

Abstract
Background and Purpose
Asthma manifests as a heterogeneous syndrome characterized by airway obstruction, inflammation and hyperresponsiveness (AHR). Although the molecular mechanisms remain unclear, activation of specific PI3K isoforms mediate inflammation and AHR. We aimed to determine whether inhibition of PI3Kδ evokes dilation of airways and to elucidate potential mechanisms.
Experimental Approach
Human precision cut lung slices from non‐asthma donors and primary human airway smooth muscle (HASM) cells from both non‐asthma and asthma donors were utilized. Phosphorylation of Akt, myosin phosphatase target subunit 1 (MYPT1) and myosin light chain (MLC) were assessed in HASM cells following either PI3K inhibitor or siRNA treatment. HASM relaxation was assessed by micro‐pattern deformation. Reversal of constriction of airways was assessed following stimulation with PI3K or ROCK inhibitors.
Key Results
Soluble inhibitors or PI3Kδ knockdown reversed carbachol‐induced constriction of human airways, relaxed agonist‐contracted HASM and inhibited pAkt, pMYPT1 and pMLC in HASM. Similarly, inhibition of Rho kinase also dilated human PCLS airways and suppressed pMYPT1 and pMLC. Baseline pMYPT1 was significantly elevated in HASM cells derived from asthma donors in comparison with non‐asthma donors. After desensitization of the β2‐adrenoceptors, a PI3Kδ inhibitor remained an effective dilator. In the presence of IL‐13, dilation by a β agonist, but not PI3K inhibitor, was attenuated.
Conclusion and Implications
PI3Kδ inhibitors act as dilators of human small airways. Taken together, these findings provide alternative approaches to the clinical management of airway obstruction in asthma.
Abbreviations
- AHR
airway hyperresponsiveness
- HASM
human airway smooth muscle
- hPCLS
human precision cut lung slices
- MLC
myosin light chain
- MYPT1
myosin phosphatase target subunit 1
- SALM
salmeterol
- TAS2 receptor
bitter taste receptor 2
- ROCK
rho‐associated protein kinase
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a, bAlexander et al., 2015a, 2015b).
Introduction
Asthma, a disorder characterized by airway inflammation and airway hyperresponsiveness (AHR), globally contributes to substantial morbidity and mortality (Vijverberg et al., 2013). Bronchodilators act either acutely as rescue treatment or chronically as maintenance therapy by reversing airway smooth muscle (ASM) shortening and dilating airways. While these agents are effective in preventing airflow obstruction, therapeutic limitations exist that include increasing sensitivity to bronchoconstrictive agents (Cooper et al., 2009), receptor tachyphylaxis (Kraan et al., 1985; Cheung et al., 1992) and heterogeneity of response imparted by β2‐adrenoceptor polymorphisms (Drazen et al., 2000). Many asthma patients remain poorly controlled with bronchodilator(s), suggesting an unmet need for alternative approaches to control asthma symptoms and morbidity (Barnes, 1995; Drazen et al., 2000).
Phosphoinositide‐3 kinase (PI3K), a family of kinases, phosphorylates membrane phospholipids. These kinases consist of regulatory subunits p85 and p101, and a catalytic subunit p110. Of the catalytic subunits, there are three classes that are differentially activated by receptor tyrosine kinases and GPCRs. Class I p110 catalytic subunits include α, β, γ and δ and are activated by GPCRs. Evidence suggests that PI3K activation helps drive the inflammation associated with allergic airways disease. Inhibition of PI3K attenuates allergen‐induced eosinophil influx, mucus production (Nashed et al., 2007), degranulation of bone marrow derived mast cells (Ali et al., 2008) and remodelling of the airways (Takeda et al., 2009). Investigators previously showed that genetic ablation or inhibition of PI3Kγ or δ isoforms substantially decreased AHR (Lee et al., 2006). In vascular smooth muscle, inhibition of p110α attenuated membrane depolarization and contraction of aortic rings by KCl. Additionally, inhibition of PI3K class I isoforms with LY294002, wortmannin or targeted knockdown of p110α also attenuated KCl‐induced phosphorylation and activation of Rho A, myosin phosphatase target subunit 1 (MYPT1) phosphorylation and myosin light chain (MLC) phosphorylation (Wang et al., 2006). Rho A and MYPT1 are components of the calcium sensitization pathway that amplifies contraction responses in smooth muscle. MYPT1, a phosphatase, reverses the phosphorylation of MLC required for smooth muscle shortening. Y27632, a Rho kinase inhibitor, also attenuated phosphorylation of MYPT1 while inhibition of PI3K p110γ attenuated acetylcholine‐induced bronchoconstriction of murine small airways and calcium flux in murine ASM (Jiang et al., 2012). We demonstrated that inhibition of PI3Kδ also attenuated TNF‐α‐induced CD38 expression, an ADP‐ribosyl transferase that mobilizes cellular calcium (Jude et al., 2012).
PI3K inhibition prior to stimulation with a contractile agonist attenuated smooth muscle contraction, calcium flux (Jiang et al., 2010) and other contractile signalling pathways (Su et al., 2004; Wang et al., 2006). For the current study, we posit that PI3Kδ inhibition directly dilates human small airways. PI3K inhibition, both inhibition of class I p110 subunits [with LY294002 (Vlahos et al., 1994)] and targeted inhibition of p110δ but not p110γ [with CAL‐101(Lannutti et al., 2011)], attenuated carbachol‐induced phosphorylation of MYPT1 and MLC in human ASM (HASM). Using human precision cut lung slices (hPCLS), we show that reversal of carbachol‐induced bronchoconstriction with PI3K inhibitors or a selective rho‐associated protein kinase (ROCK) inhibitor [Y27632 (Ishizaki et al., 2000)] is comparable with that induced by formoterol, a β2‐adrenoceptor agonist. Interestingly, PI3K inhibition attenuated phosphorylation of MYPT1 and MLC, but had little effect on generation of cAMP or agonist‐induced calcium mobilization. We show that agonist‐induced HASM contraction is reversed by both formoterol and CAL‐101 to similar levels. Modulation of MYPT1 and MLC phosphorylation by PI3K inhibitors is receptor dependent, as shown by its absence following KCl stimulation. Following salmeterol ‐induced β2‐adrenocpetor desensitization, PI3Kδ inhibition dilates human airways more effectively than a β agonist. In a Th2 type inflammatory environment consistent with that observed in asthma, we show that PI3K inhibitors promote airway dilation while formoterol‐induced airway dilation is attenuated. Additionally, we demonstrate that MYPT1 phosphorylation is elevated in HASM derived from fatal asthma patients as compared with that derived from non‐asthma patients. Taken together, these data suggest that the mechanism by which PI3K inhibition bronchodilates human airways converges at a step below an elevation in intracellular cAMP, thereby providing a complementary therapeutic strategy comparable with β agonists for reversing airflow obstruction.
