C). α-Syn protein extravasated into the nucleus of the PC12 cells subjected to OGD (Fig. 1D), and the intensity of staining was significantly greater than that for normoxic control cells (Fig. 1D,E). α-Syn siRNA treatment significantly prevented the post-OGD cell death compared with the control siRNA group measured at 24 h of reoxygenation following 6 h of OGD (by 26%; p < 0.05; Fig. 1F).
Figure 1.

A–C, α-Syn mRNA and protein levels elevated significantly when PC12 cells were subjected to 6 h of OGD and 24 h of reoxygenation. *p < 0.05 compared with the normoxic control group by Mann–Whitney U test. D, E, α-Syn protein translocated from the cytosol into nucleus in PC12 cells subjected to 6 h OGD and 24 h reoxygenation. *p < 0.05 compared with the normoxic control group by Mann–Whitney U test. F, Post-OGD cell viability was significantly higher in PC12 cells transfected with α-Syn siRNA compared with the control siRNA group. *p < 0.05 compared with the normoxic control group and #p < 0.05 compared with the control siRNA group (Kruskal–Wallis one-way ANOVA followed by Dunn's post-test). All assays were performed in triplicate. Values are mean ± SD (n = 4/group).
Ischemia induced nuclear translocation of α-Syn
Transient MCAO and 24 h of reperfusion significantly elevated α-Syn mRNA (by ∼6.1-fold; p < 0.05) and protein (by ∼5.7-fold; p < 0.05) levels in the ipsilateral cerebral cortex compared with sham control (Fig. 2A–C). In sham-operated rats, α-Syn staining was observed to be cytosolic, whereas there was progressive nuclear translocation of α-Syn between days 1 and 7 of reperfusion following transient MCAO (Fig. 3A). In the sham-operated brain, virtually no NeuN+ cells were colocalized with α-Syn (0.71%), whereas in the postischemic brain, a number of NeuN+ neurons in the peri-infarct region of the cerebral cortex were colocalized with α-Syn at both day 1 (33.6%) and day 7 (47.5%) of reperfusion (Fig. 3A). In both sham and ischemic animals, no colocalization was observed between α-Syn and GFAP (astroglial marker; Fig. 3B). To confirm that ischemia-induced increase of α-Syn levels is not limited to the in vitro or in vivo rodent stroke model, we immunostained human brain sections obtained from control and stroke patients with an antibody against human α-Syn. Similar to the ischemic rat brain, the human stroke patient showed higher α-Syn protein expression than the control subject (Fig. 3C). In control brain sections, α-Syn staining was primarily cytosolic in the neurons (NeuN+; Fig. 3C), whereas sections from the stroke patient showed translocation of α-Syn into NeuN+ neuronal nuclei similar to that in the ischemic rat brain (Fig. 3C).
Figure 2.

A–C, Transient focal ischemia induced α-Syn mRNA and protein expression in the rat cerebral cortex at 24 h of reperfusion compared with sham control. Values are mean ± SD (n = 4/group). *p < 0.05 compared with corresponding control group by Mann–Whitney U test.
Figure 3.
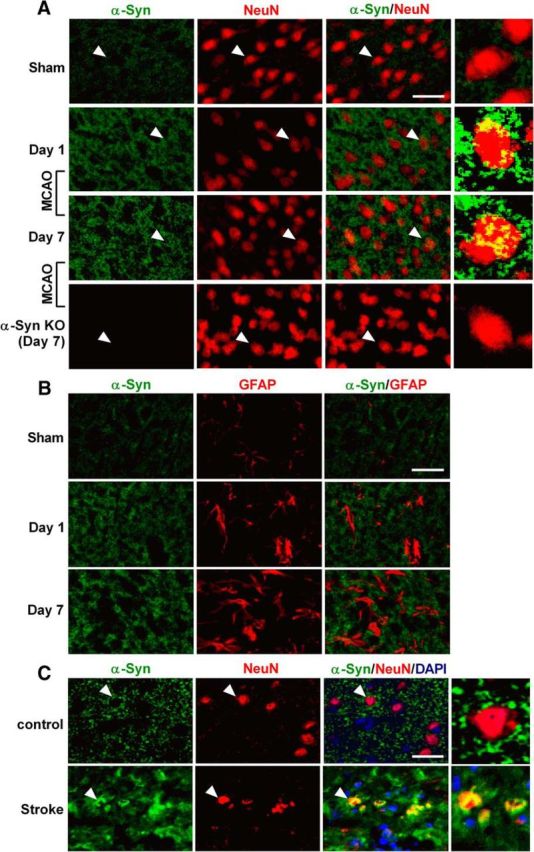
A, In the sham-operated rat cerebral cortex, the α-Syn immunoreactivity was observed in the cytosol of NeuN+ (neuronal marker) cells. At days 1 and 7 of reperfusion after transient MCAO, cytosolic α-Syn immunostaining increased and α-Syn protein translocated into the NeuN+ neuronal nuclei in the ischemic penumbral area. In the sham-operated brain, practically no NeuN+ cells were colocalized with α-Syn (0.71%), whereas in the postischemic brain, many NeuN+ neurons in the peri-infarct region of the cerebral cortex were colocalized with α-Syn at both day 1 (33.6%) and day 7 (47.5%) of reperfusion. B, No colocalization of α-Syn staining with the astroglial marker GFAP was observed in either sham or ischemic animals. C, In the human brain, punctate cytosolic staining of α-Syn was observed to be localized in NeuN+ neurons in the nonstroke subject. In sections from the stroke patient, intensity of α-Syn immunostaining is higher than in the control subject and α-Syn also translocated into neuronal nuclei. Scale bar, 30 μm. Magnified images on the right show the cells indicated with white arrows.
