

Abstract
Medulloblastoma accounts for nearly 10% of all childhood brain tumors. These tumors occur exclusively in the posterior fossa and have the potential for leptomeningeal spread. Treatment includes a combination of surgery, radiation therapy (in patients > 3 years old). Patients > 3 years old are stratified based on the volume of postoperative residual tumor and the presence or absence of metastases into “standard risk” and “high risk” categories with long term survival rates of approximately 85% and 70%, respectively. Outcomes are inferior in infants and children younger than 3 years with exception of those patients with the MBEN histologic subtype. Treatment for medulloblastoma is associated with significant morbidity, especially in the youngest patients. Recent molecular subclassification of medulloblastoma has potential prognostic and therapeutic implications. Future incorporation of molecular subgroups into treatment protocols will hopefully improve both survival outcomes and post-treatment quality of life.
Brief History of the Disorder
The name “medulloblastoma” was first introduced by Harvey Cushing and Percival Bailey in 1925. Drs. Cushing and Bailey had initially used the term “spongioblastoma cerebelli” to describe the posterior fossa tumor seen in pre-adolescents. They changed the name to medulloblastoma in order to distinguish it from a distinct glial tumor contemporaneously described as a spongioblastoma by Globus and Strauss. This new nomenclature reflected medulloblastoma’s perceived derivation from one of the five pluripotent stem cells thought to populate the primitive neural tube, although it has since been recognized that there is no embryonal cell that can be identified as a medulloblast [1-3]. Cushing, a neurosurgeon at Peter Bent Brigham Hospital, first described the defining characteristics of medulloblastoma, namely its penchant for arising in the location of the cerebellar vermis and metastasizing to distant locations, its propensity to affect younger male patients, and its comparatively short duration of signs and symptoms prior to diagnosis. Treatment of medulloblastoma patients in Cushing’s era was fraught with immediate post-operative mortality exceeding 30%. He observed that patients who underwent more extensive operative resections had a longer duration of survival as compared to those patients whose tumors were only biopsied and is credited with contributing significantly to the evolution of neurosurgical techniques in the management of medulloblastoma. Despite these advances, outcomes in medulloblastoma patients remained dismal until craniospinal irradiation was demonstrated to dramatically improve survival in a pivotal paper published by Paterson and Farr in 1953. Patients in their series received 5000 cGy to the posterior fossa and 3500 cGy to the remainder of the neuraxis and were reported to experience a 3-year survival of 65% [4]. It was subsequently recognized that this survival benefit was accompanied by significant negative impacts on younger patients in the form of neurocognitive impairment, second malignancies, and endocrine dysfunction [5]. Treatment protocols in the 1970’s began to introduce adjuvant chemotherapy to surgery and radiation in an effort to improve survival outcomes and today its use remains standard in the treatment of medulloblastoma, in some cases allowing for dose reduction or elimination of radiation [6].
In the early 1980’s, based on histological similarity between medulloblastomas and other small round blue cell tumors arising in areas outside of the posterior fossa, it was proposed that these tumors be classified together under the umbrella group of primitive neuroectodermal tumors (PNETs) [7]. However, more recent studies suggest that small round blue cell tumors of the posterior fossa are molecularly distinct from those arising in the cortex or pineal regions [8, 9]. This is reflected in the most recent WHO classification, which distinguishes medulloblastoma and its subtypes from other CNS PNETs, pineoblastomas, and atypical teratoid/rhabdoid tumors [10].
Historically, age at diagnosis and extent of disease (as measured by the modified Chang criteria) have been most strongly associated with prognosis, with younger patients and those with more advanced disease faring more poorly than older patients and those with limited disease, respectively [11]. More recently, pretreatment prognosis of medulloblastoma has been refined by histopathologic subclassification into the following variants: large-cell medulloblastoma, anaplastic medulloblastoma, desmoplastic/nodular medulloblastoma, and medulloblastoma with extensive nodularity (MBEN). The latter two variants have been shown to have a significantly superior prognosis as compared to the large cell and anaplastic variants in young children [12].
The most recent insights into the biology of medulloblastomas have occurred at the molecular level, resulting in their categorization into the following molecular subgroups: wingless (Wnt), sonic hedgehog (Shh), Group 3, and Group 4. Each subgroup is characterized by a unique set of genetics and gene expression as well as demographic and clinical features. Although this information has not yet been used for risk stratification in clinical trials, evidence suggests that prognosis is strongly associated with subgroup affiliation [13, 14].
Epidemiology
Although many sources have stated that medulloblastoma is the most common malignant brain tumor in children, the most recent data from the Central Brain Tumor Registry of the United States (CBTRUS) shows that high-grade gliomas, as a group, are slightly more prevalent. Medulloblastoma represents 9.2 percent of pediatric brain tumors in children aged 0-14 years, and approximately 338 new pediatric cases are diagnosed in the United States each year [15-18]. Up to 30 percent of cases are reported in adults, generally prior to age 40. The incidence of medulloblastoma peaks during the first decade of life with a higher incidence noted in children between 3 and 4 years of age and between 8 and 10 years of age. Fewer than 5 percent of cases are associated with the hereditary cancer predisposition syndromes familial adenomatous polyposis (FAP), historically known as Turcot syndrome, or nevoid basal cell carcinoma syndrome (NBCCS), also called Gorlin syndrome. In children, medulloblastoma is slightly more common in males, with a male to female incidence rate ratio of 0.63. There is no apparent racial or ethnic predisposition [16, 19].
Clinical Presentation
In concordance with its relatively rapid growth rate, patients with medulloblastoma commonly present with symptoms evolving over a period of weeks to months. A combination of signs and symptoms of cerebellar dysfunction and increased intracranial pressure (ICP) are frequently encountered. Classic symptoms of increased ICP include irritability, lethargy, nausea and vomiting, morning headaches, anorexia, and behavioral changes. Signs of cerebellar involvement may differ depending on the location of the lesion. Midline cerebellar tumors are more likely to result in truncal ataxia (with impaired tandem gait and Romberg testing) as compared to cerebellar hemispheric tumors that are more commonly associated with appendicular ataxia (manifesting as difficulty with rapid alternating movements, finger-nose-finger, and heel-shin testing). Additionally, cranial nerve involvement may be present either as a result of direct involvement of these nerves or as a consequence of increased ICP. Diplopia resulting from abducens nerve dysfunction is not uncommon. In cases where disease dissemination is present, symptoms related to the location of metastatic involvement may also be observed.
Differential Diagnosis
The radiographic differential diagnosis of a posterior fossa mass in children comprises ependymoma, atypical teratoid/rhabdoid tumor and pilocytic astrocytoma most commonly, with additional considerations including exophytic brainstem glioma and choroid plexus papilloma as well as teratoma in infants and hemangioblastoma in patients with Von Hippel-Lindau syndrome. In adults, metastatic disease from a distant primary site is the most frequently encountered posterior fossa lesion [20].
Medulloblastomas have distinct imaging characteristics on both computed tomography (CT) and magnetic resonance imaging (MRI). Three-quarters of medulloblastomas arise from the cerebellar vermis and tend to protrude into the fourth ventricle, although the site of origin in adults is more frequently the cerebellar hemispheres rather than the vermis. In contrast to ependymomas, medulloblastomas do not typically extend into the basal cisterns. On CT, they are most commonly seen as a hyperdense mass arising from the vermis with cyst formation or necrosis more frequently observed in older patients. Effacement of the fourth ventricle and ventricular dilatation secondary to obstructive hydrocephalus are often seen. Prominent contrast enhancement is present in 90% of cases with calcification seen in 10-20%. On MRI, medulloblastomas are hypointense to grey matter on T1-weighted imaging with heterogeneous gadolinium enhancement in 90%. They are generally iso- to hyperintense to grey matter on T2-weighted imaging and commonly appear heterogeneous due to cyst formation, calcification and necrosis. Diffusion-weighted imaging shows restricted diffusion and medulloblastomas are hyperintense to surrounding brain on fluid-attenuated inversion recovery (FLAIR) sequences. MR spectroscopy shows elevated choline peaks and decreased creatine and N-acetyl acetate peaks, with occasional elevation in lactic acid and lipid peaks [21, 22].
A recent publication by Perreault et al reported a statistically significant relationship between medulloblastoma molecular subgroups and both enhancement pattern and tumor location on MRI. In their cohort of forty-seven patients, tumors located along the cerebellar peduncle/cerebellopontine angle cistern were found to exclusively represent Wingless pathway tumors (Wnt), with three of four Wnt pathway tumors occurring in this location while one was located in the midline vermis/fourth ventricle. More than half of the thirteen sonic hedgehog (Shh) tumors were located within the cerebellar hemispheres with the remainder found in the midline vermis/fourth ventricle. Shh was the only molecular subgroup whose tumors were found in hemispheric locations. All thirty Group 3 and Group 4 tumors were encountered in the midline vermis/fourth ventricle. Group 3 tumors were more likely to demonstrate ill-defined tumor margins, while Group 4 tumors were generally characterized by minimal or absent contrast enhancement. The presence of cysts, hemorrhage/mineralization, edema, and necrosis were not specifically associated with any molecular subgroup [23]. In a series of 130 patients from the Hospital for Sick Children in Toronto, the presence of hydrocephalus necessitating CSF diversion surgery was approximately 30% in patients with Shh, Group 3, and Group 4 tumors. None of the fifteen Wnt medulloblastoma patients in this cohort required endoscopic third ventriculostomy or ventriculoperitoneal shunt placement. This finding was thought to be a result of clinical features associated with this molecular subgroup, namely older age and lack of metastatic disease at diagnosis [24]. The presence of hydrocephalus on MRI would therefore argue in favor of Shh, Group 3, or Group 4 molecular subgroups over Wnt.
