


Abstract
When analyzing single-cell RNA-seq data, constructing a pseudo-temporal path to order cells based on the gradual transition of their transcriptomes is a useful way to study gene expression dynamics in a heterogeneous cell population. Currently, a limited number of computational tools are available for this task, and quantitative methods for comparing different tools are lacking. Tools for Single Cell Analysis (TSCAN) is a software tool developed to better support in silico pseudo-Time reconstruction in Single-Cell RNA-seq ANalysis. TSCAN uses a cluster-based minimum spanning tree (MST) approach to order cells. Cells are first grouped into clusters and an MST is then constructed to connect cluster centers. Pseudo-time is obtained by projecting each cell onto the tree, and the ordered sequence of cells can be used to study dynamic changes of gene expression along the pseudo-time. Clustering cells before MST construction reduces the complexity of the tree space. This often leads to improved cell ordering. It also allows users to conveniently adjust the ordering based on prior knowledge. TSCAN has a graphical user interface (GUI) to support data visualization and user interaction. Furthermore, quantitative measures are developed to objectively evaluate and compare different pseudo-time reconstruction methods. TSCAN is available at https://github.com/zji90/TSCAN and as a Bioconductor package.
INTRODUCTION
Single-cell RNA-seq is a transformative technology that allows researchers to measure transcriptomes of individual cells (1,2). Unlike single-cell RNA-seq, conventional RNA-seq (also referred to as ‘bulk RNA-seq’) (3,4) or microarray (5,6) experiments are used to measure average gene expression of a cell population. In many applications, the cell population is heterogeneous and contains multiple cell types. As a result, the average transcriptome of the population may fail to capture important transcriptional signals in individual cells. Sometimes, using the population average to study cell type specific behavior can also be misleading due to Simpson's paradox (7,8). With the ability to measure the transcriptome of each individual cell, single-cell RNA-seq is capable of generating a higher resolution view of the gene expression landscape in a heterogeneous cell population (9–11). This can lead to a more accurate molecular characterization of a complex biological phenomenon (12).
As demonstrated by (8), one useful way to gain biological insights from single-cell RNA-seq data is to computationally order cells according to the gradual transition of their transcriptomes. For example, in a cell differentiation process, cells can evolve at different speeds. A sample of cells collected at a particular time point during differentiation can actually contain cells representing different differentiation stages. Using single-cell RNA-seq data, one may construct an ordered sequence of cells to describe the gradual transition of the single-cell transcriptome. If this in silico order is consistent with cells’ true differentiation stages, then by analyzing how gene expression changes along this ordered sequence of cells, one will be able to obtain insights on the transcriptome dynamics during the differentiation process. The process of ordering cells in silico is called pseudo-time reconstruction because it mimics a procedure that places cells on a time axis. Despite the use of the term ‘time’, ‘pseudo-time reconstruction’ can more generally refer to any cell ordering procedure regardless of whether the ordering has a time interpretation (e.g. the ordering of cells may reflect cells’ spatial order rather than their temporal order).
Several computational methods have been proposed to analyze single-cell genomic data such as single-cell mass cytometry data (13–15) and single-cell gene expression data (8,16–19). However, for pseudo-time reconstruction in single-cell RNA-seq data, there are only a limited number of methods that have been systematically tested and have easily accessible software tools. In (8), an unsupervised approach Monocle was proposed to solve this problem. Monocle uses a minimum spanning tree (MST) to describe the transition structure among cells. The backbone of the tree is extracted to serve as the pseudo-time axis to place cells in order. A similar unsupervised spanning-tree approach has also been used previously for analyzing flow cytometry data (15). As an unsupervised approach, pseudo-time reconstruction based on spanning trees does not require any prior information on cell ordering. When temporal order information is available, an alternative approach to analyzing single-cell gene expression dynamics is to use such information to supervise the analysis. An example of this supervised approach is SCUBA (16). SCUBA uses bifurcation analysis to recover biological lineages from single-cell gene expression data collected from multiple time points. Here, the multiple time points in a time course experiment are used to supervise the cell ordering and analyses of gene expression dynamics in cell differentiation processes. By using the available time information, supervised methods can be more accurate than unsupervised methods. However, in applications where time information is not available (e.g. if one needs to analyze a heterogeneous cell population from a single disease sample rather than from a time course experiment), the supervised approach is not applicable and one has to rely on unsupervised methods. For these reasons, both supervised and unsupervised methods are useful. The primary focus of this article is the unsupervised approach.
One potential limitation of Monocle is that its tree is constructed to connect individual cells. Since the cell number is large, the tree space is highly complex. Tree inference in such a complex space is associated with high variability and can be highly unstable. As a result, the optimal tree found by the algorithm may not represent cells’ true biological order. This can be illustrated using a toy example in Figure 1A–C. Here dots represent cells placed in a two dimensional space (e.g. the space corresponding to the top two principal components of the gene expression profiles), and the true biological time runs top-down vertically. The MST solution is not unique. Figure 1A and B show two possible solutions. When a slight measurement noise pushes the cell labeled by ‘*’ away from other cells, the tree in Figure 1A can easily become a better solution based on the MST algorithm. However, this solution places cells in an order different from their true biological order. One approach that may alleviate this problem is to reduce the complexity of the tree space. This is analogous to the bias-variance tradeoff in the statistics and machine learning literature. For instance, if one clusters similar cells together as in Figure 1C and then constructs a tree to connect the cluster centers, recovering the true time-axis becomes easier. In this article, we exploit this idea to develop Tools for Single Cell Analysis (TSCAN), a new tool for pseudo-time reconstruction. One additional advantage offered by clustering cells is that users can more easily adjust the order of tree nodes (i.e. cell clusters) manually if they want to do so, since the number of clusters usually is not big. By contrast, manually specifying the order of hundreds of cells is much more difficult.
Figure 1.

TSCAN Overview. (A–B) A toy example illustrating a limitation of cell-based MST. Here cells (blue circles) are placed in a two dimensional space, and the true biological time runs top-down. An MST that connects cells is not unique. Both (A) and (B) are possible solutions. (B) is more consistent with the truth. However, in reality, random measurement noise may shift the cell labeled by ‘*’ away from other cells as indicated by the arrow and dashed lines. As a result, (B) is no longer an MST. The MST in (A) on the other hand does not reflect the true order of cells. (C) The true time-axis can be found if one first groups similar cells into clusters and then constructs an MST to connect cluster centers. (D) TSCAN first constructs cluster-based MST (five clusters of cells encoded by different colors are shown as an example; numbers indicate cluster centers). The tree can have multiple paths (e.g. 1-2-3-4 or 1-2-3-5). TSCAN orders cells along each path by projecting each cell onto the tree edge. (E) The number of principal components to retain is determined by finding the best piecewise linear fit consisting of two lines (dashed).
Another limitation of existing tools is that they are mostly command-line driven and do not allow users to interactively adjust or fine-tune the analysis. For example, users often want to use their existing knowledge such as marker genes to filter out contamination cells, determine the time origin or manually change the order of certain tree nodes. However, these operations are not convenient for a command-line driven software tool such as Monocle. TSCAN addresses this limitation by providing a graphical user interface (GUI) (Figure 2). Using the GUI, users can interactively and conveniently incorporate prior biological information into the pseudo-time reconstruction analysis.
Figure 2.

TSCAN graphical user interface. Left panel contains function menus and tools for setting parameters. Right panel displays data and results. The top scatter plot shows the MST constructed for the LPS data (see Results). Cells (dots) are displayed based on their first two principal components. Clusters of cells are indicated by different colors. Numbers are cluster centers. Expression level of a marker gene BCL3 is shown for each cell. Larger marker size means higher expression. The bottom plot shows the average BCL3 expression for each tree node, standardized across all nodes to have zero mean and unit standard deviation.
