

Abstract
Amblyomma americanum (Lone star tick) is an important disease vector in the United States. It transmits several human pathogens, including the agents of human monocytic ehrlichiosis, tularemia, and southern tick-associated rash illness. Blood-feeding insects (Class Insecta) depend on bacterial endosymbionts to provide vitamins and cofactors that are scarce in blood. It is unclear how this deficiency is compensated in ticks (Class Arachnida) that feed exclusively on mammalian blood. A bacterium related to Coxiella burnetii, the agent of human Q fever, has been observed previously within cells of A. americanum. Eliminating this bacterium (CLEAA, Coxiella-like endosymbiont of A. americanum) with antibiotics reduced tick fecundity, indicating that it is an essential endosymbiont. In an effort to determine its role within this symbiosis, we sequenced the CLEAA genome. While highly reduced (656,901 bp) compared with C. burnetii (1,995,281 bp), the CLEAA genome encodes most major vitamin and cofactor biosynthesis pathways, implicating CLEAA as a vitamin provisioning endosymbiont. In contrast, CLEAA lacks any recognizable virulence genes, indicating that it is not a pathogen despite its presence in tick salivary glands. As both C. burnetii and numerous “Coxiella-like bacteria” have been reported from several species of ticks, we determined the evolutionary relationship between the two bacteria. Phylogeny estimation revealed that CLEAA is a close relative of C. burnetii, but was not derived from it. Our results are important for strategies geared toward controlling A. americanum and the pathogens it vectors, and also contribute novel information regarding the metabolic interdependencies of ticks and their nutrient-provisioning endosymbionts.
Keywords: vitamin, cofactor, nutrient provisioning, Amblyomma, endosymbiont
Introduction
Nutritional symbiosis is widespread in insects that depend on unbalanced diets (Buchner 1965). Endosymbiotic bacteria provision essential amino acids to insects that feed on plant sap that has low levels of essential amino acids (Shigenobu et al. 2000; Baumann 2005; Nakabachi et al. 2006; McCutcheon et al. 2009; McCutcheon and Moran 2010; Wernegreen 2012; Bennett and Moran 2013). Similarly, obligate blood-sucking insects, including tsetse fly, human body louse, and bedbug depend on bacterial endosymbionts for vitamins and cofactors that are lacking from mammalian blood (Akman et al. 2002; Kirkness et al. 2010; Sassera et al. 2013; Nikoh et al. 2014). These so-called primary endosymbionts live in specialized host cells and are transmitted vertically (Baumann 2005; Moran et al. 2008; Wernegreen 2012). Another form of nutritional symbiosis is also found in certain insects (e.g., termites and cockroaches) where a complex community of gut microbes provides all needed nutrients, enabling the host to live on a nutrient-poor diet, such as wood (Warnecke et al. 2007; Sabree et al. 2012). Unlike blood-feeding insects (Arthropoda; Class Insecta), it is not clear how ticks (Arthropoda; Class Arachnida) that also feed exclusively on vertebrate blood overcome a diet lacking essential vitamins and cofactors. Several bacteria that are suspected to be endosymbionts have been detected from various tick species (Rounds et al. 2012), but it is not known whether any can provide the required nutrients lacking in a bloodmeal.
Amblyomma americanum, a hard backed tick common in the southeastern United States, is an important disease vector that transmits Ehrlichia chaffeensis, Francisella tularensis, and Borrelia lonestari, the etiologic agents of human monocytic ehrlichiosis, tularemia and southern tick-associated rash illness, respectively (Childs and Paddock 2003). An intracellular “Coxiella-like bacterium” has previously been detected in various tissues (salivary gland, gut, and ovaries) of all field-caught and lab-reared Amblyomma amblyomma (Jasinskas et al. 2007; Klyachko et al. 2007). Interestingly, eliminating this bacterium with antibiotics caused severe reduction in tick fecundity and fitness, suggesting an important role for the bacterium in promoting host fitness (Zhong et al. 2007). We sequenced the whole genome of this Coxiella-like endosymbiont of A. americanum (CLEAA) and determined that it is closely related to Coxiella burnetii but was not directly derived from it, as previously assumed (Zhong 2012). Although the CLEAA genome is severely reduced, it is replete with metabolic pathways for the biosynthesis of several vitamins and cofactors that are scarce in vertebrate blood. Thus, CLEAA may impart A. americanum with essential nutrients not obtained in its diet.