α-Syn knockdown decreased infarct volume and improved motor-function recovery after stroke
α-Syn siRNA treatment significantly reduced postischemic α-Syn protein induction (by 51%; p < 0.05) compared with the control siRNA treatment group (Fig. 4A,B). In rats pretreated with α-Syn siRNA at 2 h before MCAO, the postischemic motor dysfunction was significantly curtailed compared with control siRNA-treated rats between days 3 and 7 of reperfusion as measured by the rotarod test, the beam-walk test, and the adhesive-removal test (Fig. 4C). The infarct volume measured at day 7 of reperfusion following transient MCAO was also significantly smaller in the α-Syn siRNA group compared with the control siRNA group (by 38.7%; p < 0.05; Fig. 4D,E). Postischemic treatment with α-Syn siRNA (injected at 30 min of reperfusion after 1 h transient MCAO) also significantly decreased the infarct volume (by 47.5%; p < 0.05) measured at day 7 of reperfusion compared with the control siRNA group (Fig. 4D,E).
Figure 4.
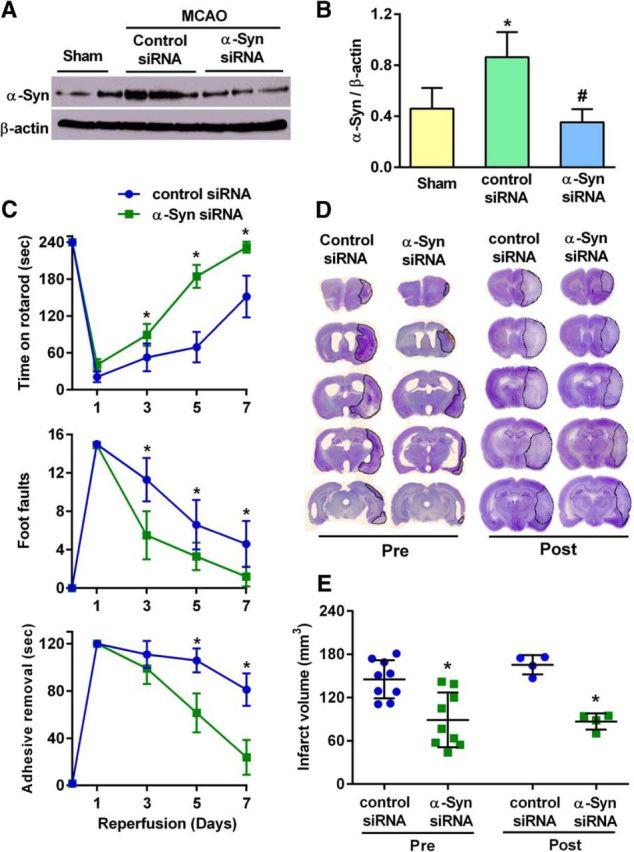
A, B, Treatment with α-Syn siRNA significantly prevented the post-MCAO increase in α-Syn protein levels in the rat cerebral cortex compared with the control siRNA treatment group. In B, values are mean ± SD (n = 4/group); *p < 0.05 compared with sham and #p < 0.05 compared with the control siRNA group by Kruskal–Wallis one-way ANOVA followed by Dunn's post-test. C, When pretreated at 2 h before MCAO with α-Syn siRNA, rats showed significantly better functional recovery than the control siRNA-treated group during 7 d period of reperfusion as assessed by the rotarod test (top), the beam-walk test (middle), and the adhesive-removal test (bottom). Rats were randomized and tested by an investigator blinded to the study groups. D, Representative cresyl violet-stained serial sections from rats subjected to transient MCAO and 7 d of reperfusion following α-Syn siRNA pretreatment (at 2 h before MCAO) or post-treatment (at 30 min of reperfusion following MCAO). Both α-Syn siRNA groups showed smaller infarct volume compared with the control siRNA groups. E, The infarct volume measured from the serial sections is significantly smaller in the α-Syn siRNA groups compared with the control siRNA groups. Values are mean ± SD in C and E (n = 9/group for pretreatment and n = 4/group for post-treatment). *p < 0.05 (repeated-measures ANOVA followed by Sidak's multiple-comparisons post-test) compared with respective reperfusion time point of the control siRNA group in C and *p < 0.05 (Mann–Whitney U test) compared with the control siRNA group in E.