Last but not least, when several different pseudo-time reconstruction methods are available, being able to evaluate and compare them to identify the best solution is important. However, how to evaluate different pseudo-time reconstruction methods is also an open problem. Objective measures for comparing different methods are still lacking. This article introduces several quantitative measures for evaluating different cell ordering methods. Using these objective measures, we show that TSCAN is capable of providing more reliable unsupervised pseudo-time reconstruction results compared to alternative methods.
MATERIALS AND METHODS
Problem formulation
Consider a representative sample of N cells drawn from a
heterogeneous cell population. Suppose the transcriptome 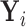 of each cell
i ∈ {1, 2, …, N} has been
profiled using single-cell RNA-seq. Here,
of each cell
i ∈ {1, 2, …, N} has been
profiled using single-cell RNA-seq. Here, 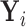 is a G dimensional vector consisting of gene expression measurements for G genes. Assume that
is a G dimensional vector consisting of gene expression measurements for G genes. Assume that 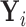 is appropriately
transformed (e.g. by taking logarithm) and normalized across cells. The single cell
ordering problem, also called pseudo-time reconstruction, is to place cells in an order
based on the gradual transition of
is appropriately
transformed (e.g. by taking logarithm) and normalized across cells. The single cell
ordering problem, also called pseudo-time reconstruction, is to place cells in an order
based on the gradual transition of 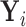 .
.
TSCAN orders cells in three steps. First, cells with similar gene expression profiles are grouped into clusters. Second, a MST is constructed to connect all cluster centers. Finally, cells are projected to the tree backbone to determine their pseudo-time and order (Figure 1D). Once cells are ordered, users may use the ordered sequence to study cell state transition and gene expression dynamics in the underlying biological process from which the cells are sampled.
Preprocessing
Before pseudo-time reconstruction, the raw gene expression data are processed as follows. First, genes with zero read count in all samples are excluded. Second, in order to alleviate the effect of drop-out events (20) on the subsequent analyses, genes with similar expression patterns are grouped into clusters by hierarchical clustering (using Euclidean distance and complete linkage). The number of clusters is set to be 5% of the total number of genes with non-zero expression. For each cluster and each cell, the expression measurements of all genes in the cluster are averaged to produce a cluster-level expression which will be used for subsequent MST construction. The drop-out event refers to the phenomenon that expressed genes, some of which are highly expressed, may have zero read count in some cells as their molecules may not be captured and amplified by chance. This is a common phenomenon in single-cell RNA-seq data. By averaging across many genes, the cluster-level expression is more stable and has smaller estimation variance compared to the measurements of individual genes. This can help to dilute the impact of drop-out events.
After gene clustering, single-cell transcriptome for cell i
becomes a H dimensional vector 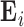 . Here,
H is the number of gene clusters.
. Here,
H is the number of gene clusters.
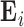 still has high dimension, and many components in this
vector are still correlated. The dimensionality makes visualization and statistical
modeling difficult. For this reason, TSCAN further reduces the dimension of
still has high dimension, and many components in this
vector are still correlated. The dimensionality makes visualization and statistical
modeling difficult. For this reason, TSCAN further reduces the dimension of
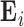 using principal component analysis (PCA). Briefly,
using principal component analysis (PCA). Briefly,
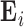 from all cells are organized into a H × N matrix
from all cells are organized into a H × N matrix
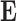 . Each row corresponds to a gene cluster. The matrix is
standardized such that expression values within each row have zero mean and unit standard
deviation. Then PCA is run on the standardized matrix, and the top K principal components (PCs) are retained. After PCA, the H dimensional vector
. Each row corresponds to a gene cluster. The matrix is
standardized such that expression values within each row have zero mean and unit standard
deviation. Then PCA is run on the standardized matrix, and the top K principal components (PCs) are retained. After PCA, the H dimensional vector 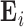 is mapped to a lower dimensional space
and becomes a K dimensional vector
is mapped to a lower dimensional space
and becomes a K dimensional vector 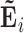 .
Here, K is much smaller than H.
.
Here, K is much smaller than H.
In order to determine K (i.e. how many PCs to retain),
TSCAN uses the following criterion. First, let λi be the data variance
explained by the ith PC. Define 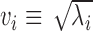 .
. 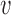 i is a
non-increasing function of i. This function can be
approximated using a continuous piecewise linear model
i is a
non-increasing function of i. This function can be
approximated using a continuous piecewise linear model 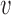 i = f(i) + ε where ε represents noise
and f(i) consists of two
regression lines (Figure 1E):
i = f(i) + ε where ε represents noise
and f(i) consists of two
regression lines (Figure 1E):
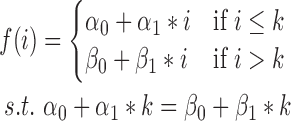 |
(1) |
TSCAN computes the least squares fit of this
model using the first 20 PCs. The fitted model varies when one changes k. TSCAN tries different k ∈ [2, 19]
and finds the k that produces the smallest squared error,
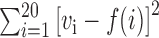 . This k will be used as the number of PCs to retain.
. This k will be used as the number of PCs to retain.
Cell clustering
After dimension reduction, cells with similar expression profiles are grouped into
clusters using the model-based clustering approach described in (21). The clustering is performed using the mclust (22) package in R which fits a
mixture of multivariate normal distributions to the data 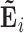 . The
variance-covariance matrix for each normal component in this mixture is designated as
‘ellipsoidal, varying volume, shape and orientation’. The number of clusters is chosen by
mclust using the Bayesian Information Criterion (BIC).
After model fitting, the posterior probability that each cell belongs to each cluster can
be computed. Cells are assigned to clusters based on the largest posterior probability.
For each cluster, the cluster mean of
. The
variance-covariance matrix for each normal component in this mixture is designated as
‘ellipsoidal, varying volume, shape and orientation’. The number of clusters is chosen by
mclust using the Bayesian Information Criterion (BIC).
After model fitting, the posterior probability that each cell belongs to each cluster can
be computed. Cells are assigned to clusters based on the largest posterior probability.
For each cluster, the cluster mean of 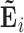 is treated as the
cluster center. Instead of using the cluster number determined by mclust based on BIC, users also have the option to specify their own cluster
number.
is treated as the
cluster center. Instead of using the cluster number determined by mclust based on BIC, users also have the option to specify their own cluster
number.
Ordering cell clusters by MST
Next, TSCAN constructs a minimum spanning tree to connect all cluster centers. In a connected and undirected graph, a spanning tree is a subgraph that is a tree and connects all the vertices (or ‘nodes’). Suppose each edge in the graph has a length equal to the Euclidean distance between the two nodes (i.e. cluster centers) connected by the edge. A MST is a spanning tree with the smallest total edge length among all possible spanning trees. Unlike the MST approach used by Monocle where the tree is constructed to connect individual cells, the MST in TSCAN is constructed to connect clusters of cells. Clustering cells reduces the variability and complexity of the tree space. The cluster level MST therefore may yield better and more stable estimates of the tree backbone which largely determines the cell ordering. Another advantage of clustering is that it dramatically reduces the number of tree nodes, so that it becomes easier for users to interactively fine-tune the analysis later (e.g. manually adjust the order of tree nodes).
A tree may have multiple branches. By default, we define the main path of the tree (solid lines in Figure 1D) as the path with the largest number of clusters. If more than one path has the same largest number of clusters, the path with the largest number of cells becomes the main path. The main path has two ends. Without other information, one end will be randomly picked up as the origin of the path. Alternatively, users can specify one end as the origin themselves using information such as marker gene expression. After the main path and its origin are determined, TSCAN will enumerate all branching paths starting from the origin. For instance, assume cluster 1 in Figure 1D is chosen as the origin, then TSCAN will report a main path 1-2-3-4 and a branching path 1-2-3-5. If the cluster order generated by the algorithm is not satisfactory to users, they have options to manually specify the paths and the order of clusters along each path.
