

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most potent and perilous diseases known, with a median survival rate of 3-5 months due to the combination of only advanced stage diagnosis and ineffective therapeutic options. Metformin (1,1-Dimethylbiguanide hydrochloride), the leading drug used for type 2 diabetes mellitus, emerges as a potential therapy for PDAC and other human cancers. Metformin exerts its anticancer action via a variety of adenosine monophosphate (AMP)-activated protein kinase (AMPK)- dependent and/or AMPK-independent mechanisms. We present data here showing that metformin down- regulated pancreatic transcription factor pancreatic duodenal homeobox-1 (PDX-1), suggesting a potential novel mechanism by which metformin exerts its anticancer action. Metformin inhibited PDX-1 expression at both protein and mRNA levels and PDX-1 transactivity as well in PDAC cells. Extracellular signal-regulated kinase (ERK) was identified as a PDX-1-interacting protein by antibody array screening in GFP-PDX-1 stable HEK293 cells. Co-transfection of ERK1 with PDX-1 resulted in an enhanced PDX-1 expression in HEK293 cells in a dose-dependent manner. Immunoprecipitation/Western blotting analysis confirmed the ERK-PDX-1 interaction in PANC-1 cells stimulated by epidermal growth factor (EGF). EGF induced an enhanced PDX-1 expression in PANC-1 cells and this stimulation was inhibited by MEK inhibitor PD0325901. Metformin inhibited EGF-stimulated PDX-1 expression with an accompanied inhibition of ERK kinase activation in PANC- 1 cells. Taken together, our studies show that PDX-1 is a potential novel target for metformin in PDAC cells and that metformin may exert its anticancer action in PDAC by down-regulating PDX-1 via a mechanism involving inhibition of ERK signaling.
Keywords: EGF, ERK, metformin, PDAC, PDX-1, signaling
Introduction
Metformin (1,1-Dimethylbiguanide hydrochloride) is the most prescribed drug for type 2 diabetes mellitus (T2DM) because of its distinctive ability to avoid causing weight gain and hypoglycaemia via suppression of hepatic glucagon production [1-4]. Beyond dominating the T2DM therapeutic field, metformin also possesses anticancer capabilities [5]. Epidemiologic studies revealed that administration of metformin, but not other anti-diabetic drugs, decreases the incidence, recurrence and mortality of pancreas cancer in patients with T2DM, thus, metformin exhibits both chemo-preventative and chemo-therapeutic activities [6-8]. As a result, there are a number of clinical trials testing metformin on breast cancer, prostate cancer, endometrial cancer, kidney cancer, lung cancer, lymphoma and colorectal cancer [5]. Pre- clinical studies have established a direct action of metformin on cancers, such as inhibition of cell proliferation [9-12], induction of apoptosis [13, 14], inhibition of migration and invasion [12] and enhancement of radiosensitivity [15]. Metformin exerts its anticancer action via a variety of AMPK-dependeng [16] and/or AMPK-independent mechanisms depending upon the cellular context. These mechanisms include the inhibition of the mTOR signaling [17, 18], the ERK signaling [10, 16, 19] and the IGF-I signaling [14], suppression of sonic hedgehog expression [20], down- regulation of specificity protein (Sp) transcription factors [21], alteration of the expression profiles of microRNAs [12, 22], and attenuation of cancer stem cell (CSC) functions [12, 18, 23] via increasing reactive oxygen species production in CSC and reducing their mitochondrial transmembrane potential [23]. However, the molecular mechanism(s) by which metformin exerts anticancer effects remains incompletely understood [5]. Pancreatic duodenal homeobox-1 (PDX-1) is a homeodomain-containing transcription factor essential for normal pancreatic development, β-cell differentiation, and maintenance of mature β-cell functions [24, 25]. In contrast, most exocrine cells express PDX-1 only at very low levels. However, several lines of evidence support the notion that PDX-1 is closely associated with pancreatic tumorigenesis. Specifically, 1) PDX-1 expression is markedly elevated in Pancreatic ductal adenocarcinoma (PDAC) [26-28]; 2) elevated PDX-1 expression levels are significantly correlated with metastasis and histological grade in patients with PDAC [26, 27] and PDX-1 expression in PDAC is an independent survival factor [26, 29]; 3) persistent expression of PDX-1 induces metaplasia [30]; 4) PDX- 1 is required for K-RasG12D to induce the development of pancreatic intraepithelial neoplasia (PanIN), metaplasia and PDAC [31]; and 5) PDX-1 enhances cell proliferation, colony formation, invasion and tumor growth of malignant tumor cells [32]. Silencing PDX-1 efficiently inhibits PDAC cell proliferation in vitro and tumor growth in vivo [33, 34], thus, PDX-1 is a potential therapeutic target in PDAC treatment.
In the present work, we determined the effect of metformin on PDX-1 expression and function in PDAC cells and elucidated the role of the ERK signaling in the regulation of PDX-1 expression. Our results indicate that treatment of PDAC cells with metformin markedly inhibited PDX-1 expression and function via inhibition of ERK signaling.