α-Syn knockdown curtailed postischemic mitochondrial dysfunction, apoptosis, oxidative stress, and autophagy
In rats treated with α-Syn siRNA, there was a significant reduction in postischemic protein levels of Drp1 and pDrp1 (markers for mitochondrial fragmentation; Fig. 5A–C), cleaved caspase-3 (marker for apoptosis; Fig. 5A,D), 3-NT (marker for oxidative stress; Fig. 5A,E), and the LC3-II/I ratio (marker for autophagy; Fig. 5A,F), compared with the control siRNA-treated group at day 3 of reperfusion following transient MCAO. Immunohistochemical analysis confirmed above Western blot results and further showed colocalization of pDrp1 (Fig. 5G), cleaved caspase-3 (Fig. 5H), 3-NT (Fig. 5I), and LC-3 II/I (Fig. 5J) with the neuronal marker NeuN after transient MCAO.
Figure 5.
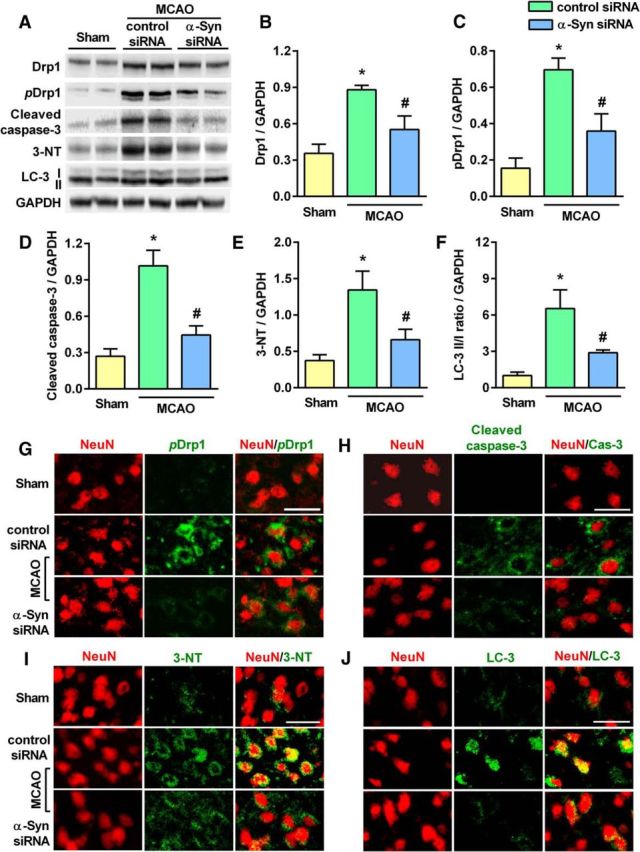
A–F, At day 3 of reperfusion following transient MCAO, protein levels of markers of mitochondrial fragmentation (Drp1 and pDrp1; (A–C), apoptosis (cleaved caspase-3; A, D), oxidative stress (3-NT; A, E), and autophagy (LC-3 II/I ratio; A, F) were significantly decreased in the penumbra of the ipsilateral cortex of α-Syn siRNA-treated rats compared with control siRNA-treated rats. The 3-NT blot shows only 1 representative protein band (∼45 kDa) that showed increased nitrotyrosination in the control siRNA/ischemia group that was significantly lower in the α-Syn siRNA/ischemia group. However, there are other proteins (lower and higher molecular weights) that also showed the same pattern. Values are mean ± SD (n = 4/group). *p < 0.05 compared with sham and #p < 0.05 compared with the control siRNA group by Kruskal–Wallis one-way ANOVA followed by Dunn's post-test. G–J, Decreased levels of immunostaining of pDrp1 (G), cleaved caspase-3 (H), 3-NT (I), and LC-3 (J) in the NeuN+ cells (neuronal) were observed in the penumbral cortex of the α-Syn siRNA group compared with the control siRNA group at day 7 of reperfusion following transient MCAO. Scale bar, 30 μm.
α-Syn knock-out mice are less susceptible to cerebral ischemia
To further confirm that postischemic induction of α-Syn mediates secondary brain damage, we induced transient MCAO in α-Syn knock-out mice (C57BL/6N-Sncatm1Mjff/J). At day 7 of reperfusion following 90 min transient MCAO, the mortality rate was 50% for the α-Syn+/+ cohort (11 of 22 died) compared with 7% for the α-Syn−/− cohort (1 of 14 died; Fig. 6A). Motor-function recovery assessed with the rotarod test and the beam-walk test was also significantly better between days 3 and 7 of reperfusion in α-Syn−/− mice compared with α-Syn+/+ mice (Fig. 6B). In addition, α-Syn−/− mice showed significantly decreased infarct volume measured at day 7 of reperfusion following transient MCAO, compared with α-Syn+/+ mice (by 43.8%; p < 0.05; Fig. 6C,D).
Figure 6.