Cell ordering and pseudo-time calculation
Once the cluster-level ordering is determined, individual cells are projected onto tree
edges to create cell-level ordering along the main path and each branching path. For each
path, all clusters on the path are collected. All cells in these clusters will be ordered
along the path as follows. Let Ci (i = 1, 2, ...,
M) indicate the ordered clusters, where M is the number of clusters on the ordered path. Suppose
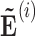 and
and 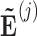 are the cluster centers for two neighboring clusters Ci and Cj in the path, and
suppose Ci precedes Cj in the ordering. The edge that connects the two clusters is
determined by
are the cluster centers for two neighboring clusters Ci and Cj in the path, and
suppose Ci precedes Cj in the ordering. The edge that connects the two clusters is
determined by 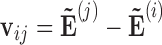 , and
the projection of cell k to the edge is determined by the
inner product
, and
the projection of cell k to the edge is determined by the
inner product 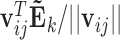 where ||.||
is the l2-norm of a vector. Cells in cluster
C1 are all projected onto the edge that
connects C1 and C2. Cells in cluster CM
are all projected onto the edge that connects CM −
1 and CM. Cells from an intermediate
cluster Cm(1 < m < M) are divided into two groups according to whether they are closer to the
center of cluster Cm − 1 or to the center of
cluster Cm + 1 in terms of Euclidean distances.
Cells closer to the center of cluster Cm − 1 are
projected onto the edge that connects clusters Cm −
1 and Cm, while cells closer to the
center of cluster Cm + 1 are mapped to the edge
connecting clusters Cm and Cm + 1.
where ||.||
is the l2-norm of a vector. Cells in cluster
C1 are all projected onto the edge that
connects C1 and C2. Cells in cluster CM
are all projected onto the edge that connects CM −
1 and CM. Cells from an intermediate
cluster Cm(1 < m < M) are divided into two groups according to whether they are closer to the
center of cluster Cm − 1 or to the center of
cluster Cm + 1 in terms of Euclidean distances.
Cells closer to the center of cluster Cm − 1 are
projected onto the edge that connects clusters Cm −
1 and Cm, while cells closer to the
center of cluster Cm + 1 are mapped to the edge
connecting clusters Cm and Cm + 1.
Cell orderings are determined in three steps. First, for cells which are in the same cluster and are projected onto the same edge, their order is determined by the projected values on the edge. Second, within each cluster, the order of cells projected onto different edges is determined by the order of edges, which is given by the cluster-level ordering. Third, the order of cells in different clusters is determined by the order of clusters. In this way, all cells can be placed in order.
Once cells are ordered, pseudo-time is computed for each ordered path. For a given path, the order of a cell on the path is set to be its pseudo-time. For instance, the pseudo-time for the kth cell on a path is set to k. The pseudo-time is constructed separately for the main path and each branching path.
Detecting differentially expressed genes
After cells are ordered, one can detect differentially expressed genes following the approach in Monocle (8). A generalized additive model (GAM, effective degrees of freedom = 3) (23) is fitted for each gene to describe the functional relationship between its expression and pseudo-time. The GAM is fitted using the mgcv (23) package in R. The model is then compared to a null model that assumes constant expression along the pseudo-temporal path. The P-value is computed using a likelihood ratio test and then converted to false discovery rate (FDR) using the method in (24). By default, genes with FDR <0.05 are reported as differential. As in Monocle, the P-value and FDR are computed based on assuming that cell ordering is given. They do not consider uncertainties in cell ordering and that, instead of being determined by experiment design, cell ordering is derived from the same data used for analyzing differential expression. We note that how to evaluate statistical significance that further accounts for these additional uncertainties remains an open problem. It requires development of more sophisticated methods and a systematic investigation of how these additional uncertainties affect different methods (e.g. how P-values change when one treats cell ordering as an unknown parameter inferred from the data). These investigations are beyond the scope of the current study as the main focus of this article is how to improve and evaluate cell ordering.
Method evaluation
We use three methods to evaluate cell ordering performance. The first approach evaluates cell ordering accuracy based on the ordering expected by independent sources of information. It is assumed that external information not used in pseudo-time reconstruction is available to evaluate the pairwise order of cells. Formally, let π denote an ordered path of Nπ cells produced by a particular pseudo-time reconstruction method. Let g(π, i, j) be a score that characterizes how well the order of the ith and jth cells in the ordered path π matches their expected order based on the external information. We define pseudo-temporal ordering score (POS) for cell ordering π as the sum of g(π, i, j) for all pairs of cells:
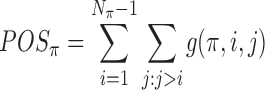 |
(2) |
Cell orderings π produced by different pseudo-time reconstruction methods can then be compared based on the POS score.
As a concrete example, suppose one has single-cell RNA-seq data collected from a time
course experiment. In such an experiment, the data collection time is known. For the
purpose of evaluating unsupervised pseudo-time reconstruction methods, one can pool cells
from all time points together, pretend that the data collection time for each cell is
unknown, and apply different methods to reconstruct pseudo-time. Different methods will
then be evaluated by comparing their cell ordering results to the order of cells based on
the true data collection time. For instance, if one has N
cells collected at V time points during a differentiation
process. Among the N cells, N cells are from time T
cells are from time T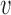 (T1 <
T2 < ⋅⋅⋅ < TV). Consider the ith cell and the jth cell in the
ordered path π where i precedes j (i.e. i < j).
One can define the pairwise score g(π, i, j) as follows:
(T1 <
T2 < ⋅⋅⋅ < TV). Consider the ith cell and the jth cell in the
ordered path π where i precedes j (i.e. i < j).
One can define the pairwise score g(π, i, j) as follows:
If the two cells are originally collected at the same time point (e.g. they are both from T
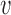 ), then g(π, i, j) = 0.
), then g(π, i, j) = 0.Otherwise, if the ith cell is collected from time point T
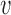 and the jth cell is collected from time point Tu, then g(π, i, j) = (u −
and the jth cell is collected from time point Tu, then g(π, i, j) = (u − 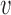 )/Dπ.
The value u −
)/Dπ.
The value u − 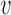 is positive if
is positive if
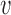 represents an earlier time point, or negative if
represents an earlier time point, or negative if
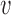 represents a time later than u.
represents a time later than u.
The denominator Dπ above is chosen to normalize POS so that POSπ ∈ [ − 1, 1] (i.e. the maximal and minimal POS among all possible orderings of cells within each path π is 1 and −1, respectively). Based on this definition, a cell ordering more consistent with the known data collection time will have higher POS score. POSπ = 1 indicates that the order of cells produced by pseudo-time reconstruction perfectly matches the order determined by the data collection time. POSπ = −1 indicates that the order of cells produced by pseudo-time reconstruction is in the opposite direction compared to the order determined by the data collection time. Using POS to evaluate cell ordering is based on assuming that the external information (i.e. the true data collection time in this example) can roughly reflect the true biological order of cells (e.g. the differentiation stage of cells). In reality, since cells collected at each time point are heterogeneous, it is possible that some cells collected at an earlier (less differentiated) time point in the differentiation time course are actually more differentiated than certain cells collected at a later time point. Despite this, it is often reasonable to expect that cells collected at the earlier time point ‘on average’ should be less differentiated than cells collected at the later time point. Therefore, the external information (i.e. the data collection time) used here can still roughly reflect the true biological order of cells and can be used as a surrogate to evaluate the cell ordering performance.
The second approach evaluates robustness of cell ordering by perturbing the original single-cell RNA-seq data set (see below). Each cell ordering method is applied to both the original data set and the perturbed data. Cell orderings produced by the original and perturbed data are then compared. To quantify the similarity between cell orderings in two pseudo-temporal paths π1 and π2, let A be the union of cells in π1 and π2, let |A| be the cardinality of A (i.e. the number of distinct cells in π1 and π2), and define the similarity score between π1 and π2 as:
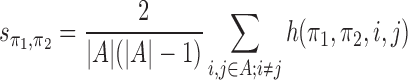 |
(3) |
Here, h(π1, π2, i, j) = 1 if the order of two cells i and j remains the same in π1 and π2 (i.e. i appears before or after j in both orderings), and h(π1, π2, i, j) = 0 otherwise. If either i or j occurs only in one path (e.g. i is in π1 but not π2), the orderings between i and j in π1 and π2 are viewed as inconsistent, and h(π1, π2, i, j) is also set to zero. A higher similarity score indicates that the two orderings π1 and π2 are more similar to each other, whereas a lower score indicates a larger deviation between the two orderings.
In this article, two different approaches were used to perturb data: cell-level
perturbation and expression-level perturbation. For cell-level perturbation, x percent (x = 95%, 90% or 75%) of
cells were randomly sampled from the original data set to serve as the perturbed data. The
gene expression profile of each cell remained unchanged. For expression-level
perturbation, we retained all cells in the original data set but added simulated noise to
their gene expression profiles (i.e. 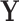 ). To generate noise, the
average expression value of each gene across all cells was computed and then subtracted
from the gene's expression value in each cell. Residuals obtained in this way were scaled
by multiplying with a scaling factor κ (κ = 5%, 10% or 25%). The scaled residuals were
then permuted and added back to the original expression values of the gene. For each
perturbation method and parameter value (x or κ), the
original data were independently perturbed 100 times to generate 100 perturbed data sets.
For each perturbed data set, similarity score between the original and perturbed orderings
was computed. Finally, the average similarity score from the 100 perturbations was
calculated to measure the robustness of each pseudo-time reconstruction method.
). To generate noise, the
average expression value of each gene across all cells was computed and then subtracted
from the gene's expression value in each cell. Residuals obtained in this way were scaled
by multiplying with a scaling factor κ (κ = 5%, 10% or 25%). The scaled residuals were
then permuted and added back to the original expression values of the gene. For each
perturbation method and parameter value (x or κ), the
original data were independently perturbed 100 times to generate 100 perturbed data sets.
For each perturbed data set, similarity score between the original and perturbed orderings
was computed. Finally, the average similarity score from the 100 perturbations was
calculated to measure the robustness of each pseudo-time reconstruction method.
The third approach evaluates the ability of a cell ordering method to detect known differentially expressed genes along the ordered cell path. Given a test data set, one can collect genes known to be differentially expressed along the biologically ordered sequence of cells and treat them as the gold standard. One can then detect differential genes along the pseudo-time axis and compare different methods based on how they rank gold standard genes.
TSCAN package and GUI
TSCAN is implemented as a Bioconductor package using the statistical programming language R. It can be run both in a command-line mode and through a GUI. The GUI is developed using the shiny package in R. It allows users to conveniently construct, visualize and tune cell ordering. For example, one can use the GUI to interactively trim unwanted cells based on expression levels of user-specified marker genes. One can also change the cluster-level ordering and then recompute the pseudo-time. TSCAN is open source, and it is freely available at https://github.com/zji90/TSCAN. Its bioconductor package can be downloaded from http://www.bioconductor.org/packages/release/bioc/html/TSCAN.html. An installation guide is provided in Supplementary Materials.
Data sets
Three data sets were compiled from the literature to evaluate TSCAN. The first data set
consists of single-cell RNA-seq samples from differentiating human skeletal muscle
myoblasts (HSMM) (8). It contains 271 cells
collected at 0, 24, 48 and 72 h after switching human myoblasts to low serum. The second
data set consists of single-cell RNA-seq samples collected after stimulating
bone-marrow-derived dendritic cells by lipopolysaccharide (LPS) (25). A total of 306 cells collected at 1, 2, 4 and 6 h after the
stimulation were used for our analysis. The third data set consists of single-cell RNA-seq
samples from hippocampal quiescent neural stem cells (qNSC) (26). It contains 172 cells collected from the same cell population. For
all data sets, the normalized gene expression values (fragments per kilo base pairs per
million total reads for HSMM and transcripts per million total reads for LPS and qNSC)
were log2 transformed after adding a pseudo-count of 1. After the raw data
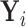 were processed to
were processed to 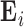 ,
,
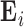 was used as input for different methods (i.e. TSCAN,
Monocle, Waterfall, SCUBA and Wanderlust below) to construct pseudo-time. The normalized
data for
was used as input for different methods (i.e. TSCAN,
Monocle, Waterfall, SCUBA and Wanderlust below) to construct pseudo-time. The normalized
data for 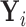 and
and 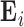 are available at the
TSCAN GitHub website (https://github.com/zji90/TSCANdata). The correspondence between sample
identifiers and sample collection time in the experiment is provided in Supplementary
Table S1.
are available at the
TSCAN GitHub website (https://github.com/zji90/TSCANdata). The correspondence between sample
identifiers and sample collection time in the experiment is provided in Supplementary
Table S1.
Comparisons with other methods
Supplementary Table S2 compares TSCAN with a number of other single cell data analysis methods. Among these methods, MARS-seq (17) and SINCE-PCR (19) do not have associated software for others to use. SPADE (15) and viSNE (13) are developed for analyzing mass cytometry or flow cytometry data, and they do not provide a cell ordering function. Diffusion map (27) is a dimension reduction technique used to define differentiation trajectories. It cannot perform cell ordering itself. The scLVM method (18) primarily focuses on identifying cell subpopulations. Again, it cannot order cells. For the above reasons, these methods are not compared with TSCAN in our subsequent data analyses.
Among the remaining methods, Monocle is designed to handle unsupervised cell ordering of single-cell RNA-seq and has a software package. Wanderlust (14) is originally developed for mass or flow cytometry data. It uses a graph-based trajectory detection algorithm to order cells under the assumption that there is no branch. We tailored its MATLAB code to allow it to take single-cell RNA-seq data as input. SCUBA (16), as discussed before, is a supervised approach. However, the SCUBA package also provides an option for unsupervised cell ordering which is based on fitting a principal curve to the data and then mapping cells onto the curve. Waterfall is a data analysis pipeline used by (26) to construct pseudo-time for their qNSC data. Similar to TSCAN, Waterfall first groups cells using k-means clustering before pseudo-time reconstruction. However, as an in-house data analysis pipeline, Waterfall does not have an associated software tool, and the pipeline cannot be directly used to analyze other data sets without manually editing the code. Also, an objective evaluation of the effects of cell clustering on cell ordering was not provided in (26). A systematic comparison among different pseudo-time reconstruction methods discussed above is still lacking. In order to benchmark the unsupervised cell ordering performance of TSCAN, we compared it with Monocle, Wanderlust, unsupervised SCUBA and Waterfall in our subsequent data analyses.
RESULTS
We evaluated TSCAN using the three data sets, HSMM, LPS and qNSC, described above. HSMM and LPS data sets contain cells collected from multiple time points in time course experiments. The actual data collection time provides important external information for evaluating cell orderings produced by unsupervised pseudo-time reconstruction methods. In our evaluation, cells from different time points were pooled together. We pretended that their data collection time were unknown. We applied different pseudo-time reconstruction methods to order these cells. Methods were then compared in terms of their accuracy, robustness and ability to detect known differentially expressed genes. Accuracy was characterized by the POS score computed using cells’ actual data collection time. Robustness was characterized by the cell ordering similarity between the original and perturbed data. In the qNSC data set, all cells were collected from the same cell population. Since there was no external information such as multiple time points to calculate the POS score, we only evaluated robustness and the ability to detect known differentially expressed genes in this data set.
HSMM analysis using a priori chosen genes for pseudo-time reconstruction
We first evaluated the performance of TSCAN using the HSMM data set, originally analyzed
by (8) using Monocle. In the original Monocle
analysis conducted by (8), the pseudo-time was
constructed using 518 genes chosen a priori before ordering
the single-cell RNA-seq data. These genes were derived by comparing different
differentiation time points and therefore are known to be associated with myoblast
differentiation. They represent a strong piece of prior knowledge for pseudo-time
reconstruction. In real applications, if one has strong prior information such as these
518 genes, one can use them as the input (to replace 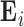 ) for TSCAN and Monocle to
construct MST. We first performed analyses in this way by using the same 518 genes for
pseudo-time reconstruction. Figure 3A and B show the cluster-level MST constructed by TSCAN.