Workup
Imaging of the entire neuraxis is indicated due to medulloblastoma’s predilection for spread along the cerebrospinal fluid (CSF) pathways with resultant drop metastases and leptomeningeal disease, present in approximately one-third of patients at time of diagnosis. In a stable patient, it is considered preferable to obtain this imaging prior to surgical intervention in order to avoid potentially confounding postoperative artifact. Lumbar puncture (LP) for CSF cytology is performed to provide additional information regarding microscopic leptomeningeal disease dissemination, although evaluation of the CSF may be negative in patients with nodular spinal cord disease. LP may be performed prior to surgery once hydrocephalus and increased intracranial pressure have been ruled out via imaging studies. As patients with posterior fossa tumors often present with symptomatic hydrocephalus, it is not uncommon for LP to be deemed unsafe at the time of presentation. When deferred until the post-operative setting, LP should be delayed by ten to fourteen days in order to avoid false positive evaluations due to surgical tumor debris [25-27]. The combination of brain/spine imaging and lumbar CSF cytology is more sensitive than either test alone; therefore, both are recommended as part of the extent of disease evaluation [28]. Extraneural spread of disease to bone and bone marrow has also rarely been reported in medulloblastoma patients. Bone scan may be indicated in symptomatic patients, and bone marrow evaluation, while not routinely performed, is considered in any patient with unexplained abnormal peripheral blood counts and occasionally in those medulloblastoma patients younger than three years of age due to their increased risk of disease dissemination outside of the CNS [26-28].
The staging criteria proposed by Chang et al in 1969 categorizes medulloblastomas by degree of metastasis. M0 disease is defined as lacking any evidence of metastatic disease by MRI of brain and spine as well as analysis of cerebrospinal fluid cytology. M1 disease is characterized by positive CSF cytology without gross leptomeningeal tumor deposits visible on imaging of the neuraxis. M2 and M3 disease are both characterized by the presence of gross nodular seeding either within the third or lateral ventricles or the cerebellar/cerebral subarachnoid space (M2) or within the spinal subarachnoid space (M3). Finally, M4 medulloblastomas are those rare cases that present with disease dissemination outside of the central nervous system [11]. The Chang staging system initially further classified medulloblastomas by the size and extent of the primary tumor. This has not shown to have prognostic significance, in contrast to the residual postoperative tumor bulk and the presence or absence of disease dissemination [29].
Histology
The 2007 WHO classification system recognizes several histopathologic variants of medulloblastoma, all of which are categorized as grade IV neoplasms within the broader grouping of embryonal neuroepithelial tumors. In addition to classic medulloblastoma, other variants include desmoplastic/nodular medulloblastoma, medulloblastoma with extensive nodularity (MBEN), anaplastic medulloblastoma, and large cell medulloblastoma [10]. The classic variant is the most frequently encountered and is characterized by both high cellularity and elevated proliferative indices. Classic medulloblastoma cells typically have small to medium-sized, round to oval-shaped hyperchromatic nuclei and minimal cytoplasm. Homer-Wright rosettes are sometimes intermingled and their presence can be associated with high mitotic activity and increased nuclear pleomorphism. Although desmoplasia may occur focally in many medulloblastomas, widespread desmoplasia is characteristic of the desmoplastic/nodular variant, which is also distinguished from the classic variant by the presence of nodular, reticulin-poor “pale islands” of neurocytic differentiation surrounded by densely packed, mitotically active cells. Compared to desmoplastic/nodular medulloblastoma, the related MBEN variant has an expanded lobular architecture with more prominent reticulin-free zones that are more elongated and rich in neutropil-like tissue. Both the desmoplastic/nodular and MBEN variants are associated with an improved prognosis as compared to classic medulloblastomas [12, 30-32]. In contrast, large cell and anaplastic medulloblastomas have a distinctly poor prognosis when compared to the classic variant. Both large cell and anaplastic variants comprise cells with large, round, vesicular nuclei and prominent nucleoli, from which the large cell variant derives its name. Like desmoplastic/nodular and MBEN medulloblastoma variants, large cell and anaplastic medulloblastomas have a significant degree of cytologic overlap and are differentiated only by the degree of anaplasia, characterized by marked nuclear pleomorphism and nuclear molding as well as atypical mitotic forms and abundant apoptotic bodies [33, 34]. Significantly inferior outcomes have been observed in patients with increasing degrees of anaplasia [35, 36]. Two additional histologic patterns have been recognized but are not considered distinct variants. Medullomyoblastomas contain rhabdomyoblastic cells that may be encountered in distinct groups or intermingled with the neuroepithelial cells of one of the aforementioned medulloblastoma variants [37]. The most rarely reported pattern is that of melanotic medulloblastoma, characterized by the accumulation of melanin in the cytoplasm of the neuroepithelial medulloblastoma cells [38].
Molecular Pathology
In 2010, an international group of medulloblastoma experts convened in Boston to come to a consensus on the molecular subgrouping of medulloblastomas. Four distinct subgroups were identified based on transcriptional profiling studies; wingless (Wnt), sonic hedgehog (Shh), Group 3, and Group 4. Each subgroup was characterized by a unique set of demographic and clinical features, genetics, and gene expression [14]. Identification of molecular subgroup more accurately predicts outcome and clinical behavior than does histopathology or clinical staging. It is hypothesized that these different medulloblastoma subgroups arise from distinct cells of origin. This is in part supported by the observation that medulloblastoma maintains its subgroup affiliation at the time of both recurrence and metastasis, a finding that differentiates medulloblastoma from other cancers which demonstrate a change in molecular subclass at recurrence or metastasis [39-41]. These molecular subgroups are distinct from the histologic subtypes, although there are some areas of substantial overlap as demonstrated by an international meta-analysis of data from seven studies with a total of 550 medulloblastoma patients. In this meta-analysis, 97% of 58 Wnt pathway tumors were classic medulloblastoma subtype by histology, and 89% of 44 desmoplastic/nodular medulloblastomas in infants belonged to the Shh molecular subgroup [42].
Interestingly, a recent study by Perreault et al demonstrated that molecular subgroup could be correctly predicted by neuroimaging in 65% of ninety-nine retrospectively reviewed medulloblastoma cases based on tumor location and enhancement pattern. In terms of location, Wnt pathway tumors were most likely to arise in the cerebellopontine angle cistern or cerebellar peduncle, while Shh pathways tumors were found in the cerebellar hemispheres. Group 3 and Group 4 tumors were the primary subgroups encountered in the midline fourth ventricle, and Group 4 tumors were characterized by absent or minimal contrast enhancement [23].
Wingless Pathway Tumors
The Wnt/β-catenin pathway is composed of secreted glycoproteins that act through signal transduction to control various aspects of embryonic development. Unregulated activation of Wnt pathway signaling leads to the accumulation of β-catenin, encoded by the CTNNB1 gene, and results in aberrant upregulation of transcription and subsequent oncogenesis. Wnt pathway tumors are the least common of the four molecular subgroups, representing approximately 10% of sporadic medulloblastomas. They are characterized genetically by the presence of monosomy 6, CTNNB1 mutations, and nuclear β-catenin positivity by immunohistochemistry. Demographically, Wnt subgroup medulloblastomas are more common in children and adults than in infants, and the slight male predominance generally seen in medulloblastomas has not been demonstrated in tumors of this subgroup. Fewer than 10% of Wnt subgroup tumors present with metastatic disease at the time of diagnosis and outcomes for these medulloblastomas are excellent, with 5-year overall survival of 95% in children and 100% in adults. TP53 mutations are found almost exclusively in Wnt and Shh subgroup medulloblastomas. In contrast to Shh subgroup tumors, the presence of mutant TP53 in Wnt subgroup tumors does not impact prognosis [43]. The association between primary CNS tumors and colorectal polyposis, historically known as Turcot syndrome, is well studied in patients with familial adenomatous polyposis (FAP). Patients with FAP have a loss of functional adenomatous polyposis coli (APC) protein as a result of an inactivating germline mutation of the APC gene. The APC protein forms part of a “destruction complex” that serves to degrade β-catenin and its loss results in the accumulation of β-catenin in the cytoplasm and nucleus. This specific disruption in the Wnt signaling pathway leads to an increased risk of medulloblastoma in FAP patients as compared to the general population [44, 45].