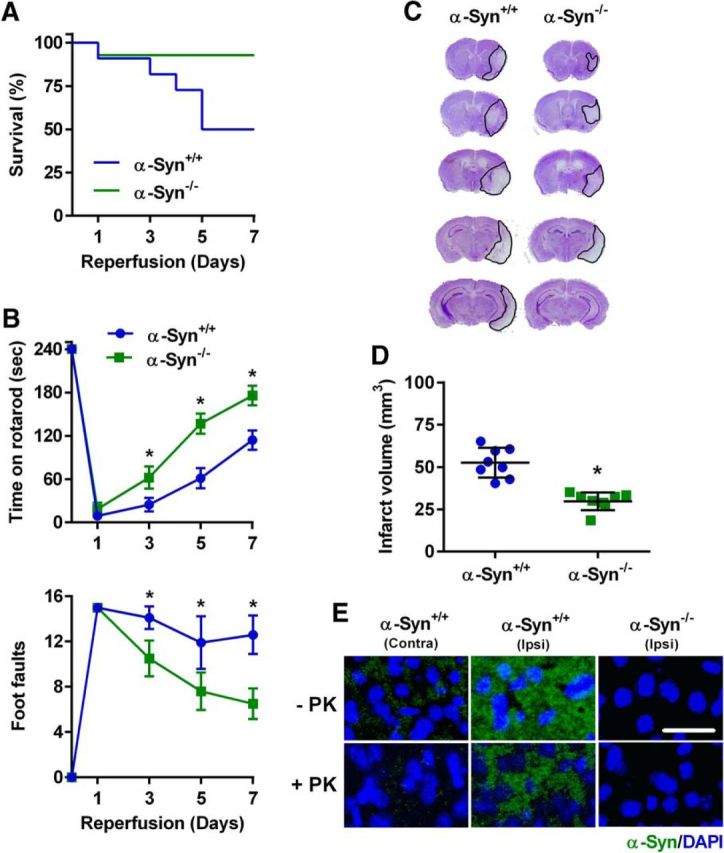
A, Kaplan–Meier curves show significantly higher rate of survival in α-Syn−/− mice compared with α-Syn+/+ mice following transient MCAO. At day 7 of reperfusion, 13 of 14 mice survived in the α-Syn−/− group, while 11 of 22 mice survived in the α-Syn+/+ group. B, α-Syn−/− mice showed significantly improved motor-function recovery compared with α-Syn+/+ mice measured by the rotarod test (top) and the beam-walk test (bottom) on days 3, 5, and 7 of reperfusion following transient MCAO. *p < 0.05 (repeated-measures ANOVA followed by Sidak's multiple-comparisons post-test) compared with respective reperfusion time point of the α-Syn+/+ group. Mice were randomized and tested by an investigator blinded to study groups. C, D, Representative cresyl violet-stained serial sections show smaller infarct volume in α-Syn−/− mice compared with α-Syn+/+ mice at day 7 of reperfusion. *p < 0.05 compared with the α-Syn+/+ group by Mann–Whitney U test. Values are mean ± SD (n = 8/group). E, At 4 months of reperfusion following transient MCAO, α-Syn immunostaining was observed to be significantly higher in the ipsilateral cortex of α-Syn+/+ mice, which resisted proteinase K treatment This indicates that α-Syn formed aggregates at this late time point in the ischemic cortex. No immunoreactivity of α-Syn was observed in α-Syn−/− mouse brain sections.
Focal ischemia leads to α-Syn aggregation
To understand whether increased expression of α-Syn protein monomers leads to the formation of aggregates over time after focal ischemia, we induced 90 min transient MCAO in cohorts of α-Syn−/− and α-Syn+/+ mice (n = 4/group) and killed them at 4 months of reperfusion. Elevated α-Syn protein levels observed at days 1 and 7 of reperfusion (Figs. 2, 3) persisted at 4 months of reperfusion (Fig. 6E). Proteinase K treatment before immunostaining is known to digest protein monomers leaving only the oligomers that form α-Syn aggregates (Fernagut et al., 2007; Tanji et al., 2010). At 4 months of reperfusion following transient MCAO, the contralateral cortex of α-Syn+/+ mice showed minimal immunostaining of α-Syn, while the ipsilateral cortex showed abundant, patchy immunostaining of α-Syn (Fig. 6E). When sections were treated with proteinase K, α-Syn immunostaining was greatly diminished (>90%) in the contralateral cortex, but mostly remained in the ipsilateral cortex (Fig. 6E). This indicates that the α-Syn protein monomers are induced in excess after stroke and aggregated in the ischemic cortex over time. Sections from α-Syn−/− mice subjected to transient MCAO and 4 months of reperfusion didn't show any α-Syn immunoreactivity in either the contralateral or ipsilateral cortex (Fig. 6E).
Ischemia induced S129 phosphorylation and oligomerization of α-Syn
S129 phosphorylation is implicated in the oligomerization and toxicity of α-Syn in PD. Transient MCAO and 24 h of reperfusion significantly increased cortical S129 phosphorylation of α-Syn compared with sham (Fig. 7A,B). Nuclear levels of α-Syn and S129 pα-Syn (Fig. 7C,D), but not their cytosolic levels (Fig. 7E,F), increased significantly in the ischemic group compared with sham. This indicates nuclear translocation of both α-Syn and S129 pα-Syn after stroke. Oligomerization of α-Syn is a hallmark of chronic neurodegenerative synucleinopathies and also leads to its aggregation over time. Western blot analysis showed 60 and 100 kDa oligomers of α-Syn in the ipsilateral cortex at day 3 of reperfusion following transient MCAO, which were minimally present in sham samples (Fig. 7G).
Figure 7.