CLEAA Is Highly Prevalent in A. americanum
A 16S rRNA gene diversity analysis of DNA isolated from four adult female A. americanum ticks revealed that over 95% of reads from all four ticks were from a bacterium closely related to C. burnetii (fig. 1). Additionally, as reported earlier, Rickettsia and Brevibacterium species were also present in all four ticks but at a significantly lower prevalence than CLEAA (Klyachko et al. 2007; Clay et al. 2008; Heise et al. 2010). Although Staphylococcus was also detected in all ticks, this common human skin bacterium was most probably introduced during tick handling or sample preparation.
Fig. 1.—
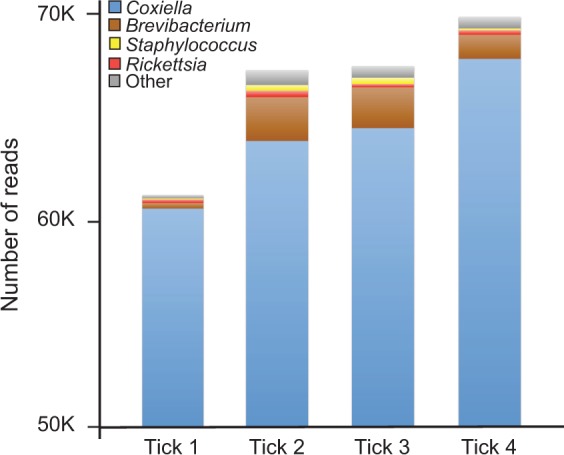
CLEAA is highly abundant in A. americanum. Abundance of Coxiella-like bacteria in four lab-reared female A. americanum ticks is depicted. y axis denotes number of sequencing reads obtained from each tick.
CLEAA Is a Sister Taxon of C. burnetii
Previous studies based on 16S rRNA analyses have indicated that CLEAA was derived from C. burnetii, which is occasionally vectored by ticks, including A. americanum (Childs and Paddock 2003; Zhong et al. 2007). Our phylogeny estimation based on 161 orthologous proteins across 34 gammaproteobacteria placed CLEAA as a sister taxon to C. burnetii (fig. 2). A phylogeny estimated from 16S rRNA genes also supported the finding that CLEAA is closely related to C. burnetii, but not directly derived from the human Q fever pathogen (fig. 3). Additionally, our analyses revealed that a large group of Coxiella-like bacteria are present in a variety of hard and soft ticks, suggestive of an ancient symbiotic relationship from which both CLEAA and C. burnetii were derived (fig. 3). Intriguingly, as shown recently for order Rickettsiales (Kang et al. 2014), a common ancestor of CLEAA and Coxiella appears to have an aquatic origin (fig. 3).
Fig. 2.—

CLEAA is a sister taxon of Coxiella. Phylogenetic tree based on 161 orthologous genes (supplementary table S3, Supplementary Material online) in CLEAA and 33 fully sequenced bacterial genomes.
Fig. 3.—

Phylogeny estimation of Coxiellaceae 16S rRNA genes. Number of taxa collapsed into each branch is shown within parentheses. Dotted lines depict branches that have been extended for clarity. Taxa colored red depict probable Coxiella-like organisms erroneously named as species of Legionella. Coxiella burnetii is highlighted in blue and CLEAA is boxed in yellow.