Sonic Hedgehog Pathway Tumors
The Shh medulloblastoma subgroup is also associated with a genetic predisposition syndrome characterized by germline mutations in the patched-1 gene (PTCH1) or the suppressor of fused gene (SUFU). Individuals with Gorlin syndrome, also known as nevoid basal cell carcinoma syndrome, suffer from the early onset of multiple nevoid basal cell carcinomas and medulloblastomas as well as developmental abnormalities. Loss of function PTCH1 and SUFU mutations result in truncation of their associated protein products, leading to failure of their tumor suppressor effects and activation of Shh signaling with subsequent tumorigenesis. Germline PTCH1 mutations have been associated with a risk of medulloblastoma of less than 2%, while the risk is substantially higher in those Gorlin syndrome patients with germline SUFU mutations, as illustrated in a small study of nine patients in which in one-third developed childhood medulloblastoma [46]. Germline Shh pathway mutations comprise only a small proportion of medulloblastomas. Somatic mutations in PTCH1 and SUFU are also associated with sporadic medulloblastomas characterized by activation of the Shh pathway, along with PTCH2, SMO, GLI1 and GLI2 mutations. As a whole, Shh subgroup tumors represent approximately 30% of medulloblastomas overall and demonstrate a bimodal distribution, being much more common in patients younger than 3 years and older than 16 years than in children between 3 and 16 years of age. Similar to Wnt pathway medulloblastomas, Shh pathway tumors are equally distributed between both genders. They are more likely to metastasize than Wnt subgroup tumors in infants and children, but less likely than Group 3 or Group 4 tumors to present with disseminated disease. Survival for Shh subgroup medulloblastomas is intermediate between that of Wnt and Group 3 medulloblastomas and varies significantly based on age and histologic subtype. Infants with desmoplastic/nodular tumors have the best prognosis amongst Shh subgroup tumors, with ten-year overall survival of 84%. The increased prevalence of desmoplastic/nodular histology in infants likely accounts for their increased overall survival at ten years of 77% as compared to children and adults, whose ten-year overall survivals are 51 and 34%, respectively. Notably, TP53 mutations are associated with significantly inferior outcomes in Shh subgroup medulloblastomas, in contrast to Wnt pathway tumors, and occur most often in patients between 5 and 18 years of age, generally representing the least frequently encountered age group in Shh subgroup tumors [42, 43].
Non-Wnt/Shh Tumors
The non-Wnt/Shh tumor subgroups comprise Group 3 and Group 4, in which the underlying genetic driver mutations have not yet been established. In contrast to the Wnt and Shh subgroups, both Group 3 and Group 4 are characterized by a male predominance of approximately 2:1 and are associated with a higher frequency of disease dissemination, with metastases detected in approximately 30% of patients at time of diagnosis. Despite these similarities, Group 3 and Group 4 subgroup medulloblastomas have distinctive clinical and genomic features. Group 3 tumors, representing approximately 30% of medulloblastomas, are more likely to have high-level expression and amplification of MYC, occur more often in infants and children, and are associated with the least favorable outcomes across all molecular subgroups, with 10-year overall survival of 39% in infants and 50% in children. Group 4 tumors form the largest molecular subgroup of medulloblastomas, comprising about 35% of medulloblastomas overall. Similar to Shh subgroups tumors, Group 4 tumors have a prognosis that is intermediate between those of Wnt and Group 3 subgroup tumors. The peak incidence for this subgroup is in late childhood and early adolescents. Within Group 4 medulloblastomas, significantly inferior outcomes have been observed in patients with metastatic disease or MYCN amplifications [42].
Treatment – General Principles
As mentioned above, medulloblastoma therapy has evolved to include surgery, radiation therapy and adjuvant chemotherapy. It is recommended that all medulloblastoma patients undergo maximal safe surgical resection. There is not a role for biopsy if the medulloblastoma diagnosis is supported by radiographic evidence. Postoperatively radiated patients in whom a gross total or near total resection is achieved have been shown to have superior overall survival as compared to patients who undergo biopsy alone [47, 48]. Beyond surgical resection, current standards of radiation therapy and medical management vary by extent of disease and age of the patient, based on the risks of recurrence and of neurocognitive effects of radiation therapy, respectively. As it has been demonstrated to significantly improve outcomes, adjuvant chemotherapy is recommended for all patients [6]. Patients who are three years of age or older are stratified as either “average-risk” or “high-risk” based on the volume of postoperative residual tumor and the presence or absence of disseminated disease. Average-risk children are defined as having <1.5 cm2 of tumor present after surgical resection and both negative CSF cytology and no evidence of disease dissemination on MRI of both brain and spine. In contrast, high-risk children are those with ≥1.5 cm2 of residual postoperative tumor and/or disease dissemination. A third group of patients, those younger than three years of age, are treated without upfront radiation therapy due to the unacceptably high risk of severe neurocognitive impairment. Outcomes in the youngest patients are often poor. The Children’s Cancer Group study CCG-9921 reported five-year event-free survival ranging from 25% in infants with metastatic disease at diagnosis to 41% in non-metastatic patients with minimum postoperative residual tumor [49]. In addition to younger age, larger residual tumor volume after surgery, and the presence of metastatic disease at diagnosis, inferior prognosis has also been associated with MYC amplification, anaplastic or large cell histology, and tumors molecularly categorized as either group 3 or sonic hedgehog pathway with TP53 mutations.
Treatment and Prognosis – Average-Risk Patients ≥ 3 Years of Age
In the post-operative setting, average-risk patients were previously treated with 36 Gy craniospinal irradiation (CSI) and a boost to the posterior fossa for a total dose of 54 Gy. The latter is indicated due to the high rate of relapse within the posterior fossa. Studies conducted by the International Society of Pediatric Oncology (SIOP) and the Children’s Oncology Group, among others, have supported a reduction in the CSI dose to 23.4-24 Gy with the addition of adjuvant chemotherapy [50, 51]. ACNS0331, a COG study of further CSI dose-reduction to 18 Gy in young children (aged 3 to 7 years) is currently underway. Similarly, several studies have demonstrated that the posterior fossa radiation volume may be safely reduced through conformal treatment to the tumor bed, allowing for sparing of normal structures, including the temporal lobes, hypothalamus, and cochleae, although this is not a universally accepted practice [51-53]. Similarly, the use of IMRT or proton beam therapy can reduce exposure to the heart and liver during CSI [54-57]. Vincristine is generally given weekly during radiation therapy. Current recommendations for post-radiation chemotherapy in average-risk patients include approximately one year of therapy consisting of eight cycles at six-week intervals of cisplatin, lomustine (CCNU), and vincristine. This regimen, first described by Packer et al in 1988, is associated with a 5-year event-free survival of approximately 80% [58]. A similar regimen substituting cyclophosphamide for lomustine has been associated with statistically equivalent outcomes [27, 59]. The St. Jude Medulloblastoma-96 trial found a similar event-free survival of 83% utilizing an alkylator-based, dose-intensive chemotherapy regimen consisting of four 4-week cycles of cyclophosphamide, cisplatin, and vincristine with autologous stem cell rescue following each cycle [50]. Delays in the initiation of radiation therapy have been associated with inferior outcomes, including those delays related to the use of neoadjuvant chemotherapy prior to irradiation [60-62].