Methods
Isolation and culture of HASM
HASM cells were derived from tracheas obtained from the National Disease Research Interchange (Philadelphia, PA, USA) and from the International Institute for the Advancement of Medicine (Edison, NJ, USA). HASM cell culture was performed as described previously (Panettieri et al., 1989a). The cells were cultured in Ham's F‐12 medium supplemented with 10% FBS, 100 U mL‐1 penicillin, 0.1 mg mL‐1 streptomycin and 2.5 mg mL‐1 amphotericin B, and this medium was replaced every 72 h. HASM cells in subculture during passages 1–5 were used, because these cells retain the expression of native contractile protein, as demonstrated by immunocytochemical staining for smooth muscle actin and myosin (Panettieri et al., 1989b). The HASM cells were derived from donors with fatal asthma or from donors who were age‐ and gender‐matched without asthma, as shown in Supporting Information Table S2.
Generation of PCLS and airway dilation assays
hPCLS were prepared as previously described (Cooper et al., 2009). Briefly, whole human lungs from non‐asthma donors were dissected and inflated using 2% (w v‐1) low melting point agarose. Once the agarose set, the lobe was sectioned, and cores of 8 mm diameter were made. The cores that contained a small airway by visual inspection were sliced at a thickness of 350 μm (Precisionary Instruments VF300 Vibratome, Greenville, NC, USA) and collected in wells containing supplemented Ham's F‐12 medium. The cores generated were randomized as to the location in the lungs they were derived from, so the slices generated came from throughout the lungs and not one specific area. This generated variation in the slices both from within a single donor, and also accounts for variation from donor to donor. Suitable airways (≤1 mm diameter) on slices were selected on the basis of the following criteria: presence of a full smooth muscle wall, presence of beating cilia and unshared muscle walls at airway branch points to eliminate possible counteracting contractile forces. Each slice contained ∼98% parenchyma tissue; hence, all airways situated on a slice had sufficient parenchymal tissue to impart basal tone. Slices containing contiguous segments of the same airway served as controls and were incubated at 37°C in a humidified air‐CO2 (95–5%) incubator. Sections were rinsed with fresh media 2–3 times on days 1 and 2 to remove agarose and endogenous substances released that variably confound the production of inflammatory mediators and/or alter airway tone (Cooper et al., 2009). Airways from each core were randomized to the different treatment groups prior to the start of the experiment. Airways were constricted to a dose response of carbachol (10−8–10−5 M), then dilated to one of the following (10−11–10−4 M): diluent (DMSO), formoterol, isoprenaline, LY294002, CAL‐101 or Y27632. DMSO alone did not induce airway dilation at the concentrations tested (data not shown).
To assess luminal area, lung slices were placed in a 12‐well plate in media and held in place using a platinum weight with nylon attachments. The airway was located using a microscope (Nikon Eclipse; model no. TE2000‐U; magnification, ×40) connected to a live video feed (Evolution QEi; model no. 32‐0074A‐130 video recorder). Airway luminal area was measured using Image‐Pro Plus software (version 6.0; Media Cybernetics) and represented in mm2 (Cooper et al., 2009). After functional studies, the area of each airway at baseline and at the end of dose the response was calculated using Image‐Pro Plus software. Maximal airway dilation (Emax), sensitivity of the airways to contractile agonist – log of the effective concentration to induce 50% airway dilation (log EC50) and the integrated response to dilatory agonist – AUC were calculated from the dose‐response curves generated. The hPCLS were derived from non‐asthma donors, as shown in Supporting Information Table S3. Airway dilation was calculated as % reversal of maximal bronchoconstriction. Time courses of airway dilation to Y27632, CAL‐101 and formoterol were performed to show the kinetics of response of the airways to reversal of carbachol‐induced airway constriction by these compounds (Supporting Information Fig. S1).
Immunoblot analysis
HASM cells were treated with LY294002, CAL‐101 or Y27632 (1 μM for time course experiments – 0–60 min, or 10−8–10−6 M for 30 min for dose response experiments) then stimulated with carbachol (10 μM – 10 min). Cells were then treated with 500 μM perchloric acid, plates scraped and cells pelleted. Pellets were solubilized in RIPA and sonicated prior to being subjected to SDS‐PAGE and transferred to nitrocellulose membranes, as previously described (Balenga et al., 2015). Phosphorylation of MYPT1, MLC and Akt were assessed, and band densities were normalized to total tubulin band density. A total of 5–7 individual donor cell lines for derived from either non‐asthma or fatal asthma donors were used in these experiments. Total protein expression of MYPT1, MLC and Akt were also assessed in the same lysates (Supporting Information Fig. S2).
Contractility measurements of individual HASM cells
Soft silicone elastomer films were micro‐patterned with fibronectin and fluorescent fibrinogen in uniform ‘X’ shapes (70 μm diagonal by 10 μm thick). These substrates were prepared using a robust sacrificial approach to facilitate covalent embedding of the extracellular matrix (ECM) molecules into the film as previously described (Tseng et al., 2014). The non‐patterned regions were blocked using 0.5% Pluronic F‐127 preventing cell adhesion outside of the fibronectin patterns. Isolated cells adhering to these ‘X’‐shaped micro‐patterns exerted tonic traction forces or stimulated contraction forces, resulting in deformations of the micro‐patterns (Figure 3A). Dimensions of contracted micro‐patterns, which correspond directly to the force applied on them by adhered cells, relative to the original unperturbed dimensions were used to assess cellular contractile responses to the tested compounds. Prior to stimulation, isolated HASM cells were seeded on the soft substrates, allowed to adhere and serum‐starved for 48 h. Cells were then stimulated with bradykinin (10−6 M, 15 min) to induce contraction and treated with 0.5% v v‐1 DMSO in medium (control), formoterol or CAL‐101 (10−10, 10−7 or 10−4 M). Each condition was performed in triplicate wells for a non‐asthmatic HASM cell line. The fluorescent micro‐patterns were imaged immediately before stimulation with bradykinin for a baseline reading, and approximately 15 min after administration of the treatments. Cell nuclei were stained with Hoechst 33342 prior to imaging, and only the patterns co‐localized with exactly one stained nucleus were used in the analysis. Following these studies, matlab (Mathworks, Natick, MA, USA) was used to measure each individual pattern and generate distributions of the relative contractions of the patterns for each treatment case. Using the medians of these distributions, the relative relaxation was calculated as the percent reversal from maximal contraction. Bradykinin was administered at 10−4 M in the movies provided in the supplemental materials (Supporting Information Movies S1 and S2), but lowered to 10−6 M in the experimental data shown.
Figure 3.
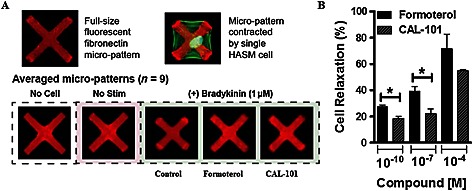
PI3K inhibition reverses bradykinin‐induced shortening in isolated HASM cells. Cells were stimulated to contract with bradykinin (10−6 M) prior to treatment with 0.5% v v‐1 DMSO control, or formoterol or CAL‐101(10−10, 10−7 and 10−4 M). (A) Representative images of fibronectin micro‐patterns on ultra‐soft silicone elastomer films. Top: typical uncontracted and contracted micro‐patterns. HASM F‐actin is shown in green. Bottom: averaged patterns for each treatment case generated from one pattern taken from each of the nine imaging sites used per sample. (B) Quantification of cell relaxation to formoterol or CAL‐101 (10−10, 10−7 and 10−4 M) following bradykinin contraction. Bars represent mean of the triplicates + SEM, with each column representing ≥720 cells analysed per triplicate well measurement from a single donor. P < 0.05 was considered significant.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). graphpad prism software (La Jolla, CA, USA) was used to determine statistical significance evaluated by Student's paired t‐test for two groups or ANOVA for multiple groups. P values of <0.05 were considered significant. For lung slice analysis (Figures 1, 5 and 8), slices were not compared with themselves for each treatment group, so repeated measures analysis was not used. Data were found to be normally distributed, and ANOVAs were used for data analysis, with Bonferroni's post test. Unpaired t‐tests with Welch's correction were used after ANOVAs had established significance to compare each inhibitor with formoterol, and conditions within a given inhibitor stimulation (for example, −/+ IL‐13 for LY294002‐induced airway dilation). For Figure 7, results were analysed by two‐way repeated measures ANOVA with desensitization pretreatment (control or salmeterol) as Factor A and bronchodilator test compound (isoprenaline or CAL‐101) as Factor B. The data passed the normality, and equal variance tests and significant effects of both factors were detected. Differences were isolated using the Bonferroni t‐test for all pairwise comparisons. Immunoblot data and single cell contractile data were analysed by Student's two‐tailed t‐tests. sigmastat (Systat, San Jose, CA, USA) and graphpad prism programmes were used in statistical analyses.
Figure 1.
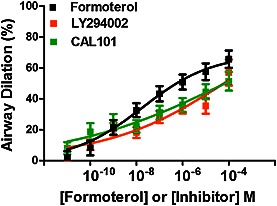
PI3K inhibition reverses carbachol‐induced bronchoconstriction in a dose‐dependent manner in hPCLS. Airways were preconstricted to carbachol (10−8–10−4 M) prior to dilation to LY294002, CAL‐101 or formoterol (10−11–10−5 M). Maximal airway dilation (Emax, ANOVA, P = 0.03) and AUC (P = 0.004) for each inhibitor was significantly different than formoterol‐induced dilation. Data are representative of n ≥ 5 donors, 26–33 slices per condition, with bars representing mean + SEM.
Figure 5.
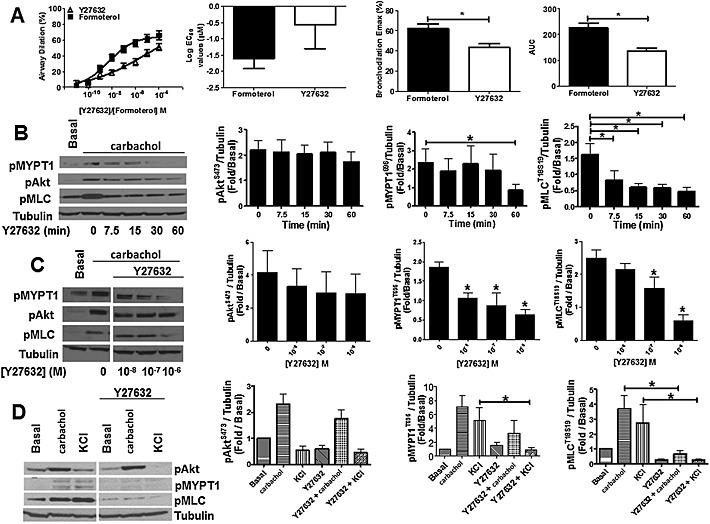
Rho Kinase inhibition induces airway dilation in hPCLS and inhibits carbachol‐induced MYPT1 and MLC phosphorylation in HASM cells. (A) hPCLS were bronchoconstricted with carbachol (n = 12 donors) and sequentially treated with 0.02% DMSO (Veh) or increasing concentrations of Y27632 (10−11 – 10−5 M). Data are representative of n ≥ 5 separate donors (two‐tailed t‐test P = 0.02 for Emax, P = 0.001 for AUC). (B) Time‐dependent inhibition of carbachol‐induced (10 min, 10 μM) phosphorylation of MYPT1 (pMYPT1) and MLC (pMLC) in HASM cells by Y27632 (1 μM). Data are representative of five different HASM donors (n = 5, mean + SEM). *P < 0.05. P values are time points compared with vehicle control. (C) Concentration‐dependent inhibition of carbachol‐induced phosphorylation of MYPT1 and MLC in HASM cells by Y27632 (30 min pretreatment, 10−8–10−6 M). Data are representative of five independent experiments. *P < 0.05. P values compared with vehicle control. (D) Y27632 (1 μM) prevents carbachol‐ (10 min, 10 μM) and KCl‐ (10 min, 50 mM) mediated pMYPT1 and pMLC, but has little effect on Akt phosphorylation in primary HASM cells. Data are representative of five different HASM donors (n = 5, mean + SEM).
Figure 8.
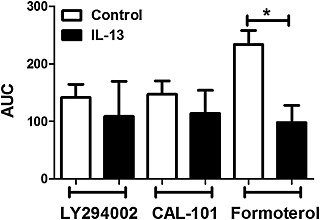
PI3K and ROCK inhibitor‐induced airway dilation in the presence and absence of IL‐13. hPCLS were incubated in the presence or absence of IL‐13 (100 ng·mL−1, 18 h), precontracted to ~60–70% of baseline luminal area and relaxed to a dose response of LY294002, CAL‐101 or the β2‐adrenoceptor agonist formoterol (10−11–10−5 M). AUC was compared between control and IL‐13 treatment for each compound used to dilate the airways. Data are representative of n ≥ 4 donors, 5–44 slices per condition, with bars representing mean + SEM, with P ≤ 0.05 considered significant (ANOVA P = 0.01; two‐tailed t‐tests: control versus IL‐13 for formoterol P = 0.009, control versus IL‐13 for CAL‐101 P = n.s., control versus IL‐13 for LY294002 P = n.s.).
Figure 7.
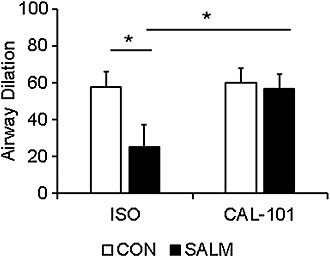
PI3K inhibition induces airway dilation of small airways despite desensitization of the β2‐adrenoceptors. hPCLS from four separate donors 79–81 slices/condition, total of 160 slices, were incubated in the presence or absence of 50 nM salmeterol (SALM; 24 h) to induce desensitization of the β2‐adrenoceptor. After washout and treatment with carbachol, isoprenaline (ISO)‐induced airway dilation was significantly attenuated by salmeterol pretreatment. CAL‐101‐induced airway dilation, however, was indistinguishable from control pretreatment and was enhanced compared with the previous treatment of β agonist‐desensitized airways. Data are presented as mean + SEM *P < 0.05.
Materials
Carbachol (carbamoyl choline chloride), formoterol (formoterol fumarate dihydrate), isoprenaline (ISO – isoproterenol hydrochloride), salmeterol (salmeterol xinafoate), bradykinin (bradykinin acetate salt) and perchloric acid were purchased from Sigma Aldrich (St. Louis, MO, USA). LY294002 was purchased from Cayman Chemical Company (Ann Arbour, MI, USA), and CAL‐101 was purchased from Selleck Chemicals (Houston, TX, USA). Y27632 was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Rho activity assay kit was purchased from Cytoskeleton (Denver, CO, USA). Constructs (siRNA) targeting PI3K p110γ and δ were purchased from Qiagen (The Netherlands). Antibodies for detection of pMYPT1‐Thr696 (5163S), pAkt (4060S), pMLC (3674S) and tubulin (3873S) were purchased from Cell Signalling Technologies (Danvers, MA, USA). Pharmacological properties of the inhibitors utilized in these studies are found in Supporting Information Table S1.
Results
PI3K inhibition reverses carbachol‐induced constriction of human small airways in a dose‐dependent manner
To determine if inhibition of PI3K dilates precontracted small human airways, PCLS were prepared from donors with no history of lung disease. PCLS airways were treated with carbachol to induce bronchoconstriction and then treated with increasing doses of LY294002, CAL‐101 or formoterol to evaluate airway dilation (Figure 1). Both LY294002 and CAL‐101 markedly reversed carbachol‐induced bronchoconstriction, but it was not to the same level as to the β agonist formoterol. Further, we evaluated ROCK and PI3K inhibitor‐induced airway dilation and detected responses as early as 5–10 min, which were sustained through 30 min following a single dose of inhibitor (Supporting information Figure S1). LY294002 and CAL‐101 also reversed bronchoconstriction induced by histamine (data not shown). Pretreatment with these inhibitors had little effect on carbachol‐induced calcium transients in HASM cells (Supporting Information Fig. S3). These data indicate that inhibition of the p110 subunit of PI3K and specifically of p110δ is bronchodilatory and that the mechanism likely involves events distal to the activation of GPCRs mediating bronchoconstriction in human airways.
PI3K inhibition attenuates carbachol‐induced phosphorylation of Akt, MYPT1 and MLC
Phosphorylation of key components of excitation‐contraction coupling pathways, including Akt (a surrogate to measure PI3K activation), MYPT1 and MLC mediate agonist‐induced HASM shortening. Conversely, dephosphorylation of these targets promotes lengthening of HASM. Accordingly, carbachol‐induced activation (phosphorylation) of Akt, MYPT1 and MLC were examined in HASM in the presence and absence of PI3K inhibition. As shown in Figure 2, selective PI3Kδ inhibition with CAL‐101 (Figure 2A) or a pan‐PI3K inhibitor LY294002 in HASM cells (Figure 2B) inhibited carbachol‐induced phosphorylation of Akt, MYPT1 and MLC in a time‐ (Figure 2A and B) and dose‐dependent manner (Figure 2C). To assess whether membrane depolarization‐induced phosphorylation of Akt, MYPT1 and MLC required PI3K activation, HASM cells were treated with KCl in presence or absence of CAL‐101 (Figure 2D). Unlike carbachol‐induced phosphorylation of MYPT1 and MLC, KCl‐dependent phosphorylation of MYPT1 and MLC was unaffected by CAL‐101 while phosphorylation of Akt was PI3K‐dependent. These data suggest that the mechanism for smooth muscle contractile responses to membrane depolarization by KCl is either parallel or it converges below PI3K and MYPT1 phosphorylation on phosphorylation of MLC.
Figure 2.
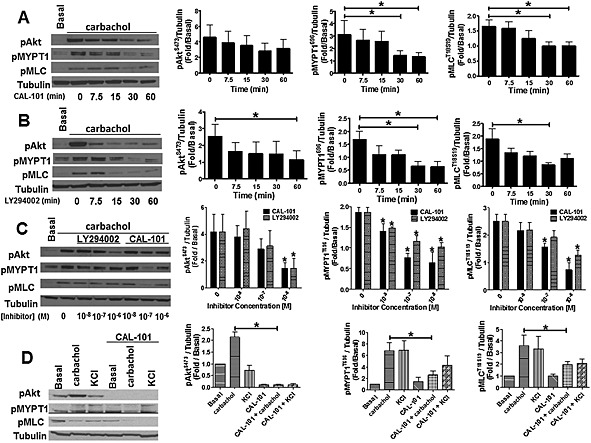
PI3K inhibition blocks carbachol‐induced MYPT1 and MLC phosphorylation in HASM cells. Time‐dependent inhibition of carbachol‐induced (10 min, 10 μM) MYPT1 and MLC phosphorylation in HASM cells by CAL‐101 (A) and LY294002 (B) (1 μM). Data are representative of five different HASM donors (n = 5 donors, mean ± SEM). *P < 0.05. P values are time points compared with basal. (C) Concentration‐dependent inhibition of carbachol‐induced MYPT1, Akt and MLC phosphorylation in HASM cells by LY294002 and CAL‐101. Data are representative of five different HASM donors. *P < 0.05. P values compared with basal (n = 5, mean ± SEM). (D) CAL‐101 (1 μM) prevents carbachol‐ (10 min, 10 μM) induced MYPT1 (pMYPT1) and MLC (pMLC) phosphorylation, but has little effect on KCl‐ (10 min, 50 mM) mediated MYPT1 and MLC phosphorylation in HASM cells. Data are representative of five different HASM donors (n = 5, mean + SEM).
PI3K inhibition reverses bradykinin‐induced shortening of single HASM cells
To determine if inhibition of PI3K reverses contractile agonist‐induced shortening of single HASM cells, cells were seeded on fluorescently micro‐patterned soft silicone elastomer substrates, stimulated with bradykinin to induce shortening and then treated with increasing doses of CAL‐101 or formoterol. Relaxation of the cells was evaluated, and we show that CAL‐101 reversed bradykinin‐induced shortening comparable with maximal levels induced by formoterol Figure 3, and Movies S1 and S2.
Suppression of PI3K p110δ protein expression inhibits –carbachol‐induced phosphorylation of MYPT1 and MLC
Murine models showed that genetic ablation of PI3K p110γ or δ attenuated airway hyperreactivity after allergen challenge (Lee et al., 2006; Jiang et al., 2010). We demonstrated that carbachol‐induced phosphorylation of contractile proteins is attenuated by pharmacological inhibition of PI3K p110, specifically p110δ. We next sought to define the requirement for p110δ in modulating phosphorylation events mediating HASM shortening by using siRNA targeting PI3K p110δ to suppress protein expression in HASM cells (Figure 4A). As a consequence, carbachol‐induced phosphorylation of both MYPT1 and MLC were significantly reduced (Figure 4B). However, knockdown of PI3K p110γ had little effect on phosphorylation of MYPT1 and MLC (Supporting Information Fig. S4). Knockdown of either PI3K p110δ or p110γ reduced carbachol‐induced Akt phosphorylation, but this was not statistically significant. The limitation to these observations is that although we detected expression of PI3Kδ, we could not detect PI3Kγ in HASM. Taken together, pharmacological inhibition and siRNA‐mediated suppression indicate that p110δ likely mediates distal events regulating ASM shortening in response to carbachol.
Figure 4.
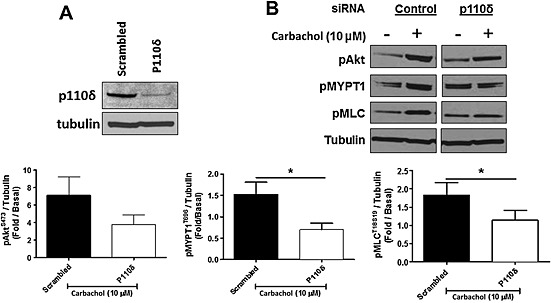
siRNA knockdown of PI3K p110δ prevents carbachol‐induced (10 min, 10 μM) MLC phosphorylation (pMLC) by suppression of ROCK in HASM cells. (A) Immunoblot analyses of HASM cells transfected with PI3K p110δ siRNA or scrambled siRNA. (B) Inhibition of carbachol‐induced phosphorylation of MYPT1 (pMYPT1) and MLC (pMLC) in HASM cells by PI3K p110δ siRNA. Data are representative of five independent experiments (n = 5, mean + SEM); *P < 0.05.
Inhibition of rho kinase with Y27632 induces airway dilation of human small airways and attenuates activation of rho kinase and phosphorylation of MLC
In vascular smooth muscle, PI3K p110α regulates Rho activation and MYPT1 phosphorylation (Wang et al., 2006). Rho kinase (ROCK) activation plays a role in the maintenance of airway muscle tone in guinea pig ileum (Sward et al., 2000) and pig trachea (Lan et al., 2015) strips. To determine if ROCK activation similarly mediates maintenance of airway tone in carbachol bronchoconstricted PCLS airways, we treated hPCLS with increasing doses of the ROCK inhibitor Y27632 and observed that Rho kinase inhibition evoked airway dilation similar to that observed with the β agonist formoterol, but not quite as robust. Like LY294002 and CAL‐101, Y27632 induced dose‐dependent airway dilation (Figure 5A). ROCK inhibition also attenuated carbachol‐induced phosphorylation of MYPT1 and MLC, but not Akt in HASM cells (Figure 5B and C). To define the role for ROCK in receptor‐independent contraction, HASM cells were also treated with KCl in the presence or absence of Y27632. As shown in Figure 5D, inhibition of ROCK activity attenuated receptor‐independent phosphorylation of MYPT1 and MLC. These data indicate that while Rho kinase activation by carbachol occurs either downstream or parallel to PI3K activation, both converge on MYPT1 and MLC phosphorylation.
Baseline rho kinase activity is increased in HASM derived from asthma donors
The present studies point to MYPT1 phosphorylation as an important mechanism mediating a procontractile phenotype. As compared with normal donors, HASM cells derived from asthma donors retain a phenotype consistent with asthma manifested by hypersecretion of inflammatory mediators, growth and proliferation (Ma et al., 2002; Benayoun et al., 2003; Roth et al., 2004; Leguillette and Lauzon, 2008; John et al., 2009; Himes et al., 2015). Therefore, we evaluated MYPT1 activation in HASM cells from non‐ and fatal asthmatic donors. As shown in Figure 6A, HASM cells derived from asthma donors exhibit greater baseline phosphorylation of MYPT1 in comparison with HASM cells derived from non‐asthma donors. In parallel, we found that ROCK activity levels were also elevated in HASM cells derived from asthma donors (Figure 6B). These data suggest that there is a greater activation of Rho kinase‐dependent pathways evoking MYPT1 phosphorylation in HASM cells from donors with asthma.
Figure 6.
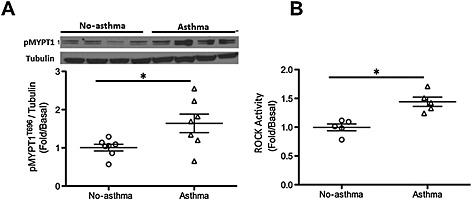
Rho kinase activation is greater in HASM cells obtained from subjects with asthma as compared to those of non‐asthmatic subjects. HASM cells obtained from fatal asthmatic and non‐asthmatic subjects were lysed and subjected to immunoblot for phosphorylated MYPT1 (A), or elisa to measure ROCK activation (B). Data represent mean + SEM fold change of MYPT1 phosphorylation as compared with tubulin control (immunoblotting) and fold compared with basal (for elisa). *P < 0.05. n = 7 asthma and seven non‐asthma for (A). n = 5 asthma and seven non‐asthma for (B).
PI3K and rho kinase inhibitors induce airway dilation after β2AR desensitization or IL‐13 stimulation
To determine the potential benefit of pharmacological inhibition of PI3K and ROCK as ‘rescue’ bronchodilators, we evaluated responses of hPCLS desensitized to a β agonist or maintained in a Th2‐like inflammatory environment, elevated levels of IL‐13. Tachyphylaxis of the β2‐adrenoceptor limits the therapeutic efficacy of β agonists in airways disease (Cooper and Panettieri, 2008; Cooper et al., 2011). Because this involves a proximal event (failure to activate of Gsα), we hypothesized that PI3K inhibition would induce airway dilation despite β2‐adrenoceptor tachyphylaxis. In hPCLS treated for 24 h with salmeterol, isoprenaline failed to reverse carbachol‐induced bronchoconstriction (Figure 7), implying tachyphylaxis. By contrast, CAL‐101 effectively dilated airways despite impaired β2‐adrenoceptor activation. Inhibition of PI3K alone or in combination with isoprenaline had no direct effect on generation of cAMP in HASM (Supporting Information Fig. S5).
We previously demonstrated that IL‐13, an important Th2 inflammatory mediator in asthma, attenuates β2‐adrenoceptor agonist‐induced airway dilation (Cooper et al., 2009), but has little effect on airway dilation to the TAS2 receptor agonist chloroquine (Robinett et al., 2014). Accordingly, we posited that IL‐13 may limit PI3K inhibitor‐mediated airway dilation of carbachol‐constricted hPCLS. While formoterol‐induced airway dilation was attenuated by IL‐13 treatment, PI3K inhibitor‐induced airway dilation was not affected significantly by IL‐13 (Figure 8). Taken together, these experiments using hPCLS define new therapeutic approaches to bronchodilation.
Discussion
Asthma, a chronic respiratory disorder marked by inflammation, recurrent reversible airflow obstruction and AHR, remains a significant cause of morbidity and mortality (Vijverberg et al., 2013). Despite efficacy of both long‐ and short‐acting bronchodilators, a significant number of asthma patients remain uncontrolled due to a number of factors including tachyphylaxis of the receptor (Kraan et al., 1985; Cheung et al., 1992), genetic polymorphisms altering receptor function or heterogeneity of patient responses (Barnes, 1995; Drazen et al., 2000). There exists an unmet need for alternative methods to induce bronchodilation that are independent of the β2‐adrenoceptor.
Studies in murine models of allergen challenge showed a role for class I PI3K p110γ and δ isoforms in trafficking/activating inflammatory cells, decreasing mucus production and attenuating AHR following allergen challenge (Nashed et al., 2007; Ali et al., 2008; Takeda et al., 2009). Interestingly, inhibition of PI3K p110δ, either by genetic ablation (Nashed et al., 2007) or by pretreatment with a δ selective pharmacological inhibitor (Jiang et al., 2010), had little effect on baseline methacholine‐induced bronchoconstriction in vivo or in ex vivo murine lung slices. A PI3Kγ selective inhibitor attenuated acetylcholine‐induced bronchoconstriction of murine precision cut lung slices and calcium flux in murine ASM cells (Jiang et al., 2010). We now show that pretreatment of HASM cells with either LY294002 or CAL‐101 had little effect on calcium flux in HASM cells (Supporting Information Fig. S3). We provide evidence of PI3K p110δ, but not γ, isoform expression in ASM [Supporting Information Fig. S2 and (Jude et al., 2012)], validating our approach to selectively targeting PI3K p110δ in modulation of contractile pathways in HASM and airway dilation in PCLS. In addition to using human cells and PCLS, our study also focused on using PI3K inhibitors to induce airway dilation rather than on using PI3K inhibition as a bronchoprotective agent. We show that in human airways, inhibition of PI3K p110 isoforms with LY294002, as well as selective inhibition of p110δ with CAL‐101, promotes airway dilation comparable with the β agonists isoprenaline or formoterol. Clinically, the onset of action for formoterol is within 1–4 min following inhalation (Ringdal et al., 1998). In contrast, airway dilation in response to a PI3K or ROCK inhibitor is slower given these compounds act on intracellular targets rather than a cell surface receptor. We demonstrate that the time scale of inhibitor‐induced dilation of carbachol‐constricted airways is in agreement with the time course of PI3K or ROCK inhibition of carbachol‐induced phosphorylation of Akt, MYPT1 and MLC (Figures 1, 2 and 5 and Supporting Information Fig. S1). As a surrogate for airway constriction and relaxation, we utilized a novel in vitro approach measuring contractility of single HASM cells to assess the effect of PI3K inhibition on cellular dynamics of contraction and relaxation (Figure 3A). Using this system, we show that both formoterol and CAL‐101 induce relaxation of HASM cells that have been precontracted by bradykinin in a dose‐dependent manner (Figure 3B, Supporting Information Movies S1 and S2). In this system, we show that there is no significant difference in the maximal relaxation elicited by formoterol or CAL‐101. These data suggest that CAL‐101 is as effective of a HASM relaxant as formoterol at a maximal concentration. These data provide alternative mechanisms/targets for inducing airway dilation in humans that are independent of β2‐adrenoceptor activation.
In vascular smooth muscle, the PI3K p110α isoform regulates Rho activation and phosphorylation of MYPT1 and MLC in response to KCl (Wang et al., 2006). Others demonstrate that in response to histamine, phosphorylation of MLC in vascular smooth muscle is PI3K‐dependent (Su et al., 2004). To determine potential mechanisms by which inhibition of PI3K induces airway dilation, we examined carbachol‐induced phosphorylation of Akt (as a marker of PI3K activation), MYPT1 and MLC. Carbachol‐induced Akt, MYPT1 and MLC phosphorylation was inhibited by CAL‐101 and LY294002 in a time‐ and dose‐dependent manner. To assess the contribution of PI3K p110 isoforms to non‐receptor‐mediated activation of contractile pathways, we also stimulated HASM cells with KCl in the presence and absence of pharmacological inhibition of PI3K. We demonstrated that phosphorylation of MLC and MYPT1 are independent of PI3K activation in response to KCl. These data provide a mechanism by which carbachol‐induced bronchoconstriction is reversed by PI3K inhibition, but that receptor‐independent stimulation of contractile pathways with KCl is PI3K‐independent unlike in vascular smooth muscle (Su et al., 2004). The physiological relevance of KCl‐induced phosphorylation of Rho kinase and MLC remains unclear (Wang et al., 2010). Additionally, we demonstrated that knockdown of PI3K p110δ attenuates carbachol‐induced phosphorylation of MLC and MYPT1. We showed that PI3K p110δ, but not γ, is expressed in HASM cells [Supporting Information Fig. S3 and (Jude et al., 2012)] and that inhibition of PI3K isoforms has no effect on expression of levels of total Akt, MYPT1 or MLC proteins (Supporting Information Fig. S2). Additionally, we demonstrated that knockdown of PI3Kγ in HASM cells (Supporting Information Fig. S4) has little effect on carbachol‐induced phosphorylation of Akt, MYPT1, and MLC. Given the lack of specific antibodies, we cannot completely rule out the possibility that the PI3K p110γ isoform may also play role in relaxation of ASM, but given the data we have concerning lack of its expression in isolated smooth muscle, we feel that this is unlikely.
β‐adrenoceptor agonists induce bronchodilation by generating cAMP and activating protein kinase A that inhibit, in part, MLC kinase and phosphorylation of MLC. To determine whether PI3K inhibition affects the phosphorylation status of MLC through generation of cAMP, we treated HASM cells with LY294002 and CAL‐101 in the presence or absence of isoprenaline and assessed cAMP generation (Supporting Information Fig. S5). We showed that treatment with the PI3K inhibitors has little effect on cAMP generation and that pretreatment with the inhibitors prior to isoprenaline stimulation also had little effect. These data suggest that PI3K inhibitor‐induced airway dilation is independent of inducing or potentiating β2 agonist‐induced cAMP production. Additionally, we showed that PI3K inhibition does not appear to directly affect carbachol‐induced calcium transients in HASM (Supporting Information Fig. S3).
Given evidence in vascular smooth muscle linking PI3K and Rho pathways (Yoshioka et al., 2007), we sought to determine whether Rho kinase inhibition attenuated constriction of human small airways and excitation‐contraction pathways activated in HASM cells. We found that Y27632, a Rho kinase inhibitor, reversed carbachol‐induced bronchoconstriction to levels comparable with that observed using PI3K inhibitors (Figure 5A). We also showed that phosphorylation of MYPT1 and MLC, but not Akt, are inhibited by the ROCK inhibitor Y27632 in a time‐ and dose‐dependent manner (Figure 5B and C). In assessing the effect of Rho kinase inhibition on receptor‐independent activation of contractile pathways, however, we found that Y27632 attenuated KCl‐dependent phosphorylation of MYPT1 and MLC (Figure 5D). These data suggest that receptor‐dependent and receptor‐independent contractile pathways activate ROCK and MLC but that receptor‐dependent pathways also activate PI3K that requires Rho kinase to mediate contractile responses, as illustrated in Figure 9.
Figure 9.
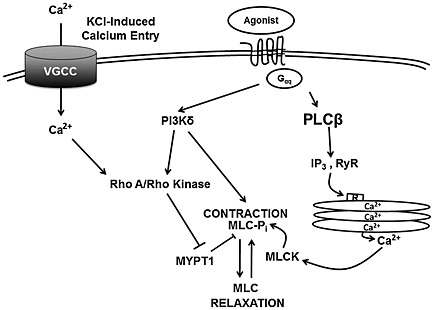
An overview of the signalling mechanisms underlying PI3K and ROCK inhibitor‐mediated airway dilation of HASM cells. IP3, inositol triphosphate; RyR, ryanodine receptor; MLCK, MLC kinase.
Given both clinical (Barnes, 1995) and experimental evidence (Cooper and Panettieri, 2008; Cooper et al., 2011) demonstrating agonist‐induced desensitization of the β2‐adrenocpetor and tachyphylaxis, we posited that inhibition of contractile pathways downstream from the receptor and independently of the β2‐adrenoceptor‐mediated pathways would induce airway dilation. While pretreatment of PCLS with salmeterol diminished airway dilation to isoprenaline, CAL‐101 retained the ability to dilate human small airways despite impaired β2‐adrenoceptor activation (Figure 7). While we presume that this is due to receptor desensitization, it is could also be a manifestation of the antagonistic properties of residual lipophilic salmeterol remaining in the tissue despite washout (McCrea and Hill, 1993; Carter and Hill, 2005). Carbachol‐induced airway constriction in these slices were comparable between the salmeterol‐treated and non‐treated airways, indicating that the bronchoprotective properties of salmeterol treatment were not retained after washing of the tissue.
The inflammatory milieu in asthma engenders AHR to contractile agonists and attenuates β2 agonist‐induced bronchodilation (Cooper et al., 2009; Albano et al., 2015; Gupta et al., 2015). Previously, we showed that exposure to IL‐13 renders airways hyposensitive to β2‐adrenoceptor agonist‐mediated airway dilation but not TAS2 receptor‐mediated airway dilation (Robinett et al., 2014). Our data show that in the presence of IL‐13, PI3K inhibitor‐dependent airway dilation retained efficacy while β2 agonist‐mediated airway dilation was partially inhibited. Farghaly et al. demonstrated that IL‐13 augmented carbachol‐induced contractility of murine tracheal rings and that this hyperresponsiveness was mediated by PI3K p110 isoforms, specifically PI3K p110δ (Farghaly et al., 2008). We previously reported that IL‐13 augments carbachol‐induced airway narrowing and attenuates β2‐adrenoceptor agonist‐induced airway dilation in human PCLS (Cooper et al., 2009; Robinett et al., 2014). Although IL‐13 attenuates β2‐adrenoceptor agonist‐induced airway (Laporte et al., 2001; Grunstein et al., 2002; Duan et al., 2004; Cooper et al., 2009; Robinett et al., 2014; Albano et al., 2015), IL‐13 has little effect on PI3K p110 inhibitor‐induced airway dilation as shown in Figure 8. We and others have shown that HASM cells derived from fatal asthma donors manifest distinct phenotypes compared to cells derived from non‐asthmatic patients, including cell proliferation (Roth et al., 2004), enzymatic function (Chambers et al., 2003; Trian et al., 2011), receptor signalling and sustained calcium transients consistent with contractility of the muscle (Ma et al., 2002; Leguillette and Lauzon, 2008; Chin et al., 2012). We now show that phosphorylation of MYPT1, a phosphatase that regulates MLC phosphorylation and HASM cell shortening, is elevated at baseline in asthma‐derived HASM cells (Figure 6). Phosphorylation of MYPT1 inactivates this key signalling event of calcium sensitization. We also demonstrated that endosomes from asthma‐derived HASM cells, as compared with non‐asthma HASM cells, manifest increased PI3Kγ activation and a reduction in protein phosphatase 2A activity that inhibits β2‐adrenoceptor resensitization (Gupta et al., 2015). Collectively, these findings have implications providing a plausible hypothesis as to why β2 agonists are less effective in Th2 airway inflammation (Duan et al., 2004; Aarthia et al., 2007; Townley, 2007). Moreover, our data suggest that in a Th2 inflammatory milieu promoting AHR, PI3K inhibitors retain efficacy as bronchodilators while β agonist responses are attenuated.
Our data suggest that both PI3K and ROCK inhibitors induce airway dilation. Because others have suggested that PI3K inhibitors may also act as anti‐inflammatory agents in asthma, PI3K activation may serve as a unique therapeutic target in asthma and chronic obstructive pulmonary disease to attenuate airway inflammation and promote airway dilation.
Author contributions
C.J.K.W., E.J.Y., G.C., I.P., R.D.D., S.B.L., R.C.K., R.A.P.J and D.D.C. contributed to the experimental design. C.J.K.W., E.J.Y., G.C., J.Z., E.P., I.P., A.A. and R.C.K. performed experiments. C.J.K.W., E.J.Y., G.C., J.Z., E.P., I.P., A.A., B.E.H. and R.C.K. analysed the data. C.J.K.W. wrote the manuscript. E.J.Y., R.D.D., S.B.L., R.C.K., R.A.P.J and D.D.C. edited the manuscript. I.P. wrote a portion of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Supporting Information
Figure S1 Time course of airway dilation to CAL‐101 and Y27632. Airways in lung slices were preconstricted to carbachol, then dilated to CAL‐101 or Y27632 (60 μM), 0–30 min. Data is representative of mean ± SEM for 4–15 slices/condition.
Figure S2 PI3K p110δ (A) and γ (B) mRNA expression in airway smooth muscle assessed by RNAseq. PI3K inhibition has little effect on expression of total protein levels (C) of MYPT1 (D), Akt (E), and MLC (F). Data is representative of n = 5–8 (A and B) or n = 2 (C‐F) HASM cell lines.
Figure S3 PI3K inhibition has little effect on agonist‐induced calcium mobilization. HASM were stimulated with 10 μM carbachol following a 30 min pretreatment with vehicle (DMSO), CAL‐101 or LY294002 (1 μM) and single cell calcium transients measured using Fluo‐8 calcium sensing dye. (A) Time course tracings of calcium flux. Both peak calcium (B) and AUC (C) for each treatment were plotted as mean ± SD.
Figure S4 siRNA knockdown of PI3K p110γ had little effect on carbachol‐induced (10 min, 10 μM) phosphorylation of MLC (pMLC), MYPT1, or Akt in HASM cells. (A) Immunoblot analyses of HASM cells transfected with PI3K p110γ siRNA or scrambled siRNA. 3 (B) Slight inhibition of carbachol‐induced phosphorylation of Akt (pAkt) and MYPT1 (pMYPT1) in HASM cells by PI3K p110δ siRNA. Data are representative of five independent experiments (n = 5, mean + SEM); *P < 0.05.
Figure S5 PI3K inhibition has little effect alone to induce or augment cAMP levels. Data are representative of 3 donors and each condition in triplicate, with bars representing mean ± SEM with a P value of <0.05 considered significant.
Table S1 Donor demographics of asthma and non‐asthma HASM cell donors.
Table S2 Donor demographics of non‐asthma hPCLS donors.
Table S3 Pharmacologic properties of the inhibitors utilized in this study. In vitro kinase assay results, IC50 values in cell based assays, and the corresponding literature citations are shown for CAL‐101, LY294002, and Y27632.
Movie S1 Formoterol‐induced relaxation of bradykinin contracted HASM. Link: https://youtu.be/51oJeQ73LEA.
Movie S2 CAL‐101‐induced relaxation of bradykinin contracted HASM. Link: https://youtu.be/2H3DKMqI‐GU.
Acknowledgements
This paper is funded by P01‐HL114471–03 (RAP/SBL/RCK), 1DP2OD007113 (DD), David and Lucile Packard Fellowship (DD).
Koziol‐White, C. J. , Yoo, E. J. , Cao, G. , Zhang, J. , Papanikolaou, E. , Pushkarsky, I. , Andrews, A. , Himes, B. E. , Damoiseaux, R. D. , Liggett, S. B. , Di Carlo, D. , Kurten, R. C. , and Panettieri, R. A. Jr. (2016) Inhibition of PI3K promotes dilation of human small airways in a rho kinase‐dependent manner. British Journal of Pharmacology, 173: 2726–2738. doi: 10.1111/bph.13542.
References
- Aarthia JJA, Pushparaj PP, Kumar SD (2007). Inhibition of phosphoinositide 3 kinase by masoprocol ameliorates IgE‐mediated anaphylaxis in vivo an ex vivo . J Allergy Clin Immunol 119: S179. [Google Scholar]
- Albano GD, Zhao J, Etling EB, Park SY, Hu H, Trudeau JB et al. (2015). IL‐13 desensitizes beta2‐adrenergic receptors in human airway epithelial cells through a 15‐lipoxygenase/G protein receptor kinase 2 mechanism. J Allergy Clin Immunol 135: 1144–1153 .e1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali K, Camps M, Pearce WP, Ji H, Ruckle T, Kuehn N et al. (2008). Isoform‐specific functions of phosphoinositide 3‐kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J Immunol 180: 2538–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balenga NA, Klichinsky M, Xie Z, Chan EC, Zhao M, Jude J et al. (2015). A fungal protease allergen provokes airway hyper‐responsiveness in asthma. Nat Commun 6: 6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (1995). Beta‐adrenergic receptors and their regulation. Am J Respir Crit Care Med 152: 838–860. [DOI] [PubMed] [Google Scholar]
- Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M (2003). Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med 167: 1360–1368. [DOI] [PubMed] [Google Scholar]
- Carter AA, Hill SJ (2005). Characterization of isoprenaline‐ and salmeterol‐stimulated interactions between beta2‐adrenoceptors and beta‐arrestin 2 using beta‐galactosidase complementation in C2C12 cells. J Pharmacol Exp Ther 315: 839–848. [DOI] [PubMed] [Google Scholar]
- Chambers LS, Black JL, Ge Q, Carlin SM, Au WW, Poniris M et al. (2003). PAR‐2 activation, PGE2, and COX‐2 in human asthmatic and nonasthmatic airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 285: L619–L627. [DOI] [PubMed] [Google Scholar]
- Cheung D, Timmers MC, Zwinderman AH, Bel EH, Dijkman JH, Sterk PJ (1992). Long‐term effects of a long‐acting beta 2‐adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. N Engl J Med 327: 1198–1203. [DOI] [PubMed] [Google Scholar]
- Chin LY, Bosse Y, Pascoe C, Hackett TL, Seow CY, Pare PD (2012). Mechanical properties of asthmatic airway smooth muscle. Eur Respir J 40: 45–54. [DOI] [PubMed] [Google Scholar]
- Cooper PR, Panettieri RA Jr (2008). Steroids completely reverse albuterol‐induced beta(2)‐adrenergic receptor tolerance in human small airways. J Allergy Clin Immunol 122: 734–740. [DOI] [PubMed] [Google Scholar]
- Cooper PR, Kurten RC, Zhang J, Nicholls DJ, Dainty IA, Panettieri RA (2011). Formoterol and salmeterol induce a similar degree of beta2‐adrenoceptor tolerance in human small airways but via different mechanisms. Br J Pharmacol 163: 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper PR, Lamb R, Day ND, Branigan PJ, Kajekar R, San Mateo L et al. (2009). TLR3 activation stimulates cytokine secretion without altering agonist‐induced human small airway contraction or relaxation. Am J Physiol Lung Cell Mol Physiol 297: L530–L537. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazen JM, Silverman EK, Lee TH (2000). Heterogeneity of therapeutic responses in asthma. Br Med Bull 56: 1054–1070. [DOI] [PubMed] [Google Scholar]
- Duan W, Chan H, Vlahos C, Wong W (2004). Anti‐inflammatory effects of LY294002, a PI3K inhibitor, in a mouse model of asthma. J Allergy Clin Immunol 113: S219. [Google Scholar]
- Farghaly HS, Blagbrough IS, Medina‐Tato DA, Watson ML (2008). Interleukin 13 increases contractility of murine tracheal smooth muscle by a phosphoinositide 3‐kinase p110delta‐dependent mechanism. Mol Pharmacol 73: 1530–1537. [DOI] [PubMed] [Google Scholar]
- Grunstein MM, Hakonarson H, Leiter J, Chen M, Whelan R, Grunstein JS et al. (2002). IL‐13‐dependent autocrine signaling mediates altered responsiveness of IgE‐sensitized airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 282: L520–L528. [DOI] [PubMed] [Google Scholar]
- Gupta MK, Asosingh K, Aronica M, Comhair S, Cao G, Erzurum S et al. (2015). Defective resensitization in human airway smooth muscle cells evokes beta‐adrenergic receptor dysfunction in severe asthma. PLoS One 10: e0125803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himes BE, Koziol‐White C, Johnson M, Nikolos C, Jester W, Klanderman B et al. (2015). Vitamin D modulates expression of the airway smooth muscle transcriptome in fatal asthma. PLoS One 10: e0134057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M et al. (2000). Pharmacological properties of Y‐27632, a specific inhibitor of rho‐associated kinases. Mol Pharmacol 57: 976–983. [PubMed] [Google Scholar]
- Jiang H, Abel PW, Toews ML, Deng C, Casale TB, Xie Y et al. (2010). Phosphoinositide 3‐kinase gamma regulates airway smooth muscle contraction by modulating calcium oscillations. J Pharmacol Exp Ther 334: 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Xie Y, Abel PW, Toews ML, Townley RG, Casale TB et al. (2012). Targeting phosphoinositide 3‐kinase gamma in airway smooth muscle cells to suppress interleukin‐13‐induced mouse airway hyperresponsiveness. J Pharmacol Exp Ther 342: 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John AE, Zhu YM, Brightling CE, Pang L, Knox AJ (2009). Human airway smooth muscle cells from asthmatic individuals have CXCL8 hypersecretion due to increased NF‐kappa B p65, C/EBP beta, and RNA polymerase II binding to the CXCL8 promoter. J Immunol 183: 4682–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude JA, Tirumurugaan KG, Kang BN, Panettieri RA, Walseth TF, Kannan MS (2012). Regulation of CD38 expression in human airway smooth muscle cells: role of class I phosphatidylinositol 3 kinases. Am J Respir Cell Mol Biol 47: 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraan J, Koeter GH, dv Mark TW, Sluiter HJ, de Vