

Abstract
Objective
To identify the genetic basis of a severe skeletal lethal dysplasia. The main clinical features of two affected fetuses included: short limbs with flared metaphyses, bowed radii, femora and tibiae, irregular ossification of hands and feet and marked platyspondyly.
Methods
Affected and non-affected family members were subjected to whole exome sequencing followed by immunoblot analysis on amniocytes isolated from one of the affected individuals.
Results
Unique compound heterozygous variants in the INPPL1 (inositol polyphosphate phosphatase-like 1) gene encoding the SHIP2 protein were identified in both affected individuals. One variant was inherited from each unaffected parent. Both allelic variants, c.[ 2327-1G>C];[1150_1151delGA], are predicted to result in premature stop codons leading to nonsense-mediated mRNA decay of the mutant alleles and no production of SHIP2. The absence of SHIP2 was confirmed by immunoblot analysis of proband amniocytes.
Conclusions
This skeletal disorder is caused by the complete absence of SHIP2 protein. INPPL1 mutations have been reported in opsismodysplasia (OPS), an autosomal recessive skeletal dysplasias with significant delayed bone formation. Our finding highlights the critical role that INPPL1/SHIP2 plays in skeletal development.
Keywords: chondrodysplasia, skeletal development, phosphatase
Clinical summary
This report describes a non-consanguineous family in which pregnancies to phenotypically normal parents resulted in two affected fetuses with a severe skeletal dysplasia. This research project was reviewed and approved by the Oregon Health and Science University Institutional Review Board and written informed consent obtained from the participants or their parents according to The Code of Federal Regulations describing the Protection of Human Subjects (45 CFR 46). Fetus #1 presented to care at 16w3d gestation due to suspicion of skeletal dysplasia on ultrasound. The pregnancy was conceived via in vitro fertilization (IVF) with fresh embryo transfer. The mother was a healthy 38-year old with a history of one miscarriage and unexplained infertility. The nuchal translucency had been increased on first trimester ultrasound. Ultrasound in the second trimester revealed short long bones and small chest circumference. There was frontal bossing with flat mid-facies as well as unilateral clubfoot. Scapulae were present and mineralization of the bones appeared normal. Amniocentesis was performed with normal female karyotype (46,XX). Based on a suspected diagnosis of Thanatophoric dysplasia, cultured amniocytes were sent for FGFR3 gene sequencing, which was normal. The parents declined additional genetic testing, but did consent to storage of DNA isolated from cultured amniocytes. The pregnancy was terminated at 17 weeks gestation. Radiographs were not performed.
Fetus #2 presented at 20w4d for ultrasound. This pregnancy was also conceived via IVF with frozen embryo transfer. Ultrasound in the second trimester revealed short long bones and a small chest circumference. As with fetus #1, there was frontal bossing with flat mid-facies. The ears also appeared low-set. The phalanges and metacarpals are very short with redundant soft tissue and there was unilateral clubfoot. The ischia are ossified. Mineralization of the bones appeared normal and, although there was mild bowing of the long bones, there were no obvious fractures by ultrasound. The skull shape was normal with normal ossification. Amniocentesis was performed with normal female karyotype (46,XX).
To test for genes commonly associated with severe chondrodysplasia, cultured amniocytes were sent for an extensive panel of genetic testing including sequencing of COL2A1, COL11A1, COL11A2, DTDST and TRIP11 and deletion/duplication analysis for COL2A1 and COL11A2. Peroxisomal plasmalogen synthesis enzymes were also assayed for chondrodysplasia punctata. All testing was normal and the pregnancy was continued. At 25w2d gestation the fetus developed neck hyperextension, polyhydramnios requiring serial amnioreductions and mild skin thickening over the skull with no evidence of hydrops. By 27w2d gestation the skin thickening had progressed to include the dorsal gluteal region and upper thigh. There was exaggerated lordosis with minimal truncal movement and mild ventriculomegaly. The ventriculomegaly continued to rapidly progress and by 31 weeks gestation the fetus had developed hydrops and was delivered stillborn.
Radiographs of fetus #2 showed hydropic changes and short limbs with flaring and metaphyseal cupping were reported. Both distal femoral metaphyses appeared irregular and the radii, femora and tibae were bowed (Figure 1). There was delayed ossification of the small bones of the hands and feet. The iliac bones were decreased in vertical height with horizontal orientation of the acetabular roofs. There was some metaphyseal translucency of the femoral metaphyses, but not undermineralization. There was severe platyspondyly throughout the spine, the sacrum was poorly ossified and the ischia and pubic bones were unossified. The neurocranium appeared large compared to the facial structures.