CLEAA Genome Is Highly Reduced and Does Not Contain Coxiella Virulence Genes
The complete genome of CLEAA is severely reduced in size when compared with that of C. burnetii (fig. 4 and supplementary fig. S1, Supplementary Material online). It consists of a circular 656,901 bp chromosome that is predicted to encode 537 protein-coding genes, 39 tRNAs, and a single rRNA operon (table 1). Similar to Coxiella, central metabolic processes such as glycolysis, nonoxidative branch of pentose phosphate pathway, TCA cycle, and nucleotide biosynthesis are largely intact in CLEAA; however, only approximately 30% of Coxiella’s gene content has been retained in the CLEAA genome. The small size, low G+C (34.6%), low gene density (83%), and the presence of 23 pseudogenes suggest that the genome has undergone reductive evolution due to CLEAA’s host-dependent life style (McCutcheon and Moran 2012).
Fig. 4.—

Alignment of C. burnetii RSA 493 and CLEAA genomes. Each contiguously colored locally collinear block (LCB) represents a region without rearrangement of the homologous backbone sequence. LCBs were calculated with the Mauve 2.1.0 aligner (Darling et al. 2010). Lines between genomes indicate orthologous LCBs. LCBs below the center in Coxiella genome represent blocks in the reverse orientation. LCB regions in Coxiella without homologs in CLEAA are indicated in white.
Table 1.
General Genome Features of CLEAA
| Feature | Value for Genome |
|---|---|
| Genome size (bp) | 656,901 |
| GC (%) | 34.6 |
| Genes (%) | 83 |
| Protein | 537 |
| rRNA | 3 |
| tRNA | 39 |
| ncRNA | 3 |
| GenBank accession | CP007541 |
Previous studies have reported a high density of CLEAA in A. americanum salivary glands, suggestive of an infective life cycle for this bacterium, as has been suggested for other tick-associated bacteria (Ahantarig et al. 2013). A dot/icm type IV secretion system (T4SS) and its secreted effectors are major contributors to the pathogenesis of C. burnetii (Beare et al. 2011) and in other pathogens in the order Legionellales (e.g., Legionella pneumophila and Rickettsiella grylli) that are closely related to C. burnetii and CLEAA. None of the genes that encode either T4SS components or effector proteins is present in CLEAA. Furthermore, no other known virulence genes or bacterial secretion systems are encoded within the CLEAA genome, but an intact Sec translocon (Stead et al. 2013) is present (supplementary table S1, Supplementary Material online).
Vitamin and Cofactor Biosynthetic Capability of CLEAA
Obligate blood-sucking insects such as tsetse fly (Glossina morsitans), body louse (Pediculus humanus humanus), and bedbug (Cimex lectularius) depend on bacterial endosymbionts to provide vitamins and cofactors that are available in trace amounts in mammalian blood (Akman et al. 2002; Kirkness et al. 2010; Sassera et al. 2013; Nikoh et al. 2014). To determine whether CLEAA can play a similar role in A. americanum, we reconstructed its vitamin and cofactor biosynthesis pathways. As shown in figure 5, CLEAA has complete (or almost complete) pathways for the biosynthesis of several vitamins and cofactors including folic acid (vitamin B9), riboflavin (B2), pantothenic acid (B5), nicotinamide (B3), pyridoxine (B6), thiamine (B1), biotin (B7), and lipoic acid—two cofactors synthesized using the fatty acid biosynthesis pathway. As shown for other endosymbionts, enzymes from either the host or from co-occurring endosymbionts might compensate for the enzymes missing from the cofactor biosynthesis pathways in CLEAA (Husnik et al. 2013; Sloan et al. 2014). Interestingly, pabA (and possibly pabB) required for the synthesis of para-aminobenzoic acid (PABA), from which folic acid is synthesized, has been acquired by CLEAA through horizontal gene transfer (HGT) from an Alphaproteobacteria (fig. 5 and supplementary fig. S2, Supplementary Material online). This scenario is similar to what has been observed in the biotin-provisioning Wolbachia endosymbiont of bedbug and in a Rickettsia endosymbiont of Ixodes scapularis where genes in the biotin biosynthesis pathway were gained through HGT (Gillespie et al. 2012; Nikoh et al. 2014). Additionally, an analysis of A. americanum expression sequence tags (ESTs) deposited in the National Center for Biotechnology Information (NCBI) dbEST database confirmed the expression within A. amblyomma of CLEAA genes from six of the eight cofactor biosynthesis pathways (supplementary table S2, Supplementary Material online), suggesting a role for vitamin and cofactor provisioning in maintaining the host–endosymbiont relationship.