Consistent with the original Monocle results reported in (8), TSCAN also detected two branches of biological process: the default main
path 1-3-5-2 and a branching path 1-3-5-4. For the main path 1-3-5-2, neither Monocle nor
TSCAN can determine whether node 1 or 2 should be the starting time point without other
information. Therefore, the path has two possible directions. By default, TSCAN randomly
picks one direction. However, if users have marker genes to inform the direction of the
pseudo-temporal path, they can use this information in TSCAN. To illustrate, ENO3 is a
marker gene for myoblast differentiation. Its expression is expected to increase as the
differentiation progresses. After providing ENO3 as a marker gene, TSCAN displays its
expression in each tree node. In this way, one can see that cluster 1 has low ENO3
expression while cluster 2 has high ENO3 expression (Figure 3C). Thus, the starting time point should be in cluster 1. As reported in (8), the branching path in the MST constructed by Monocle
was driven by contaminating interstitial mesenchymal cells, and SPHK1 is a marker gene for
these contaminating cells. Consistent with this, displaying SPHK1 expression in the TSCAN
tree nodes shows that cluster 4 in the branching path 1-3-5-4 had high SPHK1 expression
(Figure 3D), indicating that this branch was driven
by contaminating cells. Thus, the branching path 1-3-5-4 was not further analyzed.
) for TSCAN and Monocle to
construct MST. We first performed analyses in this way by using the same 518 genes for
pseudo-time reconstruction. Figure 3A and B show the cluster-level MST constructed by TSCAN.
Consistent with the original Monocle results reported in (8), TSCAN also detected two branches of biological process: the default main
path 1-3-5-2 and a branching path 1-3-5-4. For the main path 1-3-5-2, neither Monocle nor
TSCAN can determine whether node 1 or 2 should be the starting time point without other
information. Therefore, the path has two possible directions. By default, TSCAN randomly
picks one direction. However, if users have marker genes to inform the direction of the
pseudo-temporal path, they can use this information in TSCAN. To illustrate, ENO3 is a
marker gene for myoblast differentiation. Its expression is expected to increase as the
differentiation progresses. After providing ENO3 as a marker gene, TSCAN displays its
expression in each tree node. In this way, one can see that cluster 1 has low ENO3
expression while cluster 2 has high ENO3 expression (Figure 3C). Thus, the starting time point should be in cluster 1. As reported in (8), the branching path in the MST constructed by Monocle
was driven by contaminating interstitial mesenchymal cells, and SPHK1 is a marker gene for
these contaminating cells. Consistent with this, displaying SPHK1 expression in the TSCAN
tree nodes shows that cluster 4 in the branching path 1-3-5-4 had high SPHK1 expression
(Figure 3D), indicating that this branch was driven
by contaminating cells. Thus, the branching path 1-3-5-4 was not further analyzed.
Figure 3.

TSCAN analysis in HSMM data set using 518 a priori
chosen genes for pseudo-time reconstruction. (A) MST reported by TSCAN is
shown in the three-dimensional space spanned by the first three PCs of
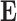 . (B) Users can display cells and MST
in chosen PCs (e.g. PC1 and PC2). (C) Mean expression level of ENO3 in
each cluster. (D) Mean expression level of SPHK1 in each cluster. Values
in (C) and (D) are both standardized across all clusters to have zero mean and unit
SD.
. (B) Users can display cells and MST
in chosen PCs (e.g. PC1 and PC2). (C) Mean expression level of ENO3 in
each cluster. (D) Mean expression level of SPHK1 in each cluster. Values
in (C) and (D) are both standardized across all clusters to have zero mean and unit
SD.
For both Monocle and TSCAN, we calculated the POS score along their reported main path. The cell ordering along each path reported by each method is provided in Supplementary Table S3. According to (8), the main path produced by Monocle in this analysis corresponds to myoblast differentiation which is the biological process of interest. Figure 4A shows the POS scores. TSCAN outperformed Monocle in terms of the POS.
Figure 4.

Evaluation results for different methods in HSMM data set where pseudo-time was constructed based on 518 a priori chosen genes. (A) POS score. (B) Robustness measured by the average similarity score from 100 independent perturbations. The heat map shows robustness of each method in each perturbation scheme. Cell Perturb: cell-level perturbation. Expr Perturb: expression-level perturbation. (C) Mean rank of gold standard genes. (D) Number of detected gold standard genes among top differential genes.
In order to understand how cell clustering affects the cell ordering performance, we
tested a modified TSCAN (nocluTSCAN) in which the cell clustering step was skipped and MST
was constructed directly to connect individual cells based on 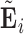 . The
analyzed path and direction were then determined as above by using SPHK1 to exclude the
contamination path and using ENO3 to determine the time origin. The comparison between
TSCAN and nocluTSCAN was well-controlled since everything was the same for these two
algorithms except for the use of cell clustering by TSCAN. By contrast, the performance
difference between Monocle and TSCAN represents a combined effect of many factors since
many of their implementation details are different. Many of these differences are
difficult to control for as they are hidden in the computer code.
. The
analyzed path and direction were then determined as above by using SPHK1 to exclude the
contamination path and using ENO3 to determine the time origin. The comparison between
TSCAN and nocluTSCAN was well-controlled since everything was the same for these two
algorithms except for the use of cell clustering by TSCAN. By contrast, the performance
difference between Monocle and TSCAN represents a combined effect of many factors since
many of their implementation details are different. Many of these differences are
difficult to control for as they are hidden in the computer code.
We also tested a marker-gene-only approach (marker) in which cells are directly ordered using the expression level of a marker gene (ENO3). Here, in order to conduct a relatively fair comparison with TSCAN, the marker-gene-only approach was only applied to cells from the analyzed TSCAN path (i.e. 1-3-5-2), and cells from the contaminated TSCAN branch (i.e. the branch with cluster 4) were excluded from this analysis. This yielded cell orderings in Supplementary Table S3. The comparison between the marker-gene-only approach and TSCAN can reveal whether the other genes used for pseudo-time reconstruction contribute additional information not provided by the marker gene (i.e. ENO3 in this example) for ordering cells.
As shown by Figure 4A, TSCAN had the best performance based on POS. It not only performed better than Monocle, but also outperformed nocluTSCAN and the marker-only approach, indicating that cell clustering and using multiple genes for ordering cells were both helpful for improving the pseudo-time reconstruction.
Next, we compared robustness of different methods based on cell ordering similarity between the original and perturbed data. Figure 4B shows the similarity scores when the perturbed data were generated by randomly subsampling 75%, 90% or 95% of cells from the original data set (cell-level perturbation) or by adding 5%, 10% or 25% random noise to the original gene expression values (expression-level perturbation). For each perturbed data set, the same protocol and marker genes as described above were used to determine the path direction and eliminate contaminating branch. Compared to Monocle and nocluTSCAN, TSCAN consistently produced higher similarity scores in all perturbation schemes (Figure 4B). This shows that cell clustering increased the stability (or equivalently, reduced the variability) of cell ordering when data were perturbed. The marker-gene-only approach was also more robust than Monocle and nocluTSCAN, and it showed similar level of robustness compared to TSCAN (Figure 4B). The robustness of the marker gene approach was not unexpected. For cell-level perturbation, genes’ expression values in each cell did not change. Consequently, the order of any pair of cells based on a marker gene's expression remained the same. The difference between the pseudo-temporal path in the original data and the path in the perturbed data in the marker gene approach mainly reflects the fact that these two paths did not contain the same set of cells. Note that not all cells in the original data were retained in the perturbed data set. Also, contaminating branches of MST constructed by TSCAN were excluded from our marker-gene-only analyses, and the contaminating branches in the original and perturbed data could contain different sets of cells. For expression-level perturbation, noises added to gene expression values represented 5–25% of the cross-cell variation of the true biological signal. Consequently, the pairwise order of many cells was still driven by the biological variation and hence remained unchanged in the marker-gene-based ordering.