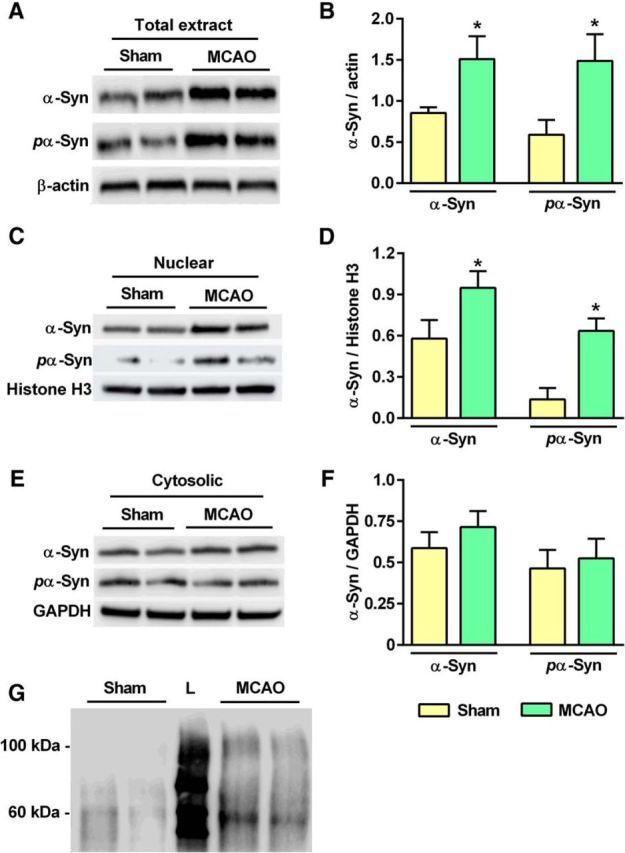
A, B, S129 pα-Syn levels were elevated significantly at 24 h of reperfusion following transient MCAO compared with sham control. C–F, The S129 pα-Syn levels were significantly elevated in the nuclear fraction (C, D), but not in the cytosolic fraction (E, F), in ischemic rats compared with sham. G, Higher molecular weight (∼60 and ∼100 kDa) oligomeric forms of α-Syn were observed in the ischemic samples collected at day 3 of reperfusion, but not in the sham samples, indicating postischemic oligomerization. L, Molecular weight ladder. Values are mean ± SD of n = 4/group. *p < 0.05 compared with sham by Mann–Whitney U test.
PLK2 knock-out mice are more resilient to cerebral ischemia
As PLK2 is the predominant kinase responsible for the S129 phosphorylation of α-Syn, we evaluated ischemic brain damage in PLK2−/− mice (129S.B6N-Plk2tm1Elan/J) compared with PLK2+/+ mice. Following 90 min transient MCAO, motor-function recovery (measured by the rotarod test and the beam-walk test) was significantly better in PLK2−/− mice compared with PLK2+/+ mice between days 3 and 7 of reperfusion (Fig. 8A). The infarct volume measured at day 7 of reperfusion was also significantly smaller in the PLK2−/− cohort compared with the PLK2+/+ cohort (by ∼36%; p < 0.05; Fig. 8B,C).
Figure 8.

A, PLK2−/− mice showed significantly improved postischemic motor-function recovery compared with PLK2+/+ mice measured by the rotarod test (top) and the beam-walk test (bottom) between days 3 and 7 of reperfusion following transient MCAO. *p < 0.05 (repeated-measures ANOVA followed by multiple Sidak's comparisons post-test) compared with respective reperfusion time point of the α-Syn+/+ group. Mice were tested by an investigator blinded to the study groups. B, Representative cresyl violet-stained serial sections showed smaller infarct in PLK2−/− mice compared with PLK2+/+ mice at day 7 of reperfusion following transient MCAO. C, The infarct volume measured from the serial sections is significantly smaller in the PLK2−/− mice compared with the PLK2+/+ mice. Values are mean ± SD (n = 5/group). *p < 0.05 compared with the PLK2+/+ mice by Mann–Whitney U test.
Discussion
In brief, results of the present study show that α-Syn, which is thought to be a chronic neurodegeneration-related protein, also mediates secondary brain damage after an acute insult to the brain, such as stroke. Transient focal cerebral ischemia induced α-Syn protein expression, nuclear translocation, oligomerization, and aggregation in the rodent brain. Increased α-Syn protein expression and nuclear translocation was also observed in the human poststroke brain. α-Syn knockdown in adult rats prevented the postischemic expression of markers of mitochondrial fragmentation, apoptosis, oxidative stress, and autophagy; decreased the infarction; and promoted better neurological recovery. Knock-out mice that lack α-Syn showed lower mortality, smaller infarcts, and fewer neurological deficits after stroke. Ischemia also induced S129 phosphorylation of α-Syn and nuclear translocation of S129 pα-Syn in the ipsilateral cortex. Stroke in knock-out mice that lack PLK2 (predominant kinase that phosphorylates α-Syn at S129) showed better neurological recovery and smaller infarcts.