Treatment and Prognosis – High-Risk Patients ≥ 3 Years of Age
The postoperative treatment of high-risk medulloblastoma in children three years of age and older typically involves the administration of “standard dose” RT (36 Gy CSI with a boost to both the posterior fossa and focal sites of metastatic disease to 55.8 Gy) as well as adjuvant chemotherapy, although the ideal chemotherapeutic regimen has not been identified to date. A phase II trial of a regimen utilizing RT with concurrent vincristine followed by maintenance chemotherapy with lomustine, vincristine, and cisplatin showed a progression-free survival of 67%, with the caveat that patients with M1 disease were analyzed together with patients with M2 or M3 disease [63]. A larger subsequent trial from the German Society of Pediatric Hematology and Oncology of an identical regimen found 3-year progression free survival in M2/M3 patients to be only 30%, and the Children’s Cancer Group (CCG) showed a similar outcome with 5-year progression-free survival of 40% using an adjuvant chemotherapy regimen of RT with concurrent vincristine followed by lomustine, prednisone and vincristine [29, 62]. Neoadjuvant chemotherapy, including the CCG’s “8-in-1” regimen (eight drugs in one day, comprising vincristine, methylprednisolone, lomustine, hydroxyurea, procarbazine, cisplatin, cyclophosphamide and cytarabine), the PNET-3 regimen tested by the International Society of Pediatric Oncology/United Kingdom Children’s Cancer Study Group (etoposide, carboplatin, cyclophosphamide, and vincristine), and the German HIT 91 trial’s “sandwich therapy” (consisting of two ifosfamide, etoposide, cisplatin, cytarabine, and high-dose methotrexate cycles prior to RT), has shown similarly poor outcomes for high-risk patients [29, 62, 64]. The current COG protocol, ACNS0332, was designed to address two specific endpoints: whether the addition of carboplatin to vincristine as a radiosensitizer during RT or concurrent retinoid therapy during maintenance chemotherapy and for six months thereafter results in improved survival. The retinoid-containing arms of this trial were recently closed after futility analysis suggested that isotretinoin therapy will not lead to a significant event-free survival advantage. The adjuvant chemotherapy used in this protocol consists of six four-week cycles of cisplatin, cyclophosphamide, and vincristine. A previous COG phase I/II trial of post-RT vincristine and cyclophosphamide with or without cisplatin and with the addition of carboplatin during RT showed 5-year overall and progression-free survival of 78% and 71%, respectively [65]. Comparable outcomes have been achieved by groups utilizing high-dose chemotherapy with autologous stem cell rescue. The St. Jude Medulloblastoma-96 trial demonstrated a 5-year overall and progression free survival of 70% in high-risk patients with the same cyclophosphamide-based, dose-intensive chemotherapy regimen utilized in average-risk patients as described above, with a percentage of patients also receiving pre-radiotherapy topotecan. There were no treatment-related mortalities observed [50]. Researchers from the Samsung Medical Center in Seoul investigated the use of high-dose chemotherapy with autologous stem cell rescue with reduced-dose radiotherapy in twenty patients, finding a 5-year event-free survival of 70% using a regimen consisting of induction chemotherapy for two cycles followed by RT and four additional cycles of induction chemotherapy prior to tandem autologous transplant. Induction chemotherapy consisted of cisplatin, etoposide, vincristine, and either cyclophosphamide or ifosfamide. CSI was initially given at a dose of 23.4 Gy but this was increased during the study period to 30.6 Gy in patients older than six years of age due to spinal cord relapse or progression in three of nine patients with metastatic disease. Focal boosts were given to the sites of primary and nodular metastatic disease at total doses of 54 Gy and 45 Gy, respectively. The first tandem transplant utilized a regimen of carboplatin, thiotepa, and etoposide, while the second comprised cyclophosphamide and melphalan. Treatment-related mortality in this study was 10% [66]. Another group has attempted to modulate radiotherapy-induced side effects through the use of hyperfractionated accelerated radiotherapy (HART). Thirty-three patients in their study received two months of postoperative induction chemotherapy with etoposide, methotrexate, cyclophosphamide, and carboplatin. HART was subsequently administered with a CSI dose of 31.2 Gy and a posterior fossa boost to 59.7 Gy in patients younger than ten years of age, and a CSI dose of 39 Gy with a posterior fossa boost to 60 Gy in those older than ten years. An additional 9 Gy was given to sites of nodular metastatic disease. Patients in complete remission (CR) after HART received maintenance chemotherapy for one year with vincristine and lomustine, while those without CR received tandem high-dose thiotepa-based autologous transplant. Five-year event-free survival rates in this study were 70% [67].
Treatment and Prognosis – Infants and Children < 3 Years of Age
The neurocognitive outcomes in children younger than three years of age who receive craniospinal irradiation are very poor. For this reason, investigations have focused on delaying or omitting radiotherapy in this population. There is evidence that regimens consisting of surgery and chemotherapy without RT can be successful in specific subsets of medulloblastoma patients, namely M0 patients in whom a gross total surgical resection has been achieved and in patients with either desmoplastic/nodular medulloblastoma or medulloblastoma with extensive nodularity (MBEN) histological subtypes. The former was investigated in medulloblastoma patients younger than five years of age without metastases or postoperative residual disease who were enrolled in a French Society of Paediatric Oncology (SFOP) trial. These patients had five-year progression-free survival of 41% as compared to 0% in those in whom only a subtotal resection was performed utilizing postoperative multiagent chemotherapy (carboplatin, vincristine, procarbazine, etoposide, and cisplatin) without radiotherapy [68]. Two chemotherapy regimens (vincristine and etoposide with either cisplatin and cyclophosphamide or carboplatin and ifosfamide) were investigated in a CCG trial that included 92 medulloblastoma patients younger than 36 months of age. Five-year event-free and overall survival for these patients were 32% and 43% with some suggestion of improved early tumor control, but not survival benefit, with the cisplatin and cyclophosphamide-containing regimen. Radiotherapy was withheld until disease progression and approximately one-quarter of M0 patients in whom a gross total resection was achieved were alive and without evidence of disease progression at the five-year mark without the use of RT [49]. Improved survival was demonstrated in several trials with the use of systemic and intraventricular methotrexate in addition to a postoperative systemic chemotherapy regimen of cyclophosphamide, carboplatin, etoposide, and vincristine. Progression-free and overall survival with this regimen in forty-eight patients younger than four years of age without disseminated disease were 57% and 80%, respectively [69, 70]. Another multi-institutional trial utilizing the same agents resulted in progression-free and overall survival of 58% and 66% in forty-three medulloblastoma patients age three years or younger with or without disseminated disease. Again, in this trial, the benefit of localized disease and gross total resection was appreciated, with five-year progression-free survival and overall survival rates up to 82% and 92%, respectively, in patients without postoperative residual tumor or evidence of metastatic disease. Unfortunately, the use intraventricular methotrexate was shown in this study to be associated with significantly lower age-matched IQ scores, although they remained significantly superior to those of children treated with radiotherapy and systemic chemotherapy [71]. The use of high-dose chemotherapy with autologous stem cell rescue, investigated in the multi-institutional “Head Start” I and II trials, demonstrated five-year progression-free and overall survival rates of 52% and 70% in twenty-one M0 patients. There was no significant impact on intellectual functioning in the 71% of surviving patients who were not treated with radiotherapy. Of note, there were four treatment-related deaths in these two trials [72]. The current COG trial, ACNS0334, is designed to investigate whether the addition of systemic methotrexate to a postoperative induction regimen of etoposide, cyclophosphamide, cisplatin, and vincristine followed by high-dose thiotepa and carboplatin with autologous stem cell rescue will improve survival in medulloblastoma patients younger than three years old with disseminated disease.
Several studies have demonstrated the prognostic implications of tumor histology in this age group. The HIT-SKK’92 trial showed five-year progression-free and overall survival of 85% and 95% in patients with desmoplastic/nodular histology [69]. A confirmatory trial performed by the same organization showed five-year progression-free and overall survival of 90% and 100% in desmoplastic/nodular and MBEN patients with M0 disease. Outcomes were significantly inferior in patients with other histologic variants [70].
Treatment and Prognosis – Patients with Relapsed Disease
Outcomes in patients with relapsed disease are generally poor, with reported five-year survival rates of approximately 25% [73, 74]. Multiple treatment strategies have been investigated, including repeat surgical resection, reirradiation, stereotactic radiosurgery, high-dose chemotherapy with autologous stem cell rescue, low-dose oral etoposide, the use of biologically targeted agents, or some combination of the above modalities. Treatment of patients with localized recurrence is more successful in general than treatment of patients with disseminated recurrence [74-84]. The current COG trial ACNS0821 for patients with recurrent medulloblastoma compares temozolomide and irinotecan therapy to the same regimen with concurrent bevacizumab.
In young children whose initial treatment did not include radiotherapy, craniospinal irradiation as part of multimodality salvage therapy has been demonstrated to be successful in small numbers of patients [82-85].
Complications of Treatment
Posterior fossa syndrome, also known as cerebellar mutism syndrome, is a well-known complication of surgical resection in medulloblastoma and other posterior fossa surgical interventions. It occurs in approximately one-quarter of medulloblastoma patients who undergo surgical resection and symptoms begin to manifest within one to two days of posterior fossa surgery. The syndrome is characterized by progressively diminished speech leading to full mutism, ataxia, emotional lability, and axial hypotonia. It appears to be associated with more aggressive surgical resections and brainstem infiltration by tumor [86-89]. Midline location of tumor and younger ago have also shown some association with the development of posterior fossa syndrome and there is a suggestion that left-handed children may be at higher risk [88, 90]. Recovery is incomplete in a significant number of affected patients, with persistence of any combination of neurocognitive deficits, speech/language impairment, and ataxia at one year following diagnosis [89]. More severely affected patients are more likely to have persistent deficits. In spite of more than two decades of research, the precise etiology of cerebellar mutism syndrome remains unclear. Relationships to postoperative edema, vasospasm-induced ischemia, or disruption of the dentatothalamocortical tracts have been proposed, and studies investigating the exact mechanism of the development of posterior fossa syndrome are ongoing [90-93].