Fig. 5.—

Cofactor biosynthetic pathways in CLEAA. Arrows represent each step catalyzed by the named enzymes in each pathway. Cofactors are within yellow boxes. Question marks indicate enzymes for which no genes were found in the CLEAA genome. Green indicates genes involved in the conversion of chorismate to PABA, with phylogeny estimation indicating pabA was acquired from Alphaproteobacteria (supplementary table S2, supplementary Material online).
Gaining new functions through bacterial endosymbionts allows eukaryotes to expand into novel niches (Moran 2007). Because all life stages of A. americanum feed exclusively on mammalian blood, which contains a limited supply of essential vitamins and cofactors, the presence of a vitamin provisioning endosymbiont enables the tick to subsist on this nutrient-poor diet. As shown recently in bedbugs (Nikoh et al. 2014), detailed functional analyses are required to determine the specific cofactor(s) provided by CLEAA to its host, but considering the potentially vital function of CLEAA in tick reproduction and development (Zhong et al. 2007), it is surprising that the endosymbiont is not retained within specialized host cells (Klyachko et al. 2007) as observed for many insect primary endosymbionts (McCutcheon and Moran 2012; Wernegreen 2012). The CLEAA–A. americanum relationship could be a fairly recent phenomenon, with CLEAA in a process of transitioning from a facultative endosymbiont to an obligate endosymbiont. However, several species of both soft and hard ticks seem to contain bacteria related to CLEAA, suggestive of an ancient relationship. In addition to providing vitamins, CLEAA might play a role in host immune system development, as shown for Wigglesworthia in tsetse flies (Weiss et al. 2011), and due to its presence in salivary glands CLEAA might be contributing certain metabolites to tick saliva, or this behavior could be a relic from an ancestral sylvatic life cycle. Functional studies of CLEAA and analyzing the genomes of other “Coxiella-like bacteria” detected in ticks from around the world may shed more light on this apparent paradox. Nevertheless, this dependency of the Lone star tick on CLEAA for reproductive success could be exploited in the field for improved control of A. americanum and the pathogens it vectors (Zhong et al. 2007) and further illuminates the complex metabolic interdependencies that exist between arthropods and their nutrient-provisioning endosymbionts.
Materials and Methods
Tick Microbiome Analysis
DNA isolated from four adult female A. americanum ticks (procured from Oklahoma State University Tick Rearing Facility) was amplified using a 16S rDNA polymerase chain reaction (PCR) primer set (V3-V4; 550 bp amplicon; Nextera DNA sample preparation kit, Illumina) and sequenced using a MiSeq system (Illumina). The sequencing reads were trimmed using Trimmomatic v0.30 (Bolger et al. 2014) and approximately 65,000 reads obtained from each sample were searched against the Greengenes 16S rRNA gene database (DeSantis et al. 2006) to identify bacterial species present in each tick.
CLEAA Genome Sequencing and Assembly
To analyze the genome of CLEAA, DNA from four female ticks was pooled and sequenced on a single lane of an Illumina HiSeq 2000, which produced approximately 152 million paired-end 100-bp reads. Sequencing reads were trimmed using Trimmomatic v0.30 (Bolger et al. 2014) and approximately 119 million paired reads were aligned to several genomes related to C. burnetii (accession numbers: NC_011527, NC_009726, NZ_AAUP01000001, NC_010115, NZ_AAYJ01000001, NC_002971.3, DQ912980.1, NC_002942.5, AY939824.1, and NZ_AAQJ01000001), to obtain a smaller set of 79,628 paired-end reads, as described earlier (Sloan and Moran 2012). These reads along with approximately 22 million pairs of original reads were assembled into six contigs using Velvet (Zerbino and Birney 2008), and SOAP2 (Li et al. 2009), and the gaps were closed using PCR. The full set of trimmed reads was mapped back to the closed genome using Bowtie2 (Langmead and Salzberg 2012) (∼4.3 million paired reads mapped uniquely) to obtain the final genome sequence, which was submitted to NCBI GenBank (accession number CP007541). The finished genome was annotated using the Joint Genome Institute IMG ER pipeline (Markowitz et al. 2009) and the NCBI Prokaryotic Genome Annotation Pipeline.