Many proteins, including α-Syn, phosphatase and tensin homolog-induced novel kinase-1 (PINK1), parkin, and amyloid-β (Aβ) play a central role in chronic neurodegenerative diseases. Of those, PINK1 has been shown to protect neurons from OGD-induced cell death (Zhao et al., 2013), while parkin depletion has been shown to contribute to neuronal death after focal ischemia (Mengesdorf et al., 2002). Furthermore, serum Aβ levels are a known predictor of short-term neurological deficits after stroke (Liu et al., 2015a) and patients with cerebral Aβ deposition show more severe and rapid cognitive decline after stroke (Liu et al., 2015b). Mutations or overexpression of α-Syn have been mostly implicated in the pathogenesis of PD, but α-Syn has also been reported to play a neuroprotective role under a cysteine-string protein-α-null-mediated neurodegenerative condition by maintaining synaptic integrity (Chandra et al., 2005; Ruiz et al., 2014). However, the role of α-Syn in poststroke brain damage was not yet studied comprehensively. A previous study showed that a mild ischemic insult (30 min transient MCAO) in adult mice leads to increased levels of α-Syn protein in the cortical cytosolic preparations at day 3 of reperfusion (Unal-Cevik et al., 2011). While we report that a moderate ischemic insult (60 min transient MCAO) in adult rats results in much higher α-Syn protein induction, as well as phosphorylation, by 6 h to day 3, and nuclear translocation of both α-Syn and S129 pα-Syn by day 7 of reperfusion, as well as formation of proteinase K-resistant aggregates by 4 months of reperfusion in mice.
We show that levels of α-Syn monomers increase by 24 h of reperfusion and levels of α-Syn oligomers increase by day 3 of reperfusion following transient MCAO in adult rats. However, it is not clear whether increased levels of α-Syn monomers are sufficient for cellular toxicity. Studies with PD models indicate that oligomeric α-Syn is the form most likely responsible for neuronal death (Danzer et al., 2007; Winner et al., 2011; Luth et al., 2014). But, studies also indicate that α-Syn monomers are neurotoxic at increased levels (Diógenes et al., 2012; Stoica et al., 2012). As secondary brain damage after transient MCAO occurs at a rapid pace during the first day of reperfusion and at a slower rate between days 1 and 3 of reperfusion, it is likely that both monomers and oligomers of α-Syn play discrete roles in neuronal death after stroke.
Pretreatment as well as post-treatment with the α-Syn-specific siRNA mixture decreased infarct volume and promoted better functional recovery after transient MCAO in adult rats, indicating that α-Syn is a mediator of ischemic brain damage. More importantly, prevention of α-Syn is therapeutically beneficial. This is further supported by the observation that α-Syn−/− mice are resilient to focal ischemia. α-Syn−/− mice showed smaller infarcts and better neurological recovery after stroke. The S129 residue in the C terminal of α-Syn that undergoes phosphorylation is highly conserved, indicating its functional importance. Under normal conditions, α-Syn predominantly exists in an unphosphorylated form, but oxidative stress and proteasome dysfunction due to pathologies promote S129 phosphorylation, which is thought to modulate α-Syn oligomerization and aggregation leading to toxicity as observed in synucleinopathies (Fujiwara et al., 2002; Visanji et al., 2011; Lashuel et al., 2013). However, the significance of S129 phosphorylation of α-Syn to functional outcome is debatable as studies using different models of PD and synucleinopathies indicate both neurotoxic and neuroprotective roles (Gorbatyuk et al., 2008; Lee et al., 2011). Furthermore, soluble oligomeric forms of α-Syn are thought to be more toxic than larger aggregated forms facilitated by S129 phosphorylation (Winner et al., 2011; Roberts and Brown, 2015). We observed that PLK2-deficient mice show neuroprotection and better functional recovery, indicating that S129 phosphorylation of α-Syn is toxic after stroke.
In PD, oxidative stress is thought to promote the nuclear translocation of α-Syn, but its significance is not clear (Monti et al., 2010). We presently observed that α-Syn protein translocated to nucleus even after focal ischemia. α-Syn translocated into neuronal nuclei following a toxic insult was shown to form complexes with histones (Goers et al., 2003), leading to reduced histone acetylation (Kontopoulos et al., 2006). This mechanism is possible even after stroke and future studies will reveal its significance. Several other mechanisms, including apoptosis, inflammation, oxidative stress, mitochondrial fragmentation, and autophagy, have been proposed for α-Syn-induced neuronal death in PD and other synucleinopathies (Dauer and Przedborski, 2003; Cheung and Ip, 2009; Tufekci et al., 2012). α-Syn overexpression is thought to increase the translocation of Drp1 as well as alter mitochondrial morphology via the extracellular signal-regulated kinase (Gui et al., 2012). Furthermore, α-Syn forms oligomers with annular, pore-like structures in the membrane that are highly permeable to calcium, leading to caspase activation and apoptotic neuronal death (Volles et al., 2001; Danzer et al., 2007). In addition, overexpression of wild-type α-Syn resulted in impaired macroautophagy via Rab1a inhibition (Winslow et al., 2010). All these studies were conducted in the context of PD. Cerebral ischemia shares many of these pathological mechanisms with PD despite the difference in pathophysiological intensity and time frame (Gill et al., 2002; Barsoum et al., 2006; Liu et al., 2010; Grohm et al., 2012). α-Syn knockdown in rats subjected to transient MCAO led to curtailed induction of pDrp1, cleaved caspase-3, 3-NT, and LC-3 II/I, indicating that α-Syn might participate in postischemic mitochondrial fragmentation, apoptosis, oxidative stress, and autophagy. Overall, our studies show that α-Syn is an attractive target for developing novel therapies to minimize poststroke brain damage and neurological dysfunction.