Figure 1.
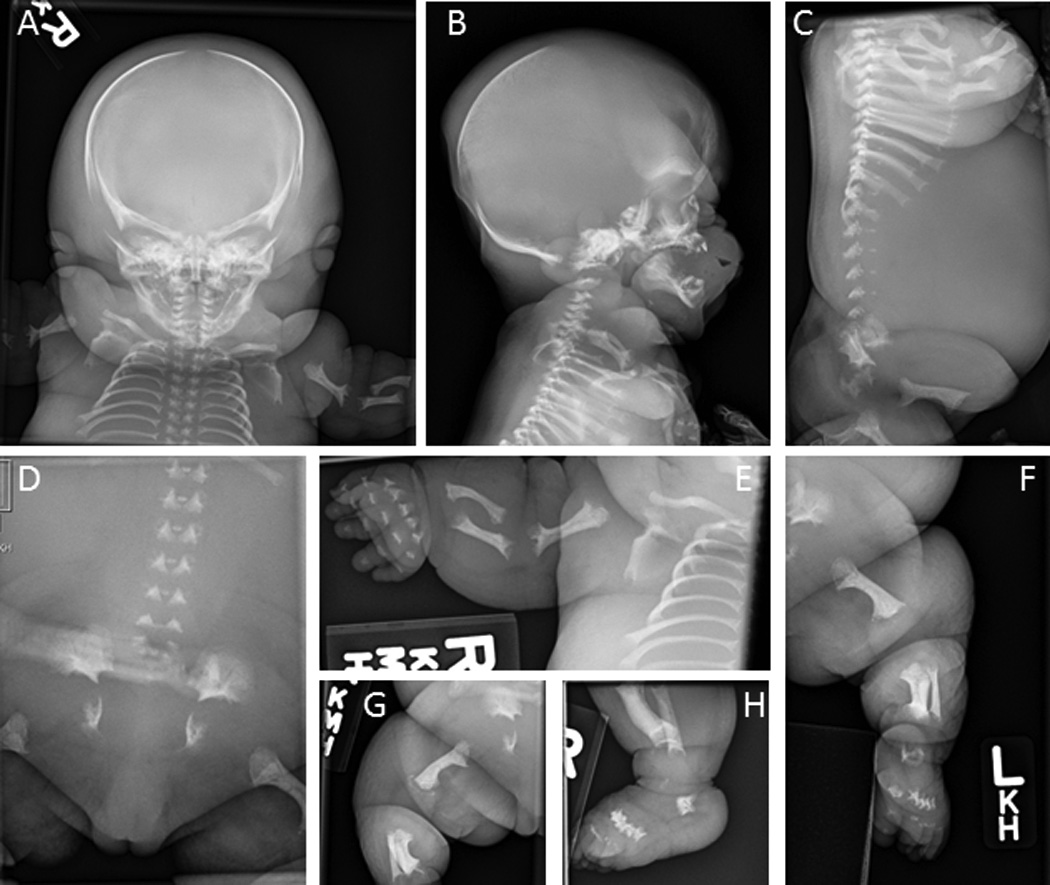
Radiographs of fetus #2 at 31 weeks gestation. A, Anterior-posterior (AP) view of head, upper spine, ribs and left arm showing prominent soft tissue consistent with hydrops. B and C, Lateral views of head and spine showing severe platyspondyly and large neurocranium. D, AP view of pelvis showing poorly ossified sacrum, ischia and pubic bones with abnormal orientation of the acetabular roof. E, AP view of right arm showing undermineralized bones of the hand with shortened metacarpals and phalanges, and bowing of the humerus, radius and ulna. F, AP view of left leg showing bowing of the femur and tibia, and flared metaphyses. G, Lateral view of right leg showing bowed femur. H, Lateral view of right foot and lower leg showing shortened, poorly ossified metatarsals.
Methods and results
Following the failure to find a genetic defect by targeted sequencing of the DNA from affected fetuses, maternal and paternal genomic DNA was collected and DNA from all four individuals subjected to exome sequencing by Ambry Genetics (Aliso Viejo, CA). The inheritance pattern of two affected individuals from phenotypically normal parents suggested an autosomal recessive model. Bioinformatic analysis of the data indicated that the two variants were present in the INPPL1 (inositol polyphosphate phosphatase-like 1) gene in the affected individuals in the pattern expected for a recessive inheritance model. These variants are absent from dbSNP and the 1000 genomes project databases and are predicted to be pathogenic.