To identify metabolic pathways, proteins from CLEAA (537) and C. burnetii RSA 493 (1,818) were annotated with metabolic information from the Kyoto Encyclopedia of Genes and Genomes (KEGG) using blastKOALA (Kanehisa et al. 2014). Proteins with existing KEGG pathway, module, or functional hierarchy (BRITE) annotations were binned into 18 general metabolic categories based on their annotation. Nonhypothetical proteins that had no significant matches in any KEGG database were searched against the Conserved Domain Database at NCBI and subsequently manually binned. Orthologs of CLEAA genes in Wigglesworthia glossinidia (NC_004344.2), Candidatus Riesia pediculicola (NC_014109.1), and Wolbachia wCle (AP013028.1) were identified by reciprocal blast, and proteins were binned into metabolic groups using the RAST server (Aziz et al. 2008) (supplementary fig. S3, Supplementary Material online).
Phylogenetic Analyses
We included 33 fully sequenced genomes to estimate a robust phylogenetic framework for placing CLEAA, a presumed member of the family Coxiellaceae (Gammaproteobacteria: Legionellales). Taxons were sampled based on a previous phylogeny of Gammaproteobacteria (Williams et al. 2010), including representatives from major groups at the base of the tree, wherein Legionellales was positioned: Xanthomonadales (Xanthomonas axonopodis pv. citri str. 306); Cardiobacteriales (Cardiobacterium hominis ATCC 15826, Dichelobacter nodosus VCS1703A); Chromatiales (Alkalilimnicola ehrlichii MLHE-1, Nitrosococcus oceani ATCC 19707); Methylococcales (Methylococcus capsulatus str. Bath); Oceanospirillales (Marinomonas sp. MWYL1); Aeromonadales (Aeromonas salmonicida subsp. salmonicida A449); and Thiotrichales (Francisella tularensis subsp. tularensis SCHU S4). Four additional species were included from derived gammaproteobacterial lineages: Alteromonadales (Shewanella oneidensis str. MR-1); Vibrionales (Vibrio cholerae O1 biovar El Tor str. N16961); Pasteurellales (Pasteurella multocida subsp. multocida str. Pm70); and Enterobacteriales (Escherichia coli str. K-12 substr. MG1655). To ensure robust sampling within Legionellales, families Legionellaceae and Coxiellaceae were adequately sampled. For Legionellaceae, Legionella longbeachae str. NSW150 and six strains of L. pneumophila (2300/99 Alcoy, Corby, Lens, Paris, 130b, and ATCC 43290) were included. For Coxiellaceae, Rickettsiella grylli, Diplorickettsia massiliensis str. 20B, and eleven strains of C. burnetti (cb109, RSA_331, CbuK Q154, Z3055, Cb175 Guyana, RSA 493, Dugway 5J108‐111, CbuG Q212, MSU Goat Q177, Q321, and Cb185) were included.