Materials and Methods
Reagents
Metformin (1,1-Dimethylbiguanide hydrochloride) and Bradford reagent were purchased from Sigma- Aldrich (St. Louis, MO). Pierce enhanced chemiluminescence (ECL) detection kit was purchased from Thermo Fisher Scientific (Waltham, MA). Immobilon-P polyvinylidene fluoride (PVDF) transfer membranes were obtained from EMD Millipore (Billerica, MA). Anti-rabbit IgG (whole molecule)- peroxidase antibody, anti-mouse IgG (Fab specific)- peroxidase antibody and monoclonal Anti-β-actin antibody were purchased from Sigma-Aldrich, while monoclonal anti-PDX-1 antibody was purchased from Cell Signaling Technology (Danvers, MA). All other unlisted chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Cell Growth
Human PDAC cells Mia PaCa-2 and PANC-1 and human embryonic kidney 293 (HEK293) cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Mia PaCa-2, PANC-1, SHIP-Luc2RFP stable Mia PaCa-2 and PANC-1, HEK293 and GFP-PDX-1 stable HEK293 cells were grown and maintained in Dulbecco's Modified Eagle's Medium (DMEM). All media were supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. 400 μg/ml G418 was added into the media for the stable cells. All the cells were incubated at conditions of 37°C and 10% carbon dioxide (CO2).
Luciferase Reporter Assays
The luciferase assay was conducted 16 hours after metformin treatment. Twenty (20) μl of the lysis supernatant from treated or untreated PDAC cells was used for the measurement of luciferase activity by a Monolight 3010 luminometer (Pharmingen). Each sample was measured twice, and all results were normalized to β-galactosidase activity.
Antibody Array Screen, Western Blotting and Transfection
Antibody array screen, immunoprecipitation, Western blotting and lipofectamine 2000 transfection were performed as described previously [35].
Quantitative Reverse Transcriptional PCR (qRT- PCR)
By using TriZol reagent (Invitrogen), total RNAs were isolated from human Mia PaCa-2 cells, mouse KC cells, and mouse KPC cells. Quantitative Reverse Transcriptional PCR was carried out with 100 ng of total RNA. A mixture of 10 μl of 2× QuantiTect SYBR Green RT-PCR Master Mix (Qiagen), 0.2 μl QuantiTect RT Mix (Qiagen), 1 μl of 10 μM forward and reverse primers, and 6.8 μl of RNase-Free Water was added to each sample for analysis by absolute quantification. qRT-PCR was performed in 96-well plates and according to Applied Biosystems protocol. The mRNA levels of PDX-1 in the samples were normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and the PDX-1 and GAPDH were measured in triplicate. The primers used were: mouse PDX-1: 5′- CCCCAGTTTACAAGCTCGCT-3′ and 5′-CTCGGTTCC ATTCGGGAAAGG-3′; human PDX-1: 5′-ATCTCCCCA TACGAAGTGCC-3′ and 5′-CGTGAGCTTTGGTGGAT TTCAT-3′; and GAPDH: 5′-ATGCCATCACTGCCACC CAGAACG-3′ and 5′-GCCAGTGAGCTTCCCGTTCA-3′. The cDNA was prepared from the total RNA using qScript cDNA SuperMix (Quanta Biosciences, Maryl- and) according to the manufacturer's protocol. PCR was performed on 1-μl aliquots from each cDNA reaction, using the PDX-1 and GDA primer sets for 45 cycles.
Statistical Analysis
The unpaired Student t test was used for the statistical analyses of PDX-1 mRNA levels and luciferase activities, with p < 0.05 indicating significant difference.
Results
Metformin Inhibits PDX-1 Expression and Function in PDAC Cells
PDX-1 has been implicated in pancreatic tumorigenesis [26-28, 30-32] and shown great promise as a target for the treatment of PDAC [33, 34]. In order to determine whether treatment with metformin decreases PDX-1 transcriptional activity, we used a synthetic human insulin promoter (SHIP)-driven Luciferase-RFP fusion reporter subject to the regulation by PDX-1 (designated as a SHIP-Luc2RFP). By using this reporter construct, we generated stably transfected Mia PaCa-2-SHIP-Luc2RFP and PANC1-SHIP-Luc2RFP PDAC cell lines, which can be used to evaluate PDX-1 gene expression and activity. To determine the effect of metformin on PDX-1 promoter activity, we performed luciferase reporter assays in Mia PaCa-2-SHIP-Luc2RFP and PANC1-SHIP-Luc2RFP PDAC cells. As shown in Fig. (1), PDX-1 transactivity was significantly inhibited by the treatment of metformin. The luminescence measurement was lowered by 45% and 53% in Mia PaCa-2 and PANC-1 cells, respectively, after treatment with metformin in comparison to untreated cells (Fig. 1A). Considering the role of PDX-1 in enhancing PDAC cell growth [32], these results imply that inhibition of PDX-1 expression and function could contribute to the mechanism by which metformin inhibits PDAC cell growth.
Fig. (1). Metformin down-regulates PDX-1 in PDAC cells.
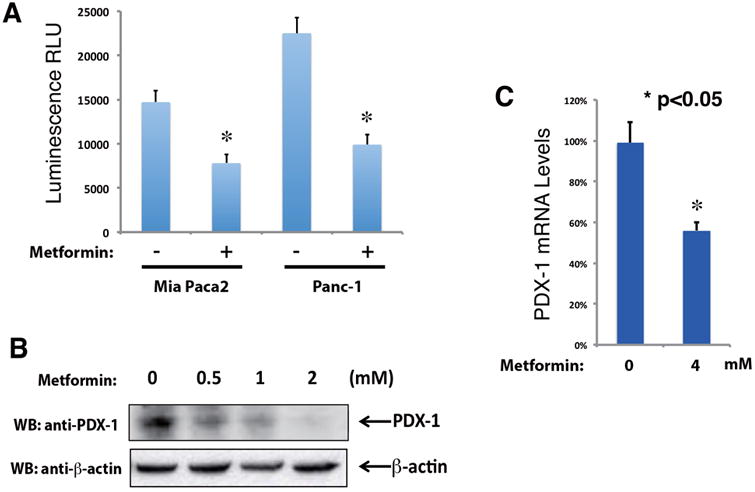
(A) Metformin inhibits PDX-1 transactivity in PDAC cells. Mia PaCa-2-SHIP-Luc2RFP and PANC-1-SHIP-Luc2RFP cells were treated with 5 mM metformin for 16 h. Luciferase reporter assays were performed to measure PDX-1 transactivity (* indicates p < 0.05 showing significant difference). (B) Metformin inhibits PDX-1 protein expression in Mia PaCa-2 cells. Mia PaCa2 cells were seeded in a density of 5 × 105 per well in a 6-well plate. Twenty-four hours later, the cells were treated with a variety of concentrations (0, 0.5, 1 and 2 mM) of metformin for 16 h. Expression levels of PDX-1 and β-actin was examined by Western blotting using an anti-PDX-1 antibody and an anti-β-actin antibody, respectively. The intensities of the PDX-1 bands were quantitated. (C) Metformin inhibits PDX-1 mRNA expression in Mia PaCa-2 cells. Mia PaCa-2 cells were treated with 4 mM metformin for 16 h. Total RNA was isolated using TriZol reagent, followed by qRT-PCR analysis. The cDNA was prepared from the total RNA using qScript cDNA SuperMix. PDX-1 mRNA levels were normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (* indicates p < 0.05 showing significant difference).
It is known that PDX-1 activates its own transcription as part of an auto-regulatory feedback mechanism. In view of the results shown in Fig. (1A), we reasoned that metformin should decrease PDX-1 mRNA and protein levels in these cells if PDX-1 regulates its own expression through a positive feedback mechanism in PDAC cells. To determine the effect of metformin on PDX-1 expression, Mia PaCa-2 cells were treated with increasing concentrations of metformin (0, 0.5, 1 and 2 mM) for 16 h. The level of PDX-1 protein was determined in cell lysates by Western blotting analysis using an antibody that detects PDX-1. As shown in Fig. (1B), exposure to metformin decreased PDX-1 protein expression in Mia PaCa-2 cells in a dose-dependent manner. To further substantiate an inhibitory effect of metformin on PDX-1 expression, we examined the impact of metformin on PDX-1 mRNA levels in Mia PaCa-2 cells. qRT-PCR analysis showed that exposure to metformin decreased mRNA level of PDX-1 in Mia PaCa-2 cells by 44% (Fig. 1C), confirming that metformin reduces the function and expression of PDX-1 in PDAC cells.
ERK1 and ERK2 Form a Physical Complex with PDX-1 in HEK293 Cells
We next focused on the mechanism(s) by which metformin regulates PDX-1 function and expression. We have first demonstrated the oncogenic function of PDX-1 in HEK293 cells by showing its stimulatory effect on cell proliferation, invasion and colony formation [32]. In an effort to investigate the oncogenic mechanism of PDX-1, we performed an antibody- based array screen in our previously generated GFP- PDX-1 stable HEK293 cells [32] to identify PDX-1- interacting proteins. Whole cell lysates from the GFP- PDX-1 stable HEK293 cells were incubated with a membrane filter spotted with antibodies against 400 different signal transduction proteins. Immunodetection was performed using an anti-GFP antibody. We found that ERK1 and ERK2 interacted with PDX-1 in this assay (Fig. 2A).
Fig. (2). ERK interacts with PDX-1 and enhances PDX-1 expression in HEK293 cells.
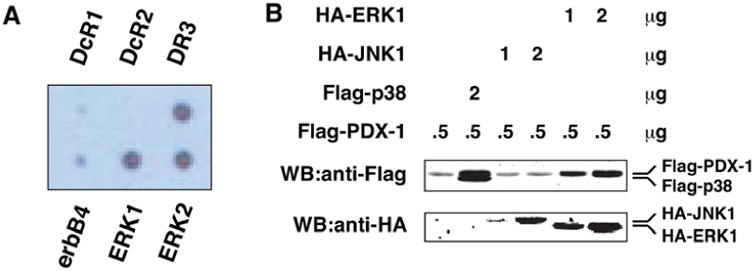
(A) ERK1 and ERK2 were identified as PDX-1-interacting proteins by antibody array screen. Cell extracts from GFP-PDX-1 stable HEK293 cells were added to a membrane filter arrayed with 400 antibodies. The presence of the antibody-antigen-GFP-PDX-1 complex was detected by horseradish peroxidase-conjugated anti-GFP antibody, followed by chemiluminescence. (B) ERK1 enhances PDX-1 expression. By using Lipofectamine 2000, HEK293T cells were transfected with Flag-PDX-1 (0.5 μg) alone, Flag-PDX-1 (0.5 μg) plus Flag-p38 (2 μg), Flag-PDX-1 (0.5 μg) plus HA-JNK1 (1 or 2 μg), or Flag-PDX-1 (0.5 μg) plus HA-ERK1 (1 or 2 μg). Expression levels of Flag-PDX-1 and Flag-p38, HA-JNK1 and HA-ERK1 were examined by Western blotting using an anti-Flag antibody and an anti-HA antibody, respectively.
To determine the functional relevance of the ERK- PDX-1 interaction, we examined the effect of ERK1 on PDX-1 expression in HEK293 cells. Flag-PDX-1 was co-transfected into these cells with Flag-p38, HA-JNK1, or HA-ERK1. In line with our recent studies [35], p38 MAP kinase enhanced PDX-1 expression (Fig. 2B, lane 2). Co-transfection of ERK1 with PDX-1 resulted in an increased PDX-1 expression in a dose-dependent manner in HEK293 cells (Fig. 2B, lanes 5 and 6). However, co-transfection of JNK1 with PDX-1 did not affect expression level of PDX-1 under the same conditions (Fig. 2B, lanes 3 and 4). Thus, PDX-1 is specifically up-regulated by ERK1, but not JNK1, corroborating that ERK1 is a positive regulator of PDX- 1 expression.
ERK Interacts with PDX-1 and Mediates EGF- Stimulated PDX-1 Expression in PDAC Cells
We next determined whether endogenous ERK and PDX-1 form a molecular complex within intact PDAC cells. To examine physical interaction between ERK and PDX-1, we performed immunoprecipitation/ Western blotting in PANC-1 cells that were treated with epidermal growth factor (EGF), a potent growth- promoting factor for PDAC cells. PDX-1 was immunoprecipitated using an anti-PDX-1 antibody, followed by Western blotting using an anti-ERK antibody. As shown in Fig. (3A), ERK was co-immunoprecipitated with PDX-1 and EGF stimulation enhanced the association between ERK and PDX-1. These data indicate that ERK is a PDX-1-interacting protein in PDAC cells and that this interaction is under growth factor regulation.
Fig. (3). ERK mediates EGF-stimulated PDX-1 expression in PANC-1 cells.
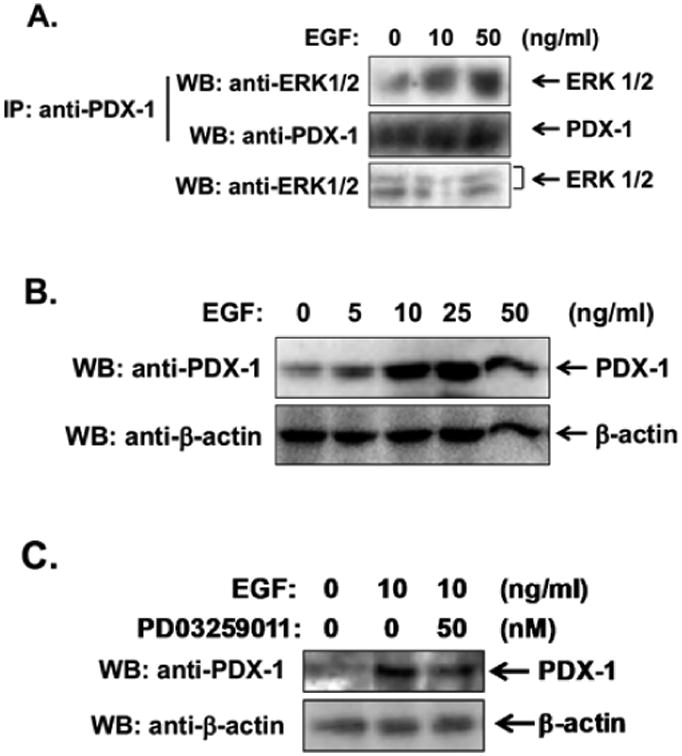
(A) EGF enhances the ERK-PDX-1 interaction in PANC-1 cells. PANC-1 cells were treated with 10 and 50 ng/ml of EGF for 30 min. PDX-1 was immunoprecipitated with an anti-PDX-1 antibody, followed by Western blotting using an anti-ERK1/2 antibody. (B) PANC-1 cells were treated with a variety of concentrations of EGF for 16 h. (C) PANC-1 cells were pretreated with 50 nM of PD0325901 for 30 min, followed by treatment with 10 ng/ml of EGF for 16 h. Expression levels of PDX-1 and β-actin were examined by Western blotting using an anti-PDX-1 antibody and an anti-β-actin antibody, respectively.
To determine whether EGF also induces PDX-1 expression in PDAC cells, we treated PANC-1 cells with increasing concentrations of this growth factor. Western blotting analysis showed that EGF markedly enhanced PDX-1 expression (Fig. 3B). To determine the role of ERK in EGF-stimulated PDX-1 expression, we pretreated PANC-1 cells with the selective MEK inhibitor PD0325901, followed by treatment with EGF. Western blotting analysis showed that EGF-stimulated PDX-1 expression was markedly inhibited by exposure to PD0325901 (Fig. 3C), indicating that ERK mediated EGF-stimulated PDX-1 expression in PDAC cells.
Metformin Inhibits EGF-Stimulated PDX-1 Expres- sion via Inhibiting ERK Signaling
Since the results presented here indicate that ERK mediates positive regulation of PDX-1 expression by EGF and metformin has recently been shown to inhibit ERK activation in PDAC cells [10,16,19], we hypothesized that the metformin inhibits PDX-1 expression by abrogating EGF-induced ERK activation. In line with this hypothesis, we found that pretreatment of PANC-1 cells with metformin markedly prevented EGF-stimulated PDX-1 expression (Fig. 4A). Metformin pretreatment also inhibited EGF-stimulated ERK kinase activation, as evidenced by decreased phosphorylation of ERK at Thr201 and Tyr204 (Fig. 4B), consistent with the notion that metformin inhibits PDX-1 expression through a mechanism involving inhibition of EGF- stimulated ERK signaling (Fig. 5).
Fig. (4). Metformin inhibits EGF-stimulated ERK kinase activation and PDX-1 expression in PANC-1 cells.
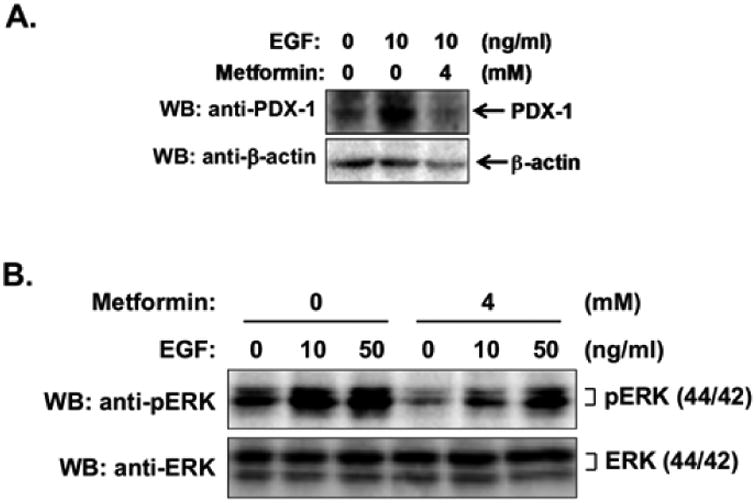
(A) PANC-1 cells were pretreated with 4 mM metformin for 1 h, followed by treatment with 10 ng/ml of EGF for 16 h. Expression levels of PDX-1 and β-actin were examined by Western blotting using an anti-PDX-1 antibody and an anti-β-actin antibody, respectively. (B) PANC-1 cells were pretreated with or without 4 mM metformin for 16 h, followed by treatment with a variety of concentrations of EGF for 15 min. ERK kinase activation was examined by Western blotting using an anti-phospho-p44/42 MAPK (ERK1/2) (Thr201/Tyr204) antibody. ERK expression was examined by Western blotting using an anti-ERK antibody.
Fig. (5). Schematic depiction of the functions of metformin in PDAC.
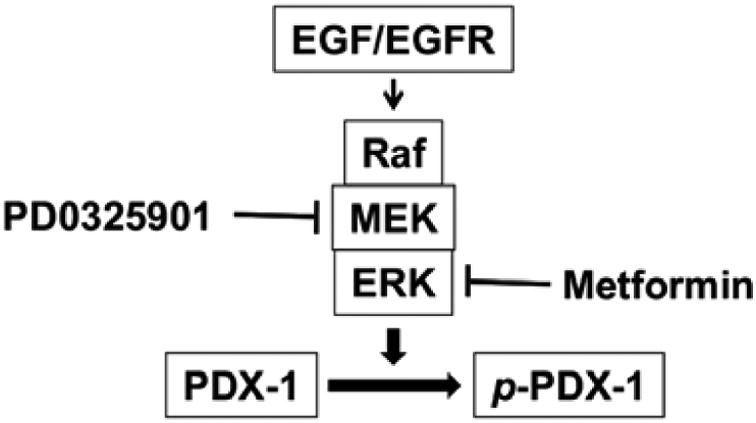
EGF stimulates kinase activation of ERK MAP kinase, which, in turn, interacts with and phosphorylates PDX-1, leading to increased expression of PDX-1. Metformin inhibits PDX-1 expression by inhibiting EGF-stimulated ERK signaling.
Discussion
Despite advances in understanding the molecular mechanisms of PDAC development, molecularly targeted therapy has not been translated into reduced mortality or improved survival in this deadly disease. Indeed, the overall 5-year survival rate is a dismal 6% and the median survival period of 4-6 months. Consequently, the focus of research, which was placed mostly on development of therapeutic compounds, is shifting gradually towards its prevention. Novel targets and agents for chemoprevention are urgently needed. The biguanide metformin is the most widely prescribed drug for the treatment of type 2 diabetes mellitus (T2DM) worldwide. Metformin is also emerging as a potential anticancer agent. Recent epidemiological studies linked administration of metformin to reduced incidence and better prognosis in T2DM oncologic patients, including PDAC. However, the underlying mechanisms by which metformin exerts its antiproliferative effect on PDAC and other human cancers remain elusive. The results presented here indicate that PDX-1, a homeodomain-containing β-cell specific transcription factor, is a novel target for metformin in PDAC. The evidence includes: 1) metformin inhibited PDX-1 transactivity; 2) metformin reduced PDX-1 protein and mRNA levels; and 3) metformin abrogated the increase in PDX-1 expression induced by EGF, a potent growth factor for PDAC cells. In pancreatic β cells, PDX-1 expression is subject to positive regulation by glucose, glucagon-like pepetide (GLP-1) [36, 37] and palmitic acid [38] as well as negative regulation by DNA damage [39], oxidative stress [40], somatostatin receptor 5 (SSTR5) agonist PRL-1980 [41] and advanced glycation end-products (AGEs) [42] via different mechanisms. It is not known whether these signals also operate in PDAC cells where PDX-1 acts as a pro-oncogenic transcription factor. Our study demonstrates that metformin is a novel inhibitor of PDX-1 function and expression in PDAC cells.
PDX-1 overexpression in PDAC cells induces proliferation accompanied by up-regulation of cyclin D, cyclin E and Cdk2 and down-regulation of p21, p27 and p53, while knockdown of PDX-1 by PDX-1 shRNA reverse the effect of PDX-1 on these cell cycle-related proteins [32]. This means that the mechanism by which PDX-1 exerts its oncogenic functions is mediated by the regulation of a specific set of genes involved in cell proliferation. The inhibition of PDX-1 by metformin as demonstrated in this study suggests that metformin inhibits tumor growth via a mechanism that disrupts PDX-1-regulated progression of cell cycle. The knowledge from these studies has potential to be used to design novel therapeutic strategies for the treatment of PDAC.
Due to the mitogenic effect of insulin and its related growth factors [11, 16, 19] and the association of pre-operational insulinemia with cancer progression rates, it has been proposed that metformin acts as a therapeutic agent for non-diabetic cancer patients based largely on its ability to systemically reduce serum insulin and glucose levels [43]. Given the critical role for PDX-1 in regulation of glucose sensing and insulin synthesis [44-48], our results have shown that metformin down-regulated PDX-1, providing a possible new insight that could be of assistance in understanding the molecular mechanism by which metformin exerts its anticancer action: metformin may inhibit insulin's mitogenic effect on cancer growth via down-regulating PDX-1 which leads to reduced insulin synthesis.
EGF has been implicated in the cellular control of PDX-1 expression [49]. ERK signaling pathway is one of the major signaling pathways that mediates EGF's actions. In this study we found that EGF enhanced PDX-1 expression in PDAC cells in an ERK-dependent manner since MEK inhibitor PD0325901 abolished EGF-stimulated PDX-1 expression. Moreover, we found that ERK interacted with PDX-1 and up-regulated PDX-1 expression in PDAC cells. Thus, ERK is a potential PDX-1 kinase. ERK is a proline-directed Ser/Thr kinase [50]. PDX-1 has three proline-directed Ser/Thr residues, Ser 61, Ser 66 and Ser 268 [51]. Although PDX-1 Ser 61 and Ser 66 can be phosphorylated by ERK in vitro [52], it is unlikely that these phosphorylation events occur in cells. First, ERK- mediated phosphorylation of PDX-1 is a positive event for PDX-1 transactivity [52]. Second, previous studies have shown that phosphorylation of Ser 61 and Ser 66 results in enhanced PDX-1 ubiquitination and degradation [40]. Therefore, ERK may target a novel phosphorylation site other than Ser 61 and Ser 66. We have recently found by mass spectrometry that mouse PDX-1 Ser 269 (corresponding to Ser 268 in human PDX-1) is phosphorylated in GFP-PDX-1 stable HEK293 cells and that Ser 269 phosphorylation contributes to stabilization of PDX-1 since mutation of Ser 269 into alanine enhances PDX-1 ubiquitination and shortened the half-life of PDX-1 [35]. Thus, it is interesting to determine if Ser 268 is the ERK phosphorylation site within PDX-1 in PDAC cells under physiological conditions.
Given that metformin inhibited EGF-stimulated ERK kinase activation, it is, thus, reasonable to speculate that metformin inhibits tumor cell proliferation through inhibiting EGF-stimulated ERK kinase activation, leading to down-regulation of PDX-1 expression at post-translational level since ERK has kinase activity towards PDX-1 (Fig. 5). Our current studies also show that metformin inhibits PDX-1 at transcriptional level, as evidenced by its inhibition of PDX-1 mRNA expression. PDX-1 is a transcription factor for PDX-1 itself [24]. Thus, it is possible that down-regulation of PDX-1 mRNA by metformin is due to the down-regulation of PDX-1 expression, which, in turn, results in decreased PDX-1 transcriptional activity. Alternatively, metformin may directly down-regulate PDX-1 mRNA stability, leading to decreased PDX-1 mRNA expression.
In summary, the present work has shown that metformin inhibits PDX-1 expression and its transactivity towards its target gene insulin, thus, acting as a negative regulator for PDX-1 expression and PDX- 1-mediated cellular functions. This study provides new insight into the understanding of the mechanism by which metformin exerts its anticancer activity by down- regulating PDX-1. Identification of PDX-1 as a target by metformin provides the foundation for a more in-depth investigation of the metformin mechanism, which may be invaluable for pancreatic cancer research.
Acknowledgments
The study was funded by The Pilot and Feasibility Study Grant from the CURE: Digestive Disease Research Center (P30DK41301) (to G. Z.), the National Institutes of Health (NIH) grants NIDDK R01- DK46441 and NCI R01-CA095731 and Ann and Jerry Moss Foundation (to F. C. B.), and P01 CA163200, P30 DK4130, R01 DK100405 and Department of Veterans Affair Grant 1I01BX001473 (to E. R.). We thank Katie Elsbury for her editorial assistance and Priscilla Massey and Jacqueline Ismen for their administrative assistance.
Abbreviations
- AMPK
Adenosine monophosphate (AMP)- activated protein kinase
- EGF
Epidermal growth factor
- ERK
Extracellular signal-regulated kinase
- HEK293
Human embryonic kidney 293 cells
- PDAC
Pancreatic ductal adenocarcinoma
- PDX-1
Pancreatic duodenal homeobox-1
Footnotes
Conflict of Interest: The authors have declared that no competing interests exist.
References
- 1.Hundal RS, Krssak M, Dufour S, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49(12):2063–9. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inzucchi SE, Maggs DG, Spollett GR, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med. 1998;338(13):867–72. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- 3.Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):42–6. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheen AJ, Paquot N. Metformin revisited: a critical review of the benefit-risk balance in at-risk patients with type 2 diabetes. Diabetes Metab. 2013;39(3):179–90. doi: 10.1016/j.diabet.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9(6):1057–64. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 6.Franciosi M, Lucisano G, Lapice E, et al. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 8(8):e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137(2):482–8. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider MB, Matsuzaki H, Haorah J, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120(5):1263–70. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 9.Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 5(3):355–64. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kisfalvi K, Moro A, Sinnett-Smith J, Eibl G, Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas. 2013;42(5):781–5. doi: 10.1097/MPA.0b013e31827aec40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinnett-Smith J, Kisfalvi K, Kui R, Rozengurt E. Metformin inhibition of mTORC1 activation, DNA synthesis and proliferation in pancreatic cancer cells: dependence on glucose concentration and role of AMPK. Biochem Biophys Res Commun. 2013;430(1):352–7. doi: 10.1016/j.bbrc.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 2012;5(3):355–64. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang LW, Li ZS, Zou DW, et al. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. 2008;14(47):7192–8. doi: 10.3748/wjg.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin- mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer. 2013;13:235. doi: 10.1186/1471-2407-13-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasih A, Elbaz HA, Huttemann M, Konski AA, Zielske SP. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res. 2014;182(1):50–9. doi: 10.1667/RR13568.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69(16):6539–45. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinnett-Smith J, Kisfalvi K, Kui R, Rozengurt E. Metformin inhibition of mTORC1 activation, DNA synthesis and proliferation in pancreatic cancer cells: dependence on glucose concentration and role of AMPK. Biochem Biophys Res Commun. 2013;430(1):352–7. doi: 10.1016/j.bbrc.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohammed A, Janakiram NB, Brewer M, et al. Antidiabetic Drug Metformin Prevents Progression of Pancreatic Cancer by Targeting in Part Cancer Stem Cells and mTOR Signaling. Transl Oncol. 2014;6(6):649–59. doi: 10.1593/tlo.13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One. 2013;8(2):e57289. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura M, Ogo A, Yamura M, Yamaguchi Y, Nakashima H. Metformin suppresses sonic hedgehog expression in pancreatic cancer cells. Anticancer Res. 2014;34(4):1765–9. [PubMed] [Google Scholar]
- 21.Nair V, Pathi S, Jutooru I, et al. Metformin inhibits pancreatic cancer cell and tumor growth and downregulates Sp transcription factors. Carcinogenesis. 2013;34(12):2870–9. doi: 10.1093/carcin/bgt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Yuan Y, Huang L, Qiao M, Zhang Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res Clin Pract. 2012;96(2):187–95. doi: 10.1016/j.diabres.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 23.Lonardo E, Cioffi M, Sancho P, et al. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS One. 2013;8(10):e76518. doi: 10.1371/journal.pone.0076518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashizawa S, Brunicardi FC, Wang XP. PDX-1 and the pancreas. Pancreas. 2004;28(2):109–20. doi: 10.1097/00006676-200403000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Kaneto H, Miyatsuka T, Shiraiwa T, et al. Crucial role of PDX-1 in pancreas development, beta-cell differentiation, and induction of surrogate beta-cells. Curr Med Chem. 2007;14(16):1745–52. doi: 10.2174/092986707781058887. [DOI] [PubMed] [Google Scholar]
- 26.Koizumi M, Doi R, Toyoda E, et al. Increased PDX-1 expression is associated with outcome in patients with pancreatic cancer. Surgery. 2003;134(2):260–6. doi: 10.1067/msy.2003.231. [DOI] [PubMed] [Google Scholar]
- 27.Liu T, Gou SM, Wang CY, et al. Pancreas duodenal homeobox-1 expression and significance in pancreatic cancer. World J Gastroenterol. 2007;13(18):2615–8. doi: 10.3748/wjg.v13.i18.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang XP, Li ZJ, Magnusson J, Brunicardi FC. Tissue MicroArray analyses of pancreatic duodenal homeobox-1 in human cancers. World J Surg. 2005;29(3):334–8. doi: 10.1007/s00268-004-7823-4. [DOI] [PubMed] [Google Scholar]
- 29.Quint K, Stintzing S, Alinger B, et al. The expression pattern of PDX-1, SHH, Patched and Gli-1 is associated with pathological and clinical features in human pancreatic cancer. Pancreatology. 2009;9(1-2):116–26. doi: 10.1159/000178882. [DOI] [PubMed] [Google Scholar]
- 30.Miyatsuka T, Kaneto H, Shiraiwa T, et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006;20(11):1435–40. doi: 10.1101/gad.1412806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gidekel Friedlander SY, Chu GC, Snyder EL, et al. Context- dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16(5):379–89. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu SH, Patel S, Gingras MC, et al. PDX-1: demonstration of oncogenic properties in pancreatic cancer. Cancer. 2011;117(4):723–33. doi: 10.1002/cncr.25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Ballian N, Belaguli NS, et al. PDX-1 acts as a potential molecular target for treatment of human pancreatic cancer. Pancreas. 2008;37(2):210–20. doi: 10.1097/MPA.0b013e31816a4a33. [DOI] [PubMed] [Google Scholar]
- 34.Liu SH, Rao DD, Nemunaitis J, et al. PDX-1 is a therapeutic target for pancreatic cancer, insulinoma and islet neoplasia using a novel RNA interference platform. PLoS One. 2012;7(8):e40452. doi: 10.1371/journal.pone.0040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou G, Wang H, Liu SH, et al. p38 MAP Kinase Interacts with and Stabilizes Pancreatic and Duodenal Homeobox-1. Curr Mol Med. 2013;13(3):377–86. [PubMed] [Google Scholar]
- 36.Buteau J, Roduit R, Susini S, Prentki M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia. 1999;42(7):856–64. doi: 10.1007/s001250051238. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Cahill CM, Pineyro MA, et al. Glucagon-like peptide-regulates the beta cell transcription factor, PDX-1, in insulinoma cells. Endocrinology. 1999;140(10):4904–7. doi: 10.1210/endo.140.10.7158. [DOI] [PubMed] [Google Scholar]
- 38.Arantes VC, Reis MA, Latorraca MQ, et al. Palmitic acid increase levels of pancreatic duodenal homeobox-1 and p38/stress-activated protein kinase in islets from rats maintained on a low protein diet. Br J Nutr. 2006;96(6):1006–12. doi: 10.1017/bjn20061950. [DOI] [PubMed] [Google Scholar]
- 39.Lebrun P, Montminy MR, Van Obberghen E. Regulation of the pancreatic duodenal dependent protein kinase. J Biol Chem. 2005;280(46):38203–10. doi: 10.1074/jbc.M504842200. [DOI] [PubMed] [Google Scholar]
- 40.Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem. 2006;281(10):6395–403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- 41.Zhou G, Gingras MC, Liu SH, et al. The hypofunctional effect of P335L single nucleotide polymorphism on SSTR5 function. World J Surg. 2011;35(8):1715–24. doi: 10.1007/s00268-010-0939-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puddu A, Storace D, Odetti P, Viviani GL. Advanced glycation end-products affect transcription factors regulating insulin gene expression. Biochem Biophys Res Commun. 2010;395(1):122–5. doi: 10.1016/j.bbrc.2010.03.152. [DOI] [PubMed] [Google Scholar]
- 43.Goodwin PJ, Pritchard KI, Ennis M, et al. Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer. 2008;8(6):501–5. doi: 10.3816/CBC.2008.n.060. [DOI] [PubMed] [Google Scholar]
- 44.Watada H, Kajimoto Y, Miyagawa J, et al. PDX-1 induces insulin and glucokinase gene expressions in alphaTC1 clone 6 cells in the presence of betacellulin. Diabetes. 1996;45(12):1826–31. doi: 10.2337/diab.45.12.1826. [DOI] [PubMed] [Google Scholar]
- 45.Bretherton-Watt D, Gore N, Boam DS. Insulin upstream factor 1 and a novel ubiquitous factor bind to the human islet amyloid polypeptide/amylin gene promoter. Biochem J. 1996;313(Pt 2):495–502. doi: 10.1042/bj3130495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carty MD, Lillquist JS, Peshavaria M, Stein R, Soeller WC. Identification of cis- and trans-active factors regulating human islet amyloid polypeptide gene expression in pancreatic beta-cells. J Biol Chem. 1997;272(18):11986–93. doi: 10.1074/jbc.272.18.11986. [DOI] [PubMed] [Google Scholar]
- 47.Serup P, Jensen J, Andersen FG, et al. Induction of insulin and islet amyloid polypeptide production in pancreatic islet glucagonoma cells by insulin promoter factor 1. Proc Natl Acad Sci U S A. 1996;93(17):9015–20. doi: 10.1073/pnas.93.17.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watada H, Kajimoto Y, Kaneto H, et al. Involvement of the homeodomain-containing transcription factor PDX-1 in islet amyloid polypeptide gene transcription. Biochem Biophys Res Commun. 1996;229(3):746–51. doi: 10.1006/bbrc.1996.1875. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe H, Saito H, Ueda J, Evers BM. Regulation of pancreatic duct cell differentiation by phosphatidylinositol-3 kinase. Biochem Biophys Res Commun. 2008;370(1):33–7. doi: 10.1016/j.bbrc.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boulton TG, Cobb MH. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul. 1991;2(5):357–71. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milewski WM, Duguay SJ, Chan SJ, Steiner DF. Conservation of PDX-1 structure, function, and expression in zebrafish. Endocrinology. 1998;139(3):1440–9. doi: 10.1210/endo.139.3.5768. [DOI] [PubMed] [Google Scholar]
- 52.Khoo S, Griffen SC, Xia Y, et al. Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J Biol Chem. 2003;278(35):32969–77. doi: 10.1074/jbc.M301198200. [DOI] [PubMed] [Google Scholar]