Each parent transmitted a different INPPL1 variant to both affected fetuses demonstrating a compound heterozygous mode of inheritance. The paternally-derived variant is a 2bp (GA dinucleotide) deletion in exon 10 [c.1150_1151delGA] leading to a change in the reading frame and a predicted premature stop codon 81 amino acids downstream. The maternal variant is a splice site mutation immediately upstream of exon 21 in the -1 position [c.2327-1G>C]. This variant predicts the skipping of exon 21 and the splicing of exon 20 to exon 22. The frameshift caused by splicing exons 20/22 is predicted to result in a premature stop codon in exon 22. All genotypes were confirmed by Sanger sequencing. Since both variants inherited by the affected fetuses are likely to have resulted in premature stop codons leading to nonsense-mediated mRNA decay, we examined fetal amniocytes from fetus #2 for SHIP2 protein (Figure 2). Protein extracts were prepared from proband and control amniocytes, and HeLa cells, electrophoresed, transferred to a solid membrane and probed with an anti-SHIP2 polyclonal antibody (Abcam cat# 166916). Control amniocytes and HeLa cell extracts contained detectable levels of SHIP2 at the correct molecular weight of 140 kDa. In contrast, no protein was detected in the proband cell extract suggesting that the mutant mRNA alleles for INPPL1 were indeed degraded and no SHIP2 protein produced. The blot was re-probed with an anti-α-tubulin antibody (Santa Cruz biotechnology cat# sc-23948) to demonstrate approximately equal loading of cell extracts.
Figure 2.
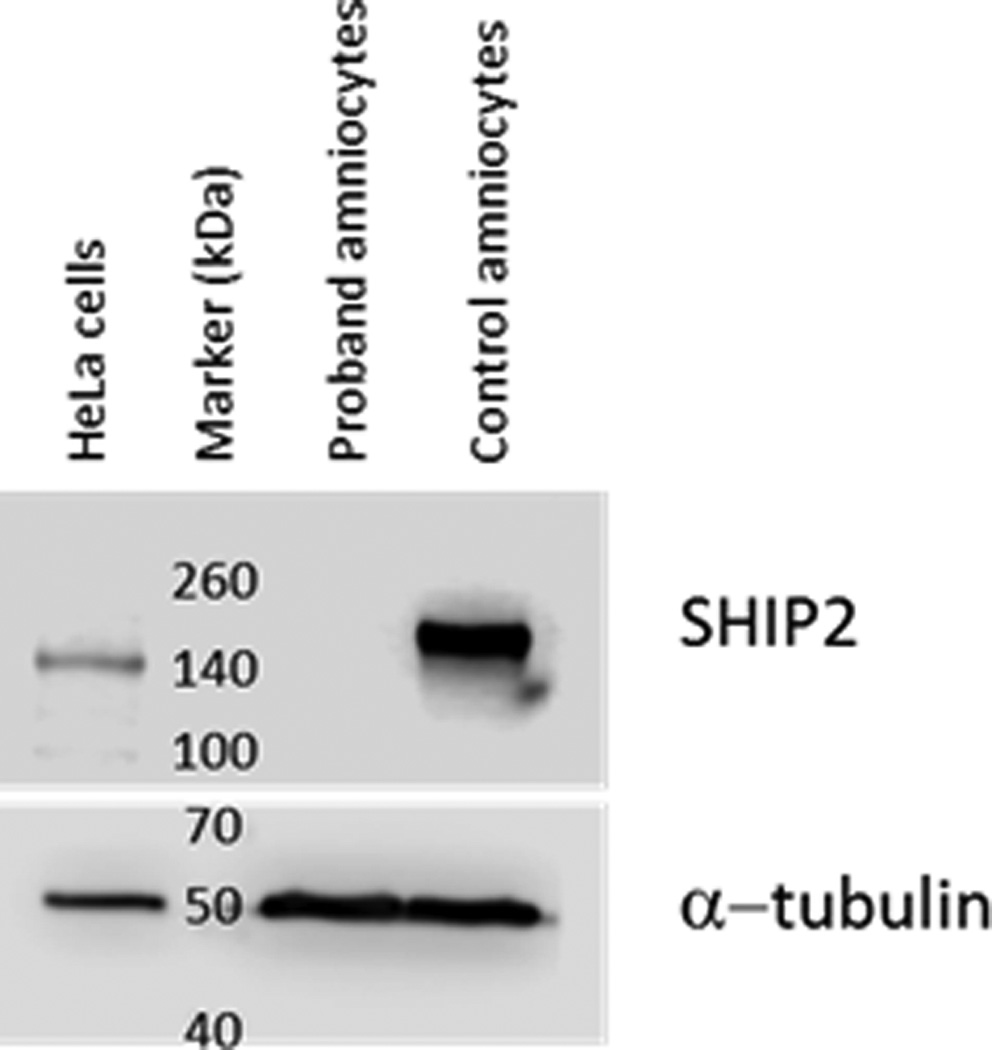
The INPPL1 gene product, SHIP2, is absent in proband amniocytes. Protein extracts from proband and control amniocytes (lanes 2 and 3) and HeLa cells (lane 1) were subjected to immunoblot analysis and probed with an anti-SHIP2 antibody. SHIP2 is present in control amniocyte and HeLa extracts but absent in proband extracts. The blot was re-probed with an anti α-tubulin antibody to show equal loading of cell extracts. Migration of molecular weight markers (kDa) are shown in lane 2.