The long-term sequelae of radiation therapy and chemotherapy are well described. Endocrinopathies as well as neurocognitive and neurosensory impairment can occur as a result of craniospinal irradiation, with their frequency and severity modulated by patient age and radiation dose [94, 95]. Cerebrovascular disease, including late-occurring stroke, steno-occlusive disease, vascular malformations, and stroke-like migraine variants, is also known to occur with increased frequency in cancer survivors with a history of cranial irradiation. Data from the Childhood Cancer Survivor Study demonstrated that the rate of late-occurring stroke is elevated in all brain tumor survivors irrespective of radiation exposure (267.6 per 100,000 person-years); however, brain tumor survivors who had been treated with cranial radiation had a significantly higher rate of stroke (339.5 per 100,000 person-years). The risk of stroke increases in a radiation dose-dependent fashion as well as with age, with an incidence of 1.3% at 10 years from diagnosis growing to 14.2% at 30 years from diagnosis in survivors of CNS tumors who were treated with ≥50 Gy of cranial radiation [96, 97]. Additionally, there is an increased risk of second malignancies in the radiation field. Chemotherapy drugs, particularly alkylating agents, are also associated with the development of treatment-related cancers. A 2013 review of 379 patients with non-disseminated medulloblastoma who were enrolled on the COG A9961 trial showed a cumulative incidence of secondary malignancies of 4.2% at ten years, somewhat higher than the estimated risk of 1-2% [98].
Vincristine is known to cause various types of neurotoxicity, including autonomic neuropathy manifesting as constipation, isolated cranial nerve involvement (most commonly the oculomotor nerve), distal paresthesias, and loss of deep tendon reflexes. These symptoms manifest with increased severity in patients with pre-existing neurologic diseases, the best described of which being Charcot-Marie-Tooth [99]. Neuropathies caused by vincristine are generally not permanent, with the exception of loss of ankle reflexes in some patients. The time to full recovery may be protracted and symptoms may continue to progress for a number of months before resolving, although children generally recover more quickly than adults [100]. Similarly, the use of cisplatin is also associated with neurotoxicity, specifically peripheral neuropathy and ototoxicity. Cisplatin-associated peripheral neuropathies are generally seen after a total cumulative dose of 300mg/m2 and may manifest with paresthesias that begin distally and spread proximally along with loss of ankle deep tendon reflexes. Autonomic symptoms are uncommon. As with vincristine-associated neuropathies, symptoms may persist or even worsen over the course of several months. Although most patients with cisplatin-related peripheral neuropathy improve, their recovery may not be complete [101]. The ototoxicity associated with cisplatin use is characterized by high-frequency sensorineural hearing loss. The risk for ototoxicity, which is generally irreversible, is highest in children younger than five years of age and is related to the cumulative dose. The impact of hearing loss on academic performance and social-emotional interactions can be substantial [102, 103].
Post-therapy quality of life and psychosocial outcomes are increasingly recognized as vital factors in decision-making regarding therapy. Historically, less emphasis was placed on these long-term outcomes than on survival. However, as survival has improved over the past decades, and will hopefully continue to improve with the development of molecularly targeted agents for medulloblastoma, the discussion regarding quality of life will become increasingly important both in the context of decision-making regarding therapy and in the design of future cooperative protocols that may consider quality of life to be a second primary endpoint along with survival [104].
Surveillance
In cases of disease recurrence, relapses encountered on surveillance imaging are associated with improved survival as compared to those detected through the emergence of clinical symptoms [105]. Although there remains a lack of consensus on the ideal frequency of surveillance evaluations, clinical and radiographic follow-up is generally recommended at three-month intervals during the first year following completion of planned therapy, at three to four-month intervals in the second year, every six months during the third year, and annually thereafter. Patterns of disease recurrence were evaluated by Perreault et al in a series that included twenty-six patients with relapsed medulloblastoma. The incidences of local, distant, and combined relapses were relatively evenly distributed [106]. It is generally agreed upon that surveillance imaging of the brain is indicated in all patients and that imaging should include full spinal MRI in patients with a history of disseminated disease at diagnosis. Isolated spinal relapse was shown to be less frequent than brain or combined brain and spine relapse in another retrospective review by Perreault et al that included eighty-nine medulloblastoma patients. Five patients in this series had an isolated spinal recurrence detected on screening MRI, two of whom did not have radiographic evidence of spinal disease at the time of diagnosis. Spinal recurrence was detected within three years in all five cases. As one patient with isolated spinal relapse in their series was treated with low-dose CSI, the authors suggested that patients treated with reduced-dose radiotherapy may benefit from closer spinal radiographic monitoring [107]. The utility of surveillance imaging of the spine has been a matter of debate. Investigators from the Hospital for Sick Children in Toronto reviewed paired brain and spine MRIs from twenty-four patients with relapse and found no isolated spinal recurrences [108]. However, in a series of sixteen relapsed patients from Massachusetts General Hospital who were treated with proton beam radiotherapy, isolated spinal recurrence was the most frequently encountered pattern of failure. Four of the six patients with relapsed spine-only disease did not have evidence of metastatic disease at the time of diagnosis [109]. There remains a lack of consensus regarding the utility and recommended frequency of spinal surveillance imaging.
Similarly, surveillance lumbar puncture is generally indicated if CSF cytology was positive at the time of diagnosis, although the frequency varies by institution and protocol. Endocrine screening and neuropsychological testing are also recommended for patients who were treated with craniospinal irradiation.
Conclusions/Future Directions
Despite marked improvements in overall survival for medulloblastoma patients over the past decades, considerable work remains to be done in order to improve survival within specific patient subgroups as well as to attenuate treatment-related morbidities and improve quality of life for survivors. There has been a recent striking increase in our understanding of the biology of medulloblastoma. Molecular subgrouping has allowed investigators to further characterize medulloblastomas in ways that have both prognostic and therapeutic significance. As increased attention is given to post-therapy quality of life and the risk of long-term treatment-related side effects, ways in which treatment intensity can be appropriately reduced as well as novel, less toxic and more targeted agents are being sought. For example, a phase I study of vismodegib, an inhibitor of the Sonic Hedgehog pathway at the level of the smoothened (SMO) protein, demonstrated that this SMO inhibitor was well tolerated in pediatric patient with recurrent or treatment-refractory medulloblastomas [110]. Incorporating this type of targeted therapy into multimodal treatment planning may allow further reduction of radiation dose and cytotoxic chemotherapy with subsequent mitigation of the negative sequelae of treatment and improved quality of life for medulloblastoma survivors. Although our understanding of the biology of medulloblastomas has evolved considerably in recent years, these advances in molecular pathology have yet to be incorporated into the treatment of medulloblastoma.
Figure 1.
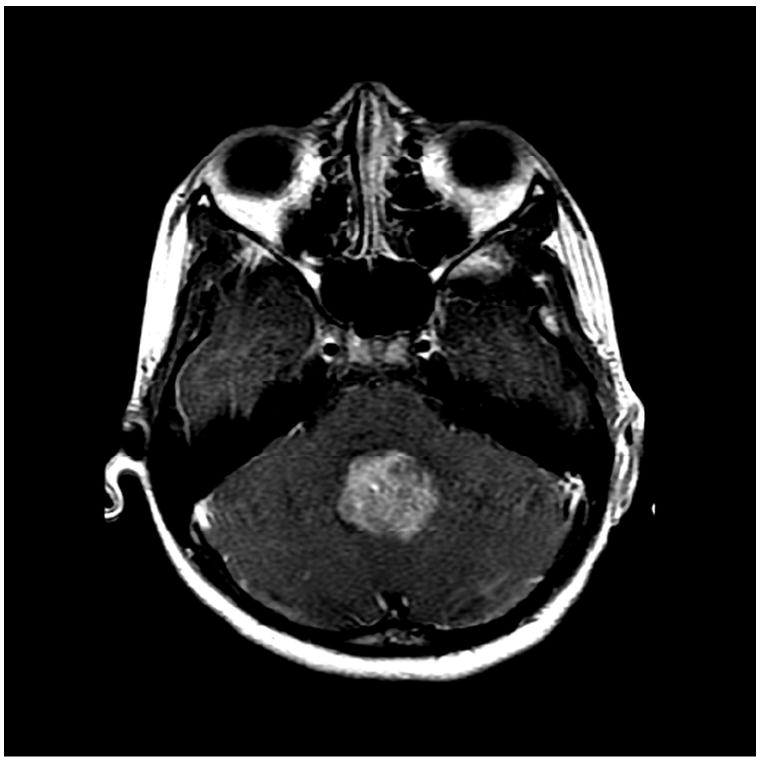
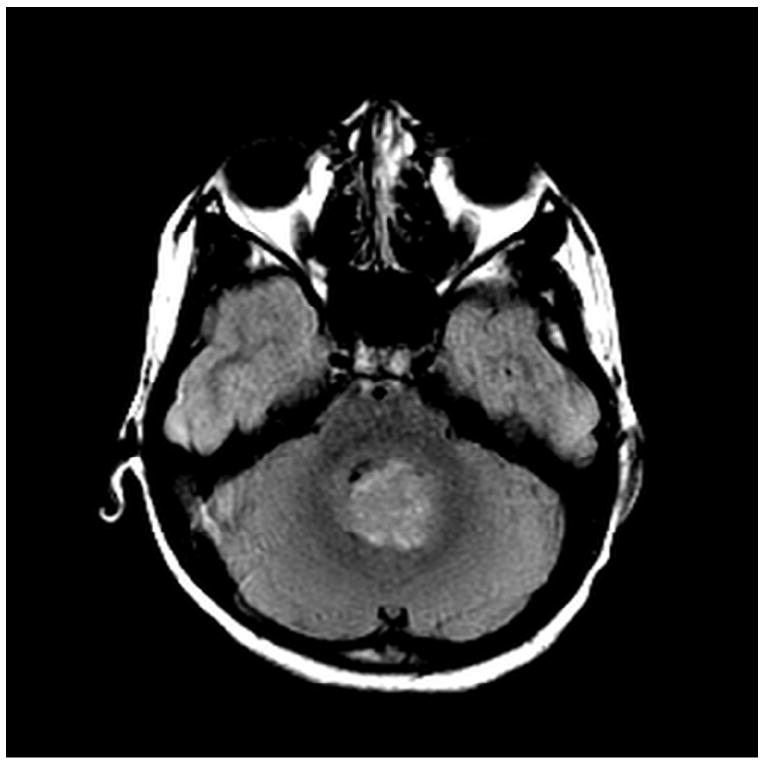
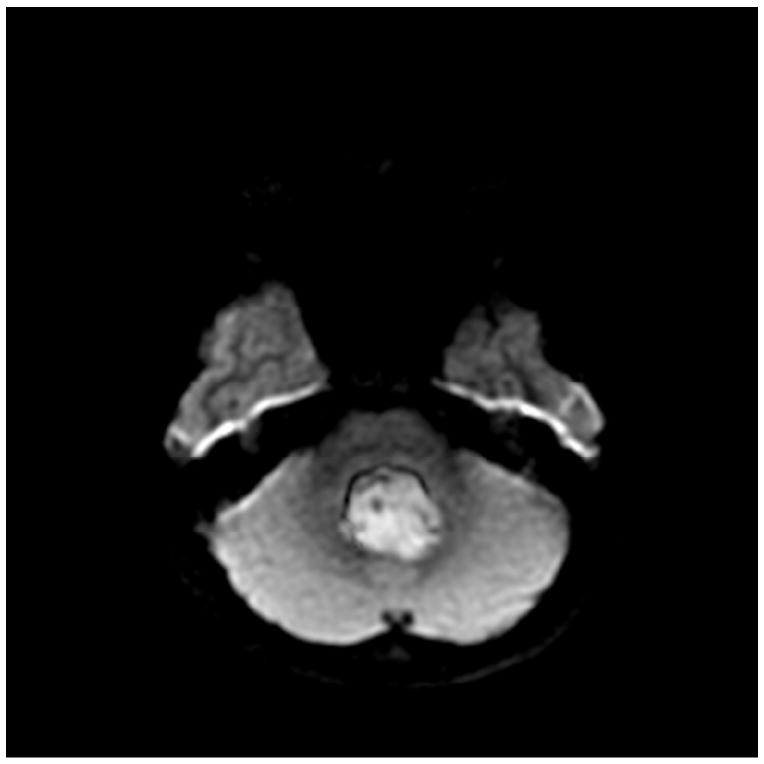
A) MRI T1 + Gadoliniun
B) MRI FLAIR
C) MRI DWI
Figure 2.
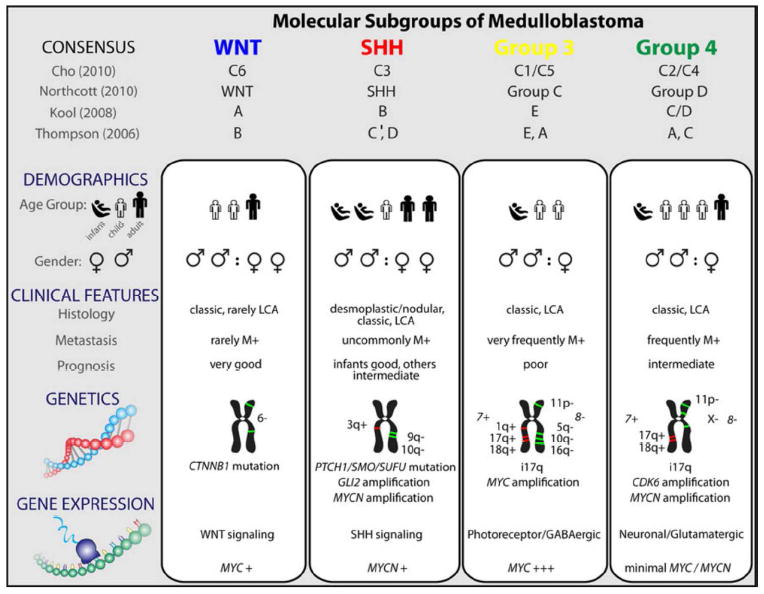
Comparison of the various subgroups of medulloblastoma including their affiliations with previously published papers on medulloblastoma molecular subgrouping
References
- 1.Bailey P, Cushing H. Medulloblastoma cerebelli: a common type of midcerebellar glioma of childhood. Arch Neurol Psychiatry. 1925;14(2):192–224. [Google Scholar]
- 2.Kunschner LJ. Harvey Cushing and medulloblastoma. Arch Neurol. 2002;59(4):642–5. doi: 10.1001/archneur.59.4.642. [DOI] [PubMed] [Google Scholar]
- 3.Globus JH, Strauss I. Spongioblastoma multiforme: a primary malignant form of brain neoplasm: its clinical and anatomic features. Arch Neurol Psychiatry. 1925;14(2):139–191. [Google Scholar]
- 4.Paterson E, Farr RF. Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta radiol. 1953;39(4):323–36. doi: 10.3109/00016925309136718. [DOI] [PubMed] [Google Scholar]
- 5.Rutka JT, Hoffman HJ. Medulloblastoma: a historical perspective and overview. J Neurooncol. 1996;29(1):1–7. doi: 10.1007/BF00165513. [DOI] [PubMed] [Google Scholar]
- 6.Tait DM, et al. Adjuvant chemotherapy for medulloblastoma: the first multicentre control trial of the International Society of Paediatric Oncology (SIOP I) Eur J Cancer. 1990;26(4):464–9. [PubMed] [Google Scholar]
- 7.Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983;42(1):1–15. [PubMed] [Google Scholar]
- 8.Pomeroy SL, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415(6870):436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 9.Spence T, et al. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol. 2014;128(2):291–303. doi: 10.1007/s00401-014-1291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis DN, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang CH, Housepian EM, Herbert C., Jr An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969;93(6):1351–9. doi: 10.1148/93.6.1351. [DOI] [PubMed] [Google Scholar]
- 12.Rutkowski S, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol. 2010;28(33):4961–8. doi: 10.1200/JCO.2010.30.2299. [DOI] [PubMed] [Google Scholar]
- 13.Shih DJ, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32(9):886–96. doi: 10.1200/JCO.2013.50.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor MD, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–72. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17(9):503–11. doi: 10.1007/s003810100496. [DOI] [PubMed] [Google Scholar]
- 16.McNeil DE, et al. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol. 2002;39(3):190–4. doi: 10.1002/mpo.10121. [DOI] [PubMed] [Google Scholar]
- 17.Ostrom QT, et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007-2011. Neuro Oncol. 2015;16(Suppl 10):x1–x36. doi: 10.1093/neuonc/nou327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostrom QT, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smoll NR, Drummond KJ. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci. 2012;19(11):1541–4. doi: 10.1016/j.jocn.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Eran A, et al. Medulloblastoma: atypical CT and MRI findings in children. Pediatr Radiol. 2010;40(7):1254–62. doi: 10.1007/s00247-009-1429-9. [DOI] [PubMed] [Google Scholar]
- 21.Koeller KK, Rushing EJ. From the archives of the AFIP: medulloblastoma: a comprehensive review with radiologic-pathologic correlation. Radiographics. 2003;23(6):1613–37. doi: 10.1148/rg.236035168. [DOI] [PubMed] [Google Scholar]
- 22.Poretti A, Meoded A, Huisman TA. Neuroimaging of pediatric posterior fossa tumors including review of the literature. J Magn Reson Imaging. 2012;35(1):32–47. doi: 10.1002/jmri.22722. [DOI] [PubMed] [Google Scholar]
- 23.Perreault S, et al. MRI surrogates for molecular subgroups of medulloblastoma. AJNR Am J Neuroradiol. 2014;35(7):1263–9. doi: 10.3174/ajnr.A3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider C, et al. Clinical implications of medulloblastoma subgroups: incidence of CSF diversion surgery. J Neurosurg Pediatr. 2015;15(3):236–42. doi: 10.3171/2014.9.PEDS14280. [DOI] [PubMed] [Google Scholar]
- 25.Gajjar A, et al. Comparison of lumbar and shunt cerebrospinal fluid specimens for cytologic detection of leptomeningeal disease in pediatric patients with brain tumors. J Clin Oncol. 1999;17(6):1825–8. doi: 10.1200/JCO.1999.17.6.1825. [DOI] [PubMed] [Google Scholar]
- 26.Packer RJ, et al. Management of children with primitive neuroectodermal tumors of the posterior fossa/medulloblastoma. Pediatr Neurosci. 1985;12(4-5):272–82. doi: 10.1159/000120264. [DOI] [PubMed] [Google Scholar]
- 27.Packer RJ, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24(25):4202–8. doi: 10.1200/JCO.2006.06.4980. [DOI] [PubMed] [Google Scholar]
- 28.Fouladi M, et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol. 1999;17(10):3234–7. doi: 10.1200/JCO.1999.17.10.3234. [DOI] [PubMed] [Google Scholar]
- 29.Zeltzer PM, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17(3):832–45. doi: 10.1200/JCO.1999.17.3.832. [DOI] [PubMed] [Google Scholar]
- 30.Sure U, et al. Staging, scoring and grading of medulloblastoma. A postoperative prognosis predicting system based on the cases of a single institute. Acta Neurochir (Wien) 1995;132(1-3):59–65. doi: 10.1007/BF01404849. [DOI] [PubMed] [Google Scholar]
- 31.Giangaspero F, et al. Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg. 1999;91(6):971–7. doi: 10.3171/jns.1999.91.6.0971. [DOI] [PubMed] [Google Scholar]
- 32.McLendon RE, et al. Diagnostic markers in paediatric medulloblastoma: a Paediatric Oncology Group Study. Histopathology. 1999;34(2):154–62. doi: 10.1046/j.1365-2559.1999.00577.x. [DOI] [PubMed] [Google Scholar]
- 33.Eberhart CG, et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94(2):552–60. doi: 10.1002/cncr.10189. [DOI] [PubMed] [Google Scholar]
- 34.McManamy CS, et al. Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol. 2003;62(6):627–32. doi: 10.1093/jnen/62.6.627. [DOI] [PubMed] [Google Scholar]
- 35.Giangaspero F, et al. Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol. 2006;112(1):5–12. doi: 10.1007/s00401-006-0064-x. [DOI] [PubMed] [Google Scholar]
- 36.Brown HG, et al. “Large cell/anaplastic” medulloblastomas: a Pediatric Oncology Group Study. J Neuropathol Exp Neurol. 2000;59(10):857–65. doi: 10.1093/jnen/59.10.857. [DOI] [PubMed] [Google Scholar]