Footnotes
This work was supported, in part, by the U.S. Department of Veterans Affairs Merit Review Grant (I01 BX002985). TaeHee Kim was supported by a predoctoral fellowship from the American Heart Association (15IRG23050015). We thank Dr. Kunlin Jin for providing the human brain sections.
The authors declare no competing financial interests.
References
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. The emerging role of autophagy in Parkinson's disease. Mol Brain. 2009;2:29. doi: 10.1186/1756-6606-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and nonneural cells. J Biol Chem. 2013;288:6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral micro RNAome. J Cereb Blood Flow Metab. 2009;29:675–687. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Nakka VP, Vemuganti R. Altered expression of PIWI RNA in the rat brain after transient focal ischemia. Stroke. 2011;42:1105–1109. doi: 10.1161/STROKEAHA.110.598391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Nakka VP, Vemuganti R. Effect of focal ischemia on long noncoding RNAs. Stroke. 2012;43:2800–2802. doi: 10.1161/STROKEAHA.112.669465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diógenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Näsström T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergström J, Ingelsson M, Quintas A, Sebastião AM, Lopes LV, Outeiro TF. Extracellular α-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci. 2012;32:11750–11762. doi: 10.1523/JNEUROSCI.0234-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraro F, Sahin G, Farran A, Soares S, Jensen PH, Kirik D, Romero-Ramos M. Ser129D mutant alpha-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson's disease. Neurobiol Dis. 2013;56:47–58. doi: 10.1016/j.nbd.2013.03.014. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gill R, Soriano M, Blomgren K, Hagberg H, Wybrecht R, Miss MT, Hoefer S, Adam G, Niederhauser O, Kemp JA, Loetscher H. Role of caspase-3 activation in cerebral ischemia-induced neurodegeneration in adult and neonatal brain. J Cereb Blood Flow Metab. 2002;22:420–430. doi: 10.1097/00004647-200204000-00006. [DOI] [PubMed] [Google Scholar]
- Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry. 2003;42:8465–8471. doi: 10.1021/bi0341152. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci. 2008;105:763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–1458. doi: 10.1038/cdd.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui YX, Wang XY, Kang WY, Zhang YJ, Zhang Y, Zhou Y, Quinn TJ, Liu J, Chen SD. Extracellular signal-regulated kinase is involved in alpha-synuclein-induced mitochondrial dynamic disorders by regulating dynamin-like protein 1. Neurobiol Aging. 2012;33:2841–2854. doi: 10.1016/j.neurobiolaging.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Inglis KJ, Chereau D, Brigham EF, Chiou SS, Schöbel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, Wright S, Chian D, Santiago P, Soriano F, Ramos C, Powell K, Goldstein JM, Babcock M, Yednock T, Bard F, et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem. 2009;284:2598–2602. doi: 10.1074/jbc.C800206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Wang X, Xie L, Mao XO, Zhu W, Wang Y, Shen J, Mao Y, Banwait S, Greenberg DA. Evidence for stroke-induced neurogenesis in the human brain. Proc Natl Acad Sci U S A. 2006;103:13198–13202. doi: 10.1073/pnas.0603512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Vemuganti R. Effect of sex and age interactions on functional outcome after stroke. CNS Neurosci Ther. 2015;21:327–336. doi: 10.1111/cns.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, Feng X, Ray N, Fernandez JR, Chao Y, Masliah E, Voronkov M, Braithwaite SP, Stock JB, Mouradian MM. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J Neurosci. 2011;31:6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68–78. doi: 10.1111/j.1471-4159.2010.06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, Cao HY, Wang YR, Jiao SS, Bu XL, Zeng F, Wang QH, Li J, Deng J, Zhou HD, Wang YJ. Serum Abeta is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox Res. 2015a;27:292–299. doi: 10.1007/s12640-015-9518-z. [DOI] [PubMed] [Google Scholar]
- Liu W, Wong A, Au L, Yang J, Wang Z, Leung EY, Chen S, Ho CL, Mok VC. Influence of amyloid-beta on cognitive decline after stroke/transient ischemic attack: three-year longitudinal study. Stroke. 2015b;46:3074–3080. doi: 10.1161/STROKEAHA.115.010449. [DOI] [PubMed] [Google Scholar]
- Lopez MS, Dempsey RJ, Vemuganti R. Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem Int. 2015;89:75–82. doi: 10.1016/j.neuint.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem. 2014;289:21490–21507. doi: 10.1074/jbc.M113.545749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010;285:2807–2822. doi: 10.1074/jbc.M109.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13:1400–1412. doi: 10.4161/cc.28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Kim T, Vemuganti R. Long noncoding RNA FosDT promotes ischemic brain injury by interacting with REST-associated chromatin-modifying proteins. J Neurosci. 2015;35:16443–16449. doi: 10.1523/JNEUROSCI.2943-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengesdorf T, Jensen PH, Mies G, Aufenberg C, Paschen W. Down-regulation of parkin protein in transient focal cerebral ischemia: A link between stroke and degenerative disease? Proc Natl Acad Sci U S A. 2002;99:15042–15047. doi: 10.1073/pnas.232588799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of alpha-synuclein. Neurotox Res. 2010;17:130–141. doi: 10.1007/s12640-009-9090-5. [DOI] [PubMed] [Google Scholar]