It is important to point out that robustness alone is not sufficient to indicate good cell ordering performance. For instance, suppose each cell has an arbitrary name. If cells are ordered based on cell name rather than gene expression profile, the order of any pair of cells will remain the same regardless of how gene expression values are perturbed. As a result, the cell ordering is robust, but it does not have any biological meaning since the cell names are arbitrary. This is similar to the well-known variance-bias tradeoff in statistics: an estimator with zero variance may have huge bias. For this reason, robustness of a pseudo-time reconstruction method needs to be interpreted in the context of whether it leads to improved cell ordering accuracy (e.g. increased POS score). Although the marker-gene-only approach was more robust than Monocle and nocluTSCAN (Figure 4B), its cell ordering accuracy was lower than Monocle and TSCAN (Figure 4A), indicating that its bias-variance tradeoff is not optimal. By contrast, TSCAN was not only more robust (Figure 4B) but also ordered cells more accurately (Figure 4A) than Monocle and nocluTSCAN.
For each method, we next detected differentially expressed genes along the ordered main path of cells. We ranked genes based on FDR, and then different methods were compared based on their ability to find genes known to be involved in the biological process in question. For the HSMM data set, we compiled 13 genes (ENO3 excluded) known to be involved in myoblast differentiation according to (8) (Supplementary Table S4). Figure 4C shows the mean rank of these gold standard genes in the differential gene analysis. A smaller mean rank indicates better performance (i.e. gold standard genes are more likely to be ranked on top). Figure 4D shows the number of gold standard genes found in the top 200, 400, …, 2000 genes ranked by each method. Monocle and TSCAN had very similar results in this analysis, and both methods outperformed nocluTSCAN and the marker gene approach.
Besides TSCAN, we investigated two other ways to perform cell-clustering-based pseudo-time reconstruction. First, we replaced mclust by k-means clustering in the cell clustering step of TSCAN while keeping all other procedures the same (k-means TSCAN). Unlike mclust which allows ellipsoidal shape of clusters, k-means clustering only allows clusters with circle shape. In order to determine the cluster number of k-means, we used an approach similar to Figure 1E, with its y-axis changed to the proportion of total data variance unexplained by the cluster structure (Supplementary Materials). Second, we tested the Waterfall algorithm (26) which also uses k-means to cluster cells before cell ordering (Supplementary Materials). Waterfall does not provide a way to choose cluster number based on the data. Its cluster number was fixed to 10 which is the default value in Waterfall codes. Both the k-means TSCAN and Waterfall produced more robust cell ordering than Monocle and nocluTSCAN (Figure 4B). However, their cell ordering accuracy did not outperform Monocle and was clearly worse than TSCAN, as indicated by the POS score (Figure 4A) and differential gene detection performance (Figure 4C and D). This suggests that although k-means TSCAN and Waterfall reduced the cell ordering variability, their bias-variance tradeoff was not optimal for improving the cell ordering accuracy.
We also tested unsupervised SCUBA (i.e. the principal-curve-based SCUBA) and Wanderlust. For SCUBA, low expression of the marker gene ENO3 was used to determine the path origin. Wanderlust was run by using the cell with the highest ENO3 gene expression as the path origin (because the lowest ENO3 expression was zero, and zero occurred in many cells, making the choice of path origin not unique). The cell ordering reported by Wanderlust was then reversed so that the reversed path had low ENO3 expression at the beginning and high ENO3 expression at the end. The same approach was also used in other test data sets below to run the Wanderlust analyses. For both methods, after cells were ordered, GAM was used to detect differentially expressed genes as in TSCAN. Both Wanderlust and SCUBA were more robust than Monocle and nocluTSCAN (Figure 4B). However, they both had lower cell ordering accuracy compared to TSCAN (Figure 4A, C and D). In fact, TSCAN produced the highest POS score (Figure 4A) and best differential gene detection performance (Figure 4C and D).
As demonstrated in (8), cell orderings based on pseudo-time may reveal gene expression patterns that cannot be discovered by bulk gene expression data. MEF2C and MYH2 are two genes involved in the HSMM differentiation. It is known that these two genes should have increasing expression during the differentiation, and the expression of MEF2C should start increasing earlier than the increase of MYH2 (8). Based on the average bulk gene expression at different time points, it was not clear that MEF2C had a monotone increasing pattern, nor was it clear which gene started to increase first (Supplementary Figure S1). By contrast, all single-cell analysis methods tested here were able to recover the overall increasing pattern of MEF2C and MYH2 along their analyzed pseudo-time axes, although in Monocle, k-means TSCAN, Waterfall, SCUBA and Wanderlust, MEF2C decreased a little before increasing (Supplementary Figure S2). Compared to the other methods, the temporal expression curves fitted by TSCAN and nocluTSCAN more clearly showed that MEF2C increased earlier than the increase of MYH2 (Supplementary Figure S2).
Based on all the analyses above, TSCAN was the method that provided the best overall performance. It offered the best cell ordering accuracy among all tested methods and improved cell ordering robustness compared to methods without using cell clustering (i.e. Monocle and nocluTSCAN).
HSMM analysis without using a priori chosen genes for pseudo-time reconstruction
In real applications, the prior information for pseudo-time reconstruction such as the
518 genes used above is not always available. When no such prior information is available,
pseudo-time reconstruction has to rely on all genes in the RNA-seq data. To evaluate the
performance of TSCAN in such a scenario, we repeated the previous analysis but constructed
pseudo-time without using the 518 a priori chosen genes.
Instead, the 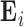 used for TSCAN was derived from all genes in the
single-cell RNA-seq data using the protocol described in Materials and Methods. We also
used
used for TSCAN was derived from all genes in the
single-cell RNA-seq data using the protocol described in Materials and Methods. We also
used 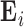 instead of
instead of 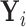 as the input for Monocle,
Waterfall, SCUBA and Wanderlust in order to make the method comparison relatively fair. Of
note, the dimensionality of
as the input for Monocle,
Waterfall, SCUBA and Wanderlust in order to make the method comparison relatively fair. Of
note, the dimensionality of 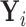 was also beyond the capacity that the
Monocle software was able to handle.
was also beyond the capacity that the
Monocle software was able to handle.
Pseudo-temporal paths generated by different methods are provided in Supplementary Table S3. The default main path given by TSCAN (Figure 5A, path 3-1-2) contained a cluster of cells with high expression in SPHK1 (Figure 5D), indicating that the main path was contaminated by interstitial mesenchymal cells and may not reflect myoblast differentiation. In such a scenario, TSCAN allows users to manually tune the analysis. For instance, with the GUI, one can conveniently visualize the expression of marker genes (Figure 5B) such as SPHK1 (Figure 5D, marker for contamination) and ENO3 (Figure 5E, marker for myoblast differentiation). Since SPHK1 is highly expressed in cluster 3, we chose to study path 2-1-4 which represents the myoblast differentiation. According to the increasing ENO3 pattern, one can specify that cluster 2 should be the path origin. Alternatively, one can also manually define a path by specifying the clusters and their order in the path (Figure 5C). In this example, both ways yielded the same path 2-1-4. Similar to TSCAN, the main path in Monocle was also contaminated by cells with high SPHK1 expression (Supplementary Table S3). However, Monocle does not provide an interface to help users conveniently incorporate such marker gene information and tune ordering. Users would need to be experienced in programming in order to adjust the analysis. In comparison, the TSCAN GUI allows users unfamiliar with programming to visualize and tune the ordering. Therefore, it lowers the bar for users to customize the pseudo-time analyses and can save them time and effort.
Figure 5.