- 37.Wright KD, et al. Isochromosome 17q, MYC amplification and large cell/anaplastic phenotype in a case of medullomyoblastoma with extracranial metastases. Pediatr Blood Cancer. 2012;59(3):561–4. doi: 10.1002/pbc.24002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li WY, et al. Melanotic medulloblastoma in children. Childs Nerv Syst. 2011;27(4):517–21. doi: 10.1007/s00381-011-1389-x. [DOI] [PubMed] [Google Scholar]
- 39.Ramaswamy V, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14(12):1200–7. doi: 10.1016/S1470-2045(13)70449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015;129(3):449–57. doi: 10.1007/s00401-015-1389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson P, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–9. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kool M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–84. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhukova N, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31(23):2927–35. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Attard TM, et al. Brain tumors in individuals with familial adenomatous polyposis: a cancer registry experience and pooled case report analysis. Cancer. 2007;109(4):761–6. doi: 10.1002/cncr.22475. [DOI] [PubMed] [Google Scholar]
- 45.Hamilton SR, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332(13):839–47. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- 46.Smith MJ, et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol. 2014;32(36):4155–61. doi: 10.1200/JCO.2014.58.2569. [DOI] [PubMed] [Google Scholar]
- 47.Hughes EN, et al. Medulloblastoma at the joint center for radiation therapy between 1968 and 1984. The influence of radiation dose on the patterns of failure and survival. Cancer. 1988;61(10):1992–8. doi: 10.1002/1097-0142(19880515)61:10<1992::aid-cncr2820611011>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 48.del Charco JO, et al. Medulloblastoma: time-dose relationship based on a 30-year review. Int J Radiat Oncol Biol Phys. 1998;42(1):147–54. doi: 10.1016/s0360-3016(98)00197-7. [DOI] [PubMed] [Google Scholar]
- 49.Geyer JR, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol. 2005;23(30):7621–31. doi: 10.1200/JCO.2005.09.095. [DOI] [PubMed] [Google Scholar]
- 50.Gajjar A, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–20. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 51.Merchant TE, et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70(3):782–7. doi: 10.1016/j.ijrobp.2007.07.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukunaga-Johnson N, et al. Patterns of failure following treatment for medulloblastoma: is it necessary to treat the entire posterior fossa? Int J Radiat Oncol Biol Phys. 1998;42(1):143–6. doi: 10.1016/s0360-3016(98)00178-3. [DOI] [PubMed] [Google Scholar]
- 53.Wolden SL, et al. Patterns of failure using a conformal radiation therapy tumor bed boost for medulloblastoma. J Clin Oncol. 2003;21(16):3079–83. doi: 10.1200/JCO.2003.11.140. [DOI] [PubMed] [Google Scholar]
- 54.St Clair WH, et al. Advantage of protons compared to conventional X-ray or IMRT in the treatment of a pediatric patient with medulloblastoma. Int J Radiat Oncol Biol Phys. 2004;58(3):727–34. doi: 10.1016/S0360-3016(03)01574-8. [DOI] [PubMed] [Google Scholar]
- 55.Yuh GE, et al. Reducing toxicity from craniospinal irradiation: using proton beams to treat medulloblastoma in young children. Cancer J. 2004;10(6):386–90. doi: 10.1097/00130404-200411000-00009. [DOI] [PubMed] [Google Scholar]
- 56.Pai Panandiker A, et al. Craniospinal irradiation with spinal IMRT to improve target homogeneity. Int J Radiat Oncol Biol Phys. 2007;68(5):1402–9. doi: 10.1016/j.ijrobp.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parker W, et al. Intensity-modulated radiotherapy for craniospinal irradiation: target volume considerations, dose constraints, and competing risks. Int J Radiat Oncol Biol Phys. 2007;69(1):251–7. doi: 10.1016/j.ijrobp.2007.04.052. [DOI] [PubMed] [Google Scholar]
- 58.Packer RJ, et al. Efficacy of adjuvant chemotherapy for patients with poor-risk medulloblastoma: a preliminary report. Ann Neurol. 1988;24(4):503–8. doi: 10.1002/ana.410240405. [DOI] [PubMed] [Google Scholar]
- 59.Packer RJ, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children’s Cancer Group Study. J Clin Oncol. 1999;17(7):2127–36. doi: 10.1200/JCO.1999.17.7.2127. [DOI] [PubMed] [Google Scholar]
- 60.Taylor RE, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21(8):1581–91. doi: 10.1200/JCO.2003.05.116. [DOI] [PubMed] [Google Scholar]
- 61.Oyharcabal-Bourden V, et al. Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol. 2005;23(21):4726–34. doi: 10.1200/JCO.2005.00.760. [DOI] [PubMed] [Google Scholar]
- 62.Kortmann RD, et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ’91. Int J Radiat Oncol Biol Phys. 2000;46(2):269–79. doi: 10.1016/s0360-3016(99)00369-7. [DOI] [PubMed] [Google Scholar]
- 63.Packer RJ, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81(5):690–8. doi: 10.3171/jns.1994.81.5.0690. [DOI] [PubMed] [Google Scholar]
- 64.Taylor RE, et al. Outcome for patients with metastatic (M2-3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur J Cancer. 2005;41(5):727–34. doi: 10.1016/j.ejca.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 65.Jakacki RI, et al. Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children’s Oncology Group Phase I/II study. J Clin Oncol. 2012;30(21):2648–53. doi: 10.1200/JCO.2011.40.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sung KW, et al. Reduced-dose craniospinal radiotherapy followed by tandem high-dose chemotherapy and autologous stem cell transplantation in patients with high-risk medulloblastoma. Neuro Oncol. 2013;15(3):352–9. doi: 10.1093/neuonc/nos304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gandola L, et al. Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J Clin Oncol. 2009;27(4):566–71. doi: 10.1200/JCO.2008.18.4176. [DOI] [PubMed] [Google Scholar]
- 68.Grill J, et al. Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol. 2005;6(8):573–80. doi: 10.1016/S1470-2045(05)70252-7. [DOI] [PubMed] [Google Scholar]
- 69.Rutkowski S, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol. 2009;11(2):201–10. doi: 10.1215/15228517-2008-084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.von Bueren AO, et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol. 2011;13(6):669–79. doi: 10.1093/neuonc/nor025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rutkowski S, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978–86. doi: 10.1056/NEJMoa042176. [DOI] [PubMed] [Google Scholar]
- 72.Dhall G, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50(6):1169–75. doi: 10.1002/pbc.21525. [DOI] [PubMed] [Google Scholar]
- 73.Bowers DC, et al. Impact of site of tumor recurrence upon survival for children with recurrent or progressive medulloblastoma. J Neurosurg. 2007;107(1 Suppl):5–10. doi: 10.3171/PED-07/07/005. [DOI] [PubMed] [Google Scholar]
- 74.Dunkel IJ, et al. High-dose carboplatin, thiotepa, and etoposide with autologous stem cell rescue for patients with previously irradiated recurrent medulloblastoma. Neuro Oncol. 2010;12(3):297–303. doi: 10.1093/neuonc/nop031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kadota RP, et al. Dose intensive melphalan and cyclophosphamide with autologous hematopoietic stem cells for recurrent medulloblastoma or germinoma. Pediatr Blood Cancer. 2008;51(5):675–8. doi: 10.1002/pbc.21655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Milker-Zabel S, et al. Results of three-dimensional stereotactically-guided radiotherapy in recurrent medulloblastoma. J Neurooncol. 2002;60(3):227–33. doi: 10.1023/a:1021184400053. [DOI] [PubMed] [Google Scholar]