Utilizing FastOrtho, an in-house modified version of OrthoMCL (Li et al. 2003), orthologous groups of 161 proteins (supplementary table S3, Supplementary Material online) were generated and processed for phylogeny estimation as previously described (Driscoll et al. 2013). Utilizing Bayesian inference for phylogeny estimation, two independent Markov chains were run in parallel using PhyloBayes MPI v.1.2e (Lartillot et al. 2013) under the CAT-GTR (general time reversible) model, with the bipartition frequencies analyzed at various time points using the bpcomp program. For tree-building, appropriate burn-in values were determined by plotting the log-likelihoods for each chain over sampled generations (time). Analyses were considered complete when the maximum difference in bipartition frequencies between the two chains was less than 0.1. Ultimately, a burn-in value of 1,000, with sampling every two trees, was used to build a consensus tree. Maximum-likelihood trees (WAG [Whelan and Goldman] and Le and Gascuel [LG] substitution models) were also generated using RAxML (Stamatakis et al. 2008) to confirm the Bayesian tree.
To better evaluate the phylogenetic position of CLEAA within Coxiellaceae, a phylogeny was estimated based on 16S rDNA sequences. The CLEAA 16S rDNA sequence was used in a BLASTN search against the NCBI nr database, with 160 sequences retrieved having 91% identity or greater. Six outgroup sequences from species of Legionella, Rickettsiella, and Diplorickettsia were also included. All 167 16S rDNA sequences were aligned using MUSCLE v3.6 (Edgar 2004) with default parameters. Ambiguously aligned positions, the majority being present within the variable regions of the small subunit rRNA structure, were culled using Gblocks (Castresana 2000; Talavera and Castresana 2007). Phylogenies of both the unmasked and masked alignments were estimated under maximum likelihood using RAxML (Stamatakis et al. 2008). The GTR substitution model was used with estimation of GAMMA and the proportion of invariable sites. Branch support was measured with bootstrapping (1,000 replications). A Bayesian tree was also generated using PhyloBayes MPI v.1.2e (Lartillot et al. 2013) as described above.
Supplementary Material
Supplementary references, figures S1–S5, and tables S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Zakee Sabree for assistance with 16S rDNA sequencing, Daniel Sloan for helpful discussions, and Christine Sislak for tick DNA extraction. This work was supported in part by Portland State University and by grants from Medical Research Foundation of Oregon and American Heart Institution to R.R. J.J.G. acknowledges support from National Institutes of Health/National Institute of Allergy and Infectious Diseases grants R01AI017828 and R01AI043006 (Abdu F. Azad, University of Maryland Baltimore, School of Medicine).
Literature Cited
- Ahantarig A, Trinachartvanit W, Baimai V, Grubhoffer L. Hard ticks and their bacterial endosymbionts (or would be pathogens) Folia Microbiol. 2013;58:419–428. doi: 10.1007/s12223-013-0222-1. [DOI] [PubMed] [Google Scholar]
- Akman L, et al. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet. 2002;32:402–407. doi: 10.1038/ng986. [DOI] [PubMed] [Google Scholar]
- Aziz RK, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol. 2005;59:155–189. doi: 10.1146/annurev.micro.59.030804.121041. [DOI] [PubMed] [Google Scholar]
- Beare PA, et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio. 2011;2:e00175–11. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GM, Moran NA. Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol Evol. 2013;5:1675–1688. doi: 10.1093/gbe/evt118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner P. Endosymbiosis of animals with plant microorganisms. New York: Interscience; 1965. [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Childs JE, Paddock CD. The ascendancy of Amblyomma americanum as a vector of pathogens affecting humans in the United States. Annu Rev Entomol. 2003;48:307–337. doi: 10.1146/annurev.ento.48.091801.112728. [DOI] [PubMed] [Google Scholar]
- Clay K, et al. Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol Ecol. 2008;17:4371–4381. doi: 10.1111/j.1365-294x.2008.03914.x. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss, and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll T, Gillespie JJ, Nordberg EK, Azad AF, Sobral BW. Bacterial DNA sifted from the Trichoplax adhaerens (Animalia: Placozoa) genome project reveals a putative rickettsial endosymbiont. Genome Biol Evol. 2013;5:621–645. doi: 10.1093/gbe/evt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, et al. A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J Bacteriol. 2012;194:376–394. doi: 10.1128/JB.06244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise SR, Elshahed MS, Little SE. Bacterial diversity in Amblyomma americanum (Acari: Ixodidae) with a focus on members of the genus Rickettsia. J Med Entomol. 2010;47:258–268. doi: 10.1603/me09197. [DOI] [PubMed] [Google Scholar]