Demonstration of GUI and TSCAN analysis of HSMM data using all genes for pseudo-time reconstruction. (A) MST constructed by TSCAN using all genes. (B) Users can choose a marker gene in GUI to visualize its expression. (C) Users can define a path by specifying the clusters to include and their ordering. (D) The average expression of SPHK1 in each cluster. (E) The average expression of ENO3 in each cluster.
After using high expression of SPHK1 to exclude the contaminating branch and using low expression of ENO3 to determine the origin of the pseudo-temporal path for each method (Supplementary Table S3), different methods were then compared.
In terms of cell ordering accuracy, TSCAN had the highest POS score (Figure 6A) and the best mean rank of gold standard genes (Figure 6C) among all methods. It also had the highest power for detecting the gold standard differential genes (Figure 6D). In terms of robustness, methods based on cell clustering (TSCAN, k-means TSCAN, Waterfall) were more robust than methods that did not use cell clustering (Monocle, nocluTSCAN), as shown by the increased similarity scores between the original and perturbed data (Figure 6B).
Figure 6.

Evaluation results for different methods in HSMM data where pseudo-time was constructed using all genes. (A) POS score. (B) Robustness measured by the average similarity score from 100 independent perturbations. (C) Mean rank of gold standard genes. (D) Number of detected gold standard genes among top differential genes.
Besides comparing cell orderings from the original and perturbed data, we also compared cell orderings constructed using and not using the 518 prior genes. To do so, similarity score between the cell ordering reported in this section and the ordering reported in the previous section was computed for each method. Supplementary Figure S3A shows that TSCAN and the marker gene approach produced higher similarity scores than other methods, suggesting that they produced the most consistent cell ordering results. For each method, we also compared the consistency of differentially expressed genes detected by using and not using the 518 prior genes for pseudo-time reconstruction. For each analysis (i.e. using or not using the 518 prior genes), we obtained the top R ranked differential genes. The number of common genes between these two analyses was then counted and plotted as a function of R in Supplementary Figure S3B. Supplementary Figure S3C shows a similar analysis with a more stringent definition of common genes. Here, any gene that did not change in the same direction along the two pseudo-temporal paths (i.e. the fitted GAM functions from the two analyses have negative correlation) was not counted as a common gene even if the gene was identified by both analyses among their top R genes. After excluding these inconsistent genes from the common gene list, the number of genes remained in the common gene list was then shown as a function of R. In both Supplementary Figures S3B and S3C, TSCAN and the marker gene approach showed higher consistency than the other methods. Compared to the marker gene approach, TSCAN cell ordering was more accurate according to the POS score and differential gene detection performance (Figure 6A, C and D). Thus, our results show that TSCAN can make the ordering results less dependent on the availability of prior genes and at the same time provide the best accuracy compared to the other methods.
When comparing the expression patterns of MEF2C and MYH2 along the pseudo-time axis, Monocle and Wanderlust failed to reveal the temporal order of MEF2C and MYH2, and the increasing pattern of these genes also became less clear (Figure 7). In Waterfall, MEF2C first decreased and then increased, and the temporal order of MEF2C and MYH2 was not very clear. By contrast, the other methods successfully revealed the increasing pattern of MEF2C and MYH2 in this analysis. Their results also more clearly show that MEF2C increased before the increase of MYH2 (Figure 7).
Figure 7.

MEF2C and MYH2 expression patterns in HSMM data set where pseudo-time was constructed using all genes. The expression of each gene in each cell is plotted as a function of cell order on the pseudo-time axis. The solid curves are the fitted GAM function. The dashed curve is the GAM fit for ENO3, the marker gene used to determine the path direction.
Overall, our analyses again show that TSCAN produced the most accurate cell ordering results, and it was more robust than methods without cell clustering.
LPS analysis
For the LPS data, we reconstructed pseudo-time without using strong prior knowledge such
as the 518 a priori chosen genes in the HSMM analysis. The
analyses were run based on 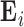 which was computed using all genes following the
protocol described in Materials and Methods. All methods only found one main path without
branching paths (Supplementary Table S3). To determine the direction of the path, we used
BCL3 as a marker gene. BCL3 is known to be involved in the response to viral and bacterial
stimulus, and its expression level is expected to increase after LPS stimulation. Figure
2 shows the expression of this marker gene in the
TSCAN GUI. Accordingly, cluster 1 was determined as the origin of the pseudo-time axis.
Comparing different methods based on POS score again shows that TSCAN had the best
accuracy (Figure 8A, BCL3 was used as the marker gene
for the marker-gene-only approach). Methods based on cell clustering (TSCAN, k-means
TSCAN, Waterfall) were more robust than those not using cell clustering (Monocle and
nocluTSCAN) (Figure 8B). To evaluate different
methods based on differentially expressed genes, we compiled 125 known marker genes (BCL3
excluded) from (25) (Supplementary Table S4).
Figure 8C and D
show the mean rank of these gold standard genes and the number of gold standard genes
found in the top ranked genes reported by each method respectively. Again, TSCAN
outperformed all other methods.
which was computed using all genes following the
protocol described in Materials and Methods. All methods only found one main path without
branching paths (Supplementary Table S3). To determine the direction of the path, we used
BCL3 as a marker gene. BCL3 is known to be involved in the response to viral and bacterial
stimulus, and its expression level is expected to increase after LPS stimulation. Figure
2 shows the expression of this marker gene in the
TSCAN GUI. Accordingly, cluster 1 was determined as the origin of the pseudo-time axis.
Comparing different methods based on POS score again shows that TSCAN had the best
accuracy (Figure 8A, BCL3 was used as the marker gene
for the marker-gene-only approach). Methods based on cell clustering (TSCAN, k-means
TSCAN, Waterfall) were more robust than those not using cell clustering (Monocle and
nocluTSCAN) (Figure 8B). To evaluate different
methods based on differentially expressed genes, we compiled 125 known marker genes (BCL3
excluded) from (25) (Supplementary Table S4).
Figure 8C and D
show the mean rank of these gold standard genes and the number of gold standard genes
found in the top ranked genes reported by each method respectively. Again, TSCAN
outperformed all other methods.
Figure 8.

Evaluation results for different methods in LPS data set. (A) POS score. (B) Robustness measured by the average similarity score from 100 independent perturbations. (C) Mean rank of gold standard genes. (D) Number of detected gold standard genes among top differential genes.
As a specific example, Figure 9 shows the expression level of a gold standard gene STAT2 for the LPS data (25). STAT2 expression is expected to increase after LPS stimulation. One can see that the TSCAN result was most consistent with the known increasing pattern of STAT2. By contrast, the increasing pattern of STAT2 was much less clear in cell orderings produced by all the other approaches. In Monocle, nocluTSCAN, k-means TSCAN, Waterfall, SCUBA and Wanderlust, STAT2 first increased and then decreased. In the marker gene approach, the increasing pattern was weak compared to the high variability of cells around the fitted curve.
Figure 9.

STAT2 expression patterns in LPS data set. STAT2 expression in each cell is plotted as a function of cell order on the pseudo-time axis. The orange curve is the fitted GAM function.
qNSC analysis
Lastly, we compared different methods using the qNSC data set. This data set does not
have multiple time points or experimental conditions. A prior gene set for cell ordering
was also not available. We therefore run the analyses based on 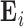 computed
using all genes as described in Materials and Methods. All methods produced one single
path without branches. To determine the path direction, we used FOXG1 as a marker gene.
FOXG1 is known to be critically involved in proliferative adult NPCs. Low expression of
FOXG1 was used to indicate the origin of the path.
computed
using all genes as described in Materials and Methods. All methods produced one single
path without branches. To determine the path direction, we used FOXG1 as a marker gene.
FOXG1 is known to be critically involved in proliferative adult NPCs. Low expression of
FOXG1 was used to indicate the origin of the path.