- 77.Wetmore C, et al. Reirradiation of recurrent medulloblastoma: does clinical benefit outweigh risk for toxicity? Cancer. 2014;120(23):3731–7. doi: 10.1002/cncr.28907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bakst RL, et al. Reirradiation for recurrent medulloblastoma. Cancer. 2011;117(21):4977–82. doi: 10.1002/cncr.26148. [DOI] [PubMed] [Google Scholar]
- 79.Yamada A, et al. Proposed strategy for the use of high-dose chemotherapy with stem cell rescue and intrathecal topotecan without whole-brain irradiation for infantile classic medulloblastoma. Pediatr Blood Cancer. 2014;61(12):2316–8. doi: 10.1002/pbc.25174. [DOI] [PubMed] [Google Scholar]
- 80.Aguilera D, et al. Response to bevacizumab, irinotecan, and temozolomide in children with relapsed medulloblastoma: a multi-institutional experience. Childs Nerv Syst. 2013;29(4):589–96. doi: 10.1007/s00381-012-2013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashley DM, et al. Response of recurrent medulloblastoma to low-dose oral etoposide. J Clin Oncol. 1996;14(6):1922–7. doi: 10.1200/JCO.1996.14.6.1922. [DOI] [PubMed] [Google Scholar]
- 82.Gururangan S, et al. Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro Oncol. 2008;10(5):745–51. doi: 10.1215/15228517-2008-044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dunkel IJ, et al. High-dose carboplatin, thiotepa, and etoposide with autologous stem-cell rescue for patients with recurrent medulloblastoma. Children’s Cancer Group. J Clin Oncol. 1998;16(1):222–8. doi: 10.1200/JCO.1998.16.1.222. [DOI] [PubMed] [Google Scholar]
- 84.Gururangan S, et al. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. J Clin Oncol. 1998;16(7):2486–93. doi: 10.1200/JCO.1998.16.7.2486. [DOI] [PubMed] [Google Scholar]
- 85.Muller K, et al. Postponed is not canceled: role of craniospinal radiation therapy in the management of recurrent infant medulloblastoma--an experience from the HIT-REZ 1997 & 2005 studies. Int J Radiat Oncol Biol Phys. 2014;88(5):1019–24. doi: 10.1016/j.ijrobp.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 86.Cochrane DD, et al. The surgical and natural morbidity of aggressive resection for posterior fossa tumors in childhood. Pediatr Neurosurg. 1994;20(1):19–29. doi: 10.1159/000120761. [DOI] [PubMed] [Google Scholar]
- 87.Doxey D, et al. Posterior fossa syndrome: identifiable risk factors and irreversible complications. Pediatr Neurosurg. 1999;31(3):131–6. doi: 10.1159/000028848. [DOI] [PubMed] [Google Scholar]
- 88.Korah MP, et al. Incidence, risks, and sequelae of posterior fossa syndrome in pediatric medulloblastoma. Int J Radiat Oncol Biol Phys. 2010;77(1):106–12. doi: 10.1016/j.ijrobp.2009.04.058. [DOI] [PubMed] [Google Scholar]
- 89.Robertson PL, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105(6 Suppl):444–51. doi: 10.3171/ped.2006.105.6.444. [DOI] [PubMed] [Google Scholar]
- 90.Law N, et al. Clinical and neuroanatomical predictors of cerebellar mutism syndrome. Neuro Oncol. 2012;14(10):1294–303. doi: 10.1093/neuonc/nos160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morris EB, et al. Proximal dentatothalamocortical tract involvement in posterior fossa syndrome. Brain. 2009;132(Pt 11):3087–95. doi: 10.1093/brain/awp241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wisoff JH, Epstein FJ. Pseudobulbar palsy after posterior fossa operation in children. Neurosurgery. 1984;15(5):707–9. doi: 10.1227/00006123-198411000-00014. [DOI] [PubMed] [Google Scholar]
- 93.Nagatani K, Waga S, Nakagawa Y. Mutism after removal of a vermian medulloblastoma: cerebellar mutism. Surg Neurol. 1991;36(4):307–9. doi: 10.1016/0090-3019(91)90094-p. [DOI] [PubMed] [Google Scholar]
- 94.Gurney JG, et al. Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors: Childhood Cancer Survivor Study. Cancer. 2003;97(3):663–73. doi: 10.1002/cncr.11095. [DOI] [PubMed] [Google Scholar]
- 95.Packer RJ, et al. Long-term neurologic and neurosensory sequelae in adult survivors of a childhood brain tumor: childhood cancer survivor study. J Clin Oncol. 2003;21(17):3255–61. doi: 10.1200/JCO.2003.01.202. [DOI] [PubMed] [Google Scholar]
- 96.Mueller S, et al. Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: a report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys. 2013;86(4):649–55. doi: 10.1016/j.ijrobp.2013.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morris B, et al. Cerebrovascular disease in childhood cancer survivors: A Children’s Oncology Group Report. Neurology. 2009;73(22):1906–13. doi: 10.1212/WNL.0b013e3181c17ea8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Packer RJ, et al. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children’s Oncology Group trial A9961. Neuro Oncol. 2013;15(1):97–103. doi: 10.1093/neuonc/nos267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trobaugh-Lotrario AD, Smith AA, Odom LF. Vincristine neurotoxicity in the presence of hereditary neuropathy. Med Pediatr Oncol. 2003;40(1):39–43. doi: 10.1002/mpo.10105. [DOI] [PubMed] [Google Scholar]
- 100.Legha SS. Vincristine neurotoxicity. Pathophysiology and management. Med Toxicol. 1986;1(6):421–7. doi: 10.1007/BF03259853. [DOI] [PubMed] [Google Scholar]
- 101.Roelofs RI, et al. Peripheral sensory neuropathy and cisplatin chemotherapy. Neurology. 1984;34(7):934–8. doi: 10.1212/wnl.34.7.934. [DOI] [PubMed] [Google Scholar]
- 102.Li Y, Womer RB, Silber JH. Predicting cisplatin ototoxicity in children: the influence of age and the cumulative dose. Eur J Cancer. 2004;40(16):2445–51. doi: 10.1016/j.ejca.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 103.Knight KR, Kraemer DF, Neuwelt EA. Ototoxicity in children receiving platinum chemotherapy: underestimating a commonly occurring toxicity that may influence academic and social development. J Clin Oncol. 2005;23(34):8588–96. doi: 10.1200/JCO.2004.00.5355. [DOI] [PubMed] [Google Scholar]
- 104.Gudrunardottir T, et al. Treatment developments and the unfolding of the quality of life discussion in childhood medulloblastoma: a review. Childs Nerv Syst. 2014;30(6):979–90. doi: 10.1007/s00381-014-2388-5. [DOI] [PubMed] [Google Scholar]
- 105.Saunders DE, et al. Surveillance neuroimaging of intracranial medulloblastoma in children: how effective, how often, and for how long? J Neurosurg. 2003;99(2):280–6. doi: 10.3171/jns.2003.99.2.0280. [DOI] [PubMed] [Google Scholar]
- 106.Perreault S, et al. Relapse patterns in pediatric embryonal central nervous system tumors. J Neurooncol. 2013;115(2):209–15. doi: 10.1007/s11060-013-1213-4. [DOI] [PubMed] [Google Scholar]
- 107.Perreault S, et al. Surveillance imaging in children with malignant CNS tumors: low yield of spine MRI. J Neurooncol. 2014;116(3):617–23. doi: 10.1007/s11060-013-1347-4. [DOI] [PubMed] [Google Scholar]
- 108.Bartels U, et al. Role of spinal MRI in the follow-up of children treated for medulloblastoma. Cancer. 2006;107(6):1340–7. doi: 10.1002/cncr.22129. [DOI] [PubMed] [Google Scholar]
- 109.Sethi RV, et al. Patterns of failure after proton therapy in medulloblastoma; linear energy transfer distributions and relative biological effectiveness associations for relapses. Int J Radiat Oncol Biol Phys. 2014;88(3):655–63. doi: 10.1016/j.ijrobp.2013.11.239. [DOI] [PubMed] [Google Scholar]
- 110.Gajjar A, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19(22):6305–12. doi: 10.1158/1078-0432.CCR-13-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]