- Husnik F, et al. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell. 2013;153:1567–1578. doi: 10.1016/j.cell.2013.05.040. [DOI] [PubMed] [Google Scholar]
- Jasinskas A, Zhong J, Barbour AG. Highly prevalent Coxiella sp. bacterium in the tick vector Amblyomma americanum. Appl Environ Microbiol. 2007;73:334–336. doi: 10.1128/AEM.02009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, et al. Extensive diversity of Rickettsiales bacteria in two species of ticks from China and the evolution of the Rickettsiales. BMC Evol Biol. 2014;14:167. doi: 10.1186/s12862-014-0167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkness EF, et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc Natl Acad Sci U S A. 2010;107:12168–12173. doi: 10.1073/pnas.1003379107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko O, Stein BD, Grindle N, Clay K, Fuqua C. Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl Environ Microbiol. 2007;73:6584–6594. doi: 10.1128/AEM.00537-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 2013;62:611–615. doi: 10.1093/sysbio/syt022. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Markowitz VM, et al. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci U S A. 2009;106:15394–15399. doi: 10.1073/pnas.0906424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Functional convergence in reduced genomes of bacterial symbionts spanning 200 million years of evolution. Genome Biol Evol. 2010;2:708–718. doi: 10.1093/gbe/evq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- Moran NA. Symbiosis as an adaptive process and source of phenotypic complexity. Proc Natl Acad Sci U S A. 2007;104:8627–8633. doi: 10.1073/pnas.0611659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Nakabachi A, et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- Nikoh N, et al. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc Natl Acad Sci U S A. 2014;111:10257–10262. doi: 10.1073/pnas.1409284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rounds MA, et al. Identification of endosymbionts in ticks by broad-range polymerase chain reaction and electrospray ionization mass spectrometry. J Med Entomol. 2012;49:843–850. doi: 10.1603/me12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabree ZL, et al. Genome shrinkage and loss of nutrient-providing potential in the obligate symbiont of the primitive termite Mastotermes darwiniensis. Appl Environ Microbiol. 2012;78:204–210. doi: 10.1128/AEM.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassera D, Epis S, Pajoro M, Bandi C. Microbial symbiosis and the control of vector-borne pathogens in tsetse flies, human lice, and triatomine bugs. Pathog Glob Health. 2013;107:285–292. doi: 10.1179/2047773213Y.0000000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenobu S, et al. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Sloan DB, et al. Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol Bio Evol. 2014;31:857–871. doi: 10.1093/molbev/msu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Moran NA. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol Biol Evol. 2012;29:3781–3792. doi: 10.1093/molbev/mss180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Stead CM, Omsland A, Beare PA, Sandoz KM, Heinzen RA. Sec-mediated secretion by Coxiella burnetii. BMC Microbiol. 2013;13:222. doi: 10.1186/1471-2180-13-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Warnecke F, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- Weiss BL, Wang J, Aksoy S. Tsetse immune system maturation requires the presence of obligate symbionts in larvae. PLoS Biol. 2011;9:e1000619. doi: 10.1371/journal.pbio.1000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen JJ. Strategies of genomic integration within insect-bacterial mutualisms. Biol Bull. 2012;223:112–122. doi: 10.1086/BBLv223n1p112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KP, et al. Phylogeny of gammaproteobacteria. J Bacteriol. 2010;192:2305–2314. doi: 10.1128/JB.01480-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J. Coxiella-like endosymbionts. Adv Exp Med Biol. 2012;984:365–379. doi: 10.1007/978-94-007-4315-1_18. [DOI] [PubMed] [Google Scholar]
- Zhong J, Jasinskas A, Barbour AG. Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS One. 2007;2:e405. doi: 10.1371/journal.pone.0000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.