In the qNSC analysis, the POS score cannot be calculated because external information such as data collection time is not available. Therefore, we only evaluated each method's robustness and its ability to detect known differential genes. For the differential gene analysis, 1999 known marker genes (excluding FOXG1) were compiled from (26) to serve as the gold standard (Supplementary Table S4). Once again, methods using cell clustering (TSCAN, k-means TSCAN, Waterfall) improved robustness of cell ordering compared to those without using cell clustering (Monocle, nocluTSCAN) (Figure 10A). TSCAN offered the best mean rank of gold standard genes among all methods (Figure 10B), and it also had the highest power for detecting the gold standard differential genes (Figure 10C). Supplementary Figure S4 shows the expression level of a gold standard gene SOX9. As a down-regulated transcription factor, SOX9 expression is expected to decrease along the pseudo-time (26). TSCAN and Waterfall results were consistent with this known decreasing pattern of SOX9, and the decreasing pattern was most evident in TSCAN. By contrast, SOX9 expression first increased and then decreased in Monocle, nocluTSCAN and SCUBA. For k-means TSCAN, SOX9 expression first decreased and then increased. For the marker-gene-only approach and Wanderlust, SOX9 expression slightly increased. Overall, TSCAN performed the best among all methods.
Figure 10.

Evaluation results for different methods in qNSC data set. (A) Robustness measured by the average similarity score from 100 independent perturbations. (B) Mean rank of gold standard genes. (C) Number of detected gold standard genes among top differential genes.
The graphical user interface (GUI)
TSCAN has a GUI. As discussed above, the GUI in TSCAN allows users to visualize marker genes and tune main paths and cluster-level orderings. Besides these functions, the GUI also provides multiple trimming criteria for users to efficiently trim unwanted cells. For example, to exclude cells with high expression in two genes PDGFRA and SPHK1 in HSMM data set, one can set up two trimming criteria such as PDGFRA > 1 and SPHK1 > 1 (Supplementary Figure S5A) and TSCAN will exclude cells meeting both criteria (Supplementary Figure S5B). Finally, the GUI can be used to visualize expression of user-specified genes along pseudo-time as heat maps. For example, Supplementary Figure S5C visualizes the expression of two genes CCNA2 and CCNB2 after obtaining the pseudo-time ordering in HSMM data. Together, these functions make the pseudo-time analyses of single-cell RNA-seq data more convenient and user-friendly.
DISCUSSION
In summary, TSCAN offers a new tool to support pseudo-time analysis of single-cell RNA-seq data. As demonstrated by our results, this approach robustly provides competitive performance based on different criteria. By comparing methods using and not using cell clustering, we have shown that cell clustering is a useful technique for reducing the variability and improving the accuracy of the MST-based pseudo-time analysis. Although the cell clustering idea has also been used previously in Waterfall, a systematic evaluation of the impact of cell clustering on cell ordering was not provided in the Waterfall study (26). Besides the development and systematic evaluation of the TSCAN algorithm, we also developed a GUI for TSCAN. The GUI of TSCAN provides users with the flexibility to interactively explore and adjust the analysis results.
In order to evaluate TSCAN and other unsupervised pseudo-time reconstruction methods, we used two time course data sets with multiple time points, HSMM and LPS, and intentionally avoided using any information on data collection time in our pseudo-time analyses. In this way, the data collection time can provide an independent source of information for evaluating the accuracy of cell ordering via POS score. Such an evaluation cannot be done if the test data set has only one time point. This explains why we used HSMM and LPS for evaluation even though in principle such data could be analyzed in other ways. For instance, one could perform supervised rather than unsupervised analysis to order cells. Alternatively, one could perform an initial analysis to identify differentially expressed genes between different data collection time points and then use them as prior genes (similar to the 518 prior genes for HSMM) to order cells. Unlike the HSMM and LPS data, the qNSC data set represents a different situation faced by many investigators. Here, single-cell RNA-seq data are collected from only one biological condition rather than from multiple time points or conditions. In such a scenario, supervised methods that use data collection time information to order cells cannot be applied, and one cannot compare different time points or conditions to find differential genes and use them as prior genes for cell ordering. It is therefore important to be able to perform unsupervised pseudo-time analysis such as TSCAN.
Besides TSCAN, this article also introduced several methods to quantitatively evaluate cell ordering performance. We expect that these evaluation methods will continue to be useful in the future for evaluating other pseudo-time reconstruction algorithms. Although TSCAN was tested using RNA-seq, in principle it should not be difficult to tailor this approach to other data types should single-cell data for those data types become available in the future.
Supplementary Material
Acknowledgments
The authors would like to thank Dr Hongjun Song and Dr Jaehoon Shin for providing qNSC data and helpful discussions on Waterfall. The authors would also like to thank Dr Ben Sherwood for his suggestions on improving the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01HG006282]. Funding for open access charge: National Institutes of Health (NIH) [R01HG006282].
Conflict of interest statement. None declared.
REFERENCES
- 1.Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A., et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 2.Tang F., Barbacioru C., Bao S., Lee C., Nordman E., Wang X., Lao K., Surani M. A. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6:468–478. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schena M., Shalon D., Davis R. W., Brown P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 6.Schulze A., Downward J. Navigating gene expression using microarrays–a technology review. Nat. Cell Biol. 2001;3:E190–E195. doi: 10.1038/35087138. [DOI] [PubMed] [Google Scholar]
- 7.Simpson E.H. The interpretation of interaction in contingency tables. J. Roy. Stat. Soc. B. 1951;13:238–241. [Google Scholar]
- 8.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Islam S., Kjllquist U., Moliner A., Zajac P., Fan J.B., Lönnerberg P., Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsköld D., Luo S., Wang Y.C., Li R., Deng Q., Faridani O.R., Daniels G.A., Khrebtukova I., Loring J.F., Laurent L.C., et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treutlein B., Brownfield D.G., Wu A.R., Neff N.F., Mantalas G.L., Espinoza F.H., Desai T.J., Krasnow M.A., Quake S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saliba A.E., Westermann A.J., Gorski S.A., Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amir el-A.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C., Shenfeld D.K., Krishnaswamy S., Nolan G.P., Pe'er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bendall S.C., Davis K.L., Amir el-A.D., Tadmor M.D., Simonds E.F., Chen T.J., Shenfeld D.K., Nolan G.P., Pe'er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu P., Simonds E.F., Bendall S.C., Gibbs K.D., Jr, Bruggner R.V., Linderman M.D., Sachs K., Nolan G.P., Plevritis S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marco E., Karp R.L., Guo G., Robson P., Hart A.H., Trippa L., Yuan G.C. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E5643–E5650. doi: 10.1073/pnas.1408993111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buettner F., Natarajan K.N., Casale F.P., Proserpio V., Scialdone A., Theis F.J., Teichmann S.A., Marioni J.C., Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 19.Dalerba P., Kalisky T., Sahoo D., Rajendran P.S., Rothenberg M.E., Leyrat A.A., Sim S., Okamoto J., Johnston D.M., Qian D., et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011;29:1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fraley C., Raftery A. E. Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. Assoc. 2002;97:611–631. [Google Scholar]
- 22.Chris F., Adrian E.R.T., Brendan M., Luca S. Technical Report. Washington D.C: Department of Statistics, University of Washington; 2012. mclust Version 4 for R: Normal mixture modeling for model-based clustering, classification, and density estimation. No. 597. [Google Scholar]
- 23.Wood S.N. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J. Roy. Stat. Soc. B. 2011;73:3–36. [Google Scholar]
- 24.Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 25.Amit I., Garber M., Chevrier N., Leite A.P., Donner Y., Eisenhaure T., Guttman M., Grenier J.K., Li W., Zuk O., et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin J., Berg D.A., Zhu Y., Shin J.Y., Song J., Bonaguidi M.A., Enikolopov G., Nauen D.W., Christian K.M., Ming G.L., et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haghverdi L., Buettner F., Theis F.J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 2015;31:2989–2998. doi: 10.1093/bioinformatics/btv325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.