Discussion
We report the identification of compound heterozygous mutations in the INPPL1 gene, encoding the SHIP2 protein, in a family with two individuals with a lethal skeletal dysplasia. INPPL1 mutations have been identified in opsismodysplasia (OPS) [OMIM 258480] (Below et al., 2013, Huber et al., 2013, Iida et al., 2013, Li et al., 2014) and, early in 2015, in Schneckenbecken Dysplasia [OMIM 269250] (Lee et al., 2015). These conditions were not initially considered as a diagnosis and other genes known to be involved in more common severe chondrodyspasias, including FGFR3, COL2A1, COL11A1, COL11A2, DTST and TRIP11, were sequenced. Following the failure to identify mutations in these genes a whole exome approach was used leading to the identification of compound heterozygous mutations in INPPL1.
OPS is a rare, autosomal recessive chondrodysplasia first described in 1977 and is primarily characterized by delayed bone maturation (Zonana et al., 1977, Maroteaux et al., 1984, Cormier-Daire et al., 2003). Affected patients have rhizomelic micromelia, small hands and feet, relative macrocephaly and characteristic craniofacial abnormalities including a prominent brow ridge. Radiographic findings reported include shortened long bones with markedly delayed epiphyseal ossification and severe platyspondyly. Histological analysis of fetal growth plate cartilage shows disorganized proliferative zones with near absent columnar organization (Cormier-Daire et al., 2003) suggesting that the poor mineralization and other bony defects are due to abnormal cartilage growth plate organization and chondrocyte differentiation. Disease severity is highly variable from neonatal death due to respiratory failure to survival through to adulthood.
Recently, a homozygous INPPL1 mutation was described in Schneckenbecken Dysplasia (SD), a severe, autosomal recessive, perinatal lethal dysplasia similar to OPS (Lee et al., 2015). SD demonstrates locus heterogeneity and had previously been shown to be caused by mutations in SLC35D1, encoding an ER-resident sugar transporter (Hiraoka et al., 2007, Furuichi et al., 2009). SD is distinguished from other severe skeletal dysplasias by the presence on radiographs of a medial projection from the ilia that resembles a snail. Since the family presented here does not appear to have this feature based on the available radiographs we suggest that a diagnosis of OPS is appropriate.
To date, 19 OPS and 1 SD families with INPPL1 mutations have been reported (Below et al., 2013, Huber et al., 2013, Iida et al., 2013, Li et al., 2014, Lee et al., 2015). SHIP2 is one of 10 mammalian inositol polyphosphate 5-phosphatases and functions to dephosphorylate the lipid second messenger phosphoinositol (3,4,5)P3 at the 5-position of the inositol ring to phosphoinositol (3,4)P2 (Pesesse et al., 1997, Habib et al., 1998). Consistent with a role for INPPL1 in skeletal development, the Inppl1−/− mice have diminished growth and craniofacial abnormalities, however, these were mild in severity. There may be non-catalytic roles of SHIP2 since, in addition to the phosphatase domain, the protein contains several motifs known to be involved in protein-protein interactions (see reviews by (Backers et al., 2003, Erneux et al., 2011)). It is unknown at the present time whether the effects on cartilage and bone development are due to catalytic or non-catalytic mechanisms involving growth factor second messenger signaling.
Future studies will be directed towards understanding why INPPL1/SHIP2 is essential for normal skeletal differentiation and development.
Acknowledgments
We thank the family for their participation and support.
Footnotes
The authors have no conflicts of interest to report.