- Nakka VP, Lang BT, Lenschow DJ, Zhang DE, Dempsey RJ, Vemuganti R. Increased cerebral protein ISGylation after focal ischemia is neuroprotective. J Cereb Blood Flow Metab. 2011;31:2375–2384. doi: 10.1038/jcbfm.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakka VP, Prakash-babu P, Vemuganti R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: potential therapeutic targets for acute CNS injuries. Mol Neurobiol. 2016;53:532–544. doi: 10.1007/s12035-014-9029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- Pacheco C, Aguayo LG, Opazo C. An extracellular mechanism that can explain the neurotoxic effects of alpha-synuclein aggregates in the brain. Front Physiol. 2012;3:297. doi: 10.3389/fphys.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandi G, Nakka VP, Dharap A, Roopra A, Vemuganti R. MicroRNA miR-29c down-regulation leading to de-repression of its target DNA methyltransferase 3a promotes ischemic brain damage. PLoS One. 2013;8:e58039. doi: 10.1371/journal.pone.0058039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th edition. San Diego: Academic; 1998. [Google Scholar]
- Roberts HL, Brown DR. Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules. 2015;5:282–305. doi: 10.3390/biom5020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R, Biea IA, Tabares L. alpha-Synuclein A30P decreases neurodegeneration and increases synaptic vesicle release probability in CSPalpha-null mice. Neuropharmacology. 2014;76:106–117. doi: 10.1016/j.neuropharm.2013.08.032. [DOI] [PubMed] [Google Scholar]
- Salvi M, Trashi E, Marin O, Negro A, Sarno S, Pinna LA. Superiority of PLK-2 as alpha-synuclein phosphorylating agent relies on unique specificity determinants. Biochem Biophys Res Commun. 2012;418:156–160. doi: 10.1016/j.bbrc.2011.12.152. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2:13. doi: 10.1186/2040-7378-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid AW, Fauvet B, Moniatte M, Lashuel HA. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol Cell Proteomics. 2013;12:3543–3558. doi: 10.1074/mcp.R113.032730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica G, Lungu G, Bjorklund NL, Taglialatela G, Zhang X, Chiu V, Hill HH, Schenk JO, Murray I. Potential role of α-synuclein in neurodegeneration: studies in a rat animal model. J Neurochem. 2012;122:812–822. doi: 10.1111/j.1471-4159.2012.07805.x. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Takeda A, Hasegawa T, Matsuzaki-Kobayashi M, Sugeno N, Kikuchi A, Itoyama Y, Furukawa K. Mechanisms of neuronal death in synucleinopathy. J Biomed Biotechnol. 2006;2006:19365. doi: 10.1155/JBB/2006/19365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji K, Mori F, Mimura J, Itoh K, Kakita A, Takahashi H, Wakabayashi K. Proteinase K-resistant alpha-synuclein is deposited in presynapses in human Lewy body disease and A53T alpha-synuclein transgenic mice. Acta Neuropathol. 2010;120:145–154. doi: 10.1007/s00401-010-0676-z. [DOI] [PubMed] [Google Scholar]
- Tenreiro S, Eckermann K, Outeiro TF. Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci. 2014;7:42. doi: 10.3389/fnmol.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufekci KU, Meuwissen R, Genc S, Genc K. Inflammation in Parkinson's disease. Adv Protein Chem Struct Biol. 2012;88:69–132. doi: 10.1016/B978-0-12-398314-5.00004-0. [DOI] [PubMed] [Google Scholar]
- Unal-Cevik I, Gursoy-Ozdemir Y, Yemisci M, Lule S, Gurer G, Can A, Müller V, Kahle PJ, Dalkara T. Alpha-synuclein aggregation induced by brief ischemia negatively impacts neuronal survival in vivo: a study in [A30P]alpha-synuclein transgenic mouse. J Cereb Blood Flow Metab. 2011;31:913–923. doi: 10.1038/jcbfm.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, Wislet-Gendebien S, Oschipok LW, Zhang G, Aubert I, Fraser PE, Tandon A. Effect of Ser-129 phosphorylation on interaction of alpha-synuclein with synaptic and cellular membranes. J Biol Chem. 2011;286:35863–35873. doi: 10.1074/jbc.M111.253450. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Characterization of kinases involved in the phosphorylation of aggregated α-synuclein. J Neurosci Res. 2011;89:231–247. doi: 10.1002/jnr.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan YP, Sailor KA, Lang BT, Park SW, Vemuganti R, Dempsey RJ. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:1213–1224. doi: 10.1038/sj.jcbfm.9600432. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Chen F, Chen S, Liu X, Cui M, Dong Q. The Parkinson's disease-associated gene PINK1 protects neurons from ischemic damage by decreasing mitochondrial translocation of the fission promoter Drp1. J Neurochem. 2013;127:711–722. doi: 10.1111/jnc.12340. [DOI] [PubMed] [Google Scholar]