References
- Backers K, Blero D, Paternotte N, Zhang J, Erneux C. The termination of PI3K signalling by SHIP1 and SHIP2 inositol 5-phosphatases. Adv Enzyme Regul. 2003;43:15–28. doi: 10.1016/s0065-2571(02)00043-2. [DOI] [PubMed] [Google Scholar]
- Below JE, Earl DL, Shively KM, Mcmillin MJ, Smith JD, Turner EH, Stephan MJ, Al-Gazali LI, Hertecant JL, Chitayat D, Unger S, Cohn DH, Krakow D, Swanson JM, Faustman EM, Shendure J, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian, G Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am J Hum Genet. 2013;92:137–143. doi: 10.1016/j.ajhg.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier-Daire V, Delezoide AL, Philip N, Marcorelles P, Casas K, Hillion Y, Faivre L, Rimoin DL, Munnich A, Maroteaux P, Le Merrer M. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J Med Genet. 2003;40:195–200. doi: 10.1136/jmg.40.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erneux C, Edimo WE, Deneubourg L, Pirson I. SHIP2 multiple functions: a balance between a negative control of PtdIns(3,4,5)P(3) level, a positive control of PtdIns(3,4)P(2) production, and intrinsic docking properties. J Cell Biochem. 2011;112:2203–2209. doi: 10.1002/jcb.23146. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Kayserili H, Hiraoka S, Nishimura G, Ohashi H, Alanay Y, Lerena JC, Aslanger AD, Koseki H, Cohn DH, Superti-Furga A, Unger S, Ikegawa S. Identification of loss-of-function mutations of SLC35D1 in patients with Schneckenbecken dysplasia, but not with other severe spondylodysplastic dysplasias group diseases. J Med Genet. 2009;46:562–568. doi: 10.1136/jmg.2008.065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib T, Hejna JA, Moses RE, Decker SJ. Growth factors and insulin stimulate tyrosine phosphorylation of the 51C/SHIP2 protein. J Biol Chem. 1998;273:18605–18609. doi: 10.1074/jbc.273.29.18605. [DOI] [PubMed] [Google Scholar]
- Hiraoka S, Furuichi T, Nishimura G, Shibata S, Yanagishita M, Rimoin DL, Superti-Furga A, Nikkels PG, Ogawa M, Katsuyama K, Toyoda H, Kinoshita-Toyoda A, Ishida N, Isono K, Sanai Y, Cohn DH, Koseki H, Ikegawa S. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat Med. 2007;13:1363–1367. doi: 10.1038/nm1655. [DOI] [PubMed] [Google Scholar]
- Huber C, Faqeih EA, Bartholdi D, Bole-Feysot C, Borochowitz Z, Cavalcanti DP, Frigo A, Nitschke P, Roume J, Santos HG, Shalev SA, Superti-Furga A, Delezoide AL, Le Merrer M, Munnich A, Cormier-Daire V. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am J Hum Genet. 2013;92:144–149. doi: 10.1016/j.ajhg.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida A, Okamoto N, Miyake N, Nishimura G, Minami S, Sugimoto T, Nakashima M, Tsurusaki Y, Saitsu H, Shiina M, Ogata K, Watanabe S, Ohashi H, Matsumoto N, Ikegawa S. Exome sequencing identifies a novel INPPL1 mutation in opsismodysplasia. J Hum Genet. 2013;58:391–394. doi: 10.1038/jhg.2013.25. [DOI] [PubMed] [Google Scholar]
- Lee H, Nevarez L, Lachman RS, Wilcox WR, Krakow D, Cohn DH, University of Washington Center for Mendelian, G A second locus for schneckenbecken dysplasia identified by a mutation in the gene encoding inositol polyphosphate phosphatase-like 1 (INPPL1) Am J Med Genet A. 2015 doi: 10.1002/ajmg.a.37173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Krakow D, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian, G. Chang Y, Lachman RS, Yilmaz A, Kayserili H, Cohn DH. Opsismodysplasia resulting from an insertion mutation in the SH2 domain, which destabilizes INPPL1. Am J Med Genet A. 2014;164A:2407–2411. doi: 10.1002/ajmg.a.36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux P, Stanescu V, Stanescu R, Le Marec B, Moraine C, Lejarraga H. Opsismodysplasia: a new type of chondrodysplasia with predominant involvement of the bones of the hand and the vertebrae. Am J Med Genet. 1984;19:171–182. doi: 10.1002/ajmg.1320190117. [DOI] [PubMed] [Google Scholar]
- Pesesse X, Deleu S, De Smedt F, Drayer L, Erneux C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem Biophys Res Commun. 1997;239:697–700. doi: 10.1006/bbrc.1997.7538. [DOI] [PubMed] [Google Scholar]
- Zonana J, Rimoin DL, Lachman RS, Cohen AH. A unique chondrodysplasia secondary to a defect in chondroosseous transformation. Birth Defects Orig Artic Ser. 1977;13:155–163. [PubMed] [Google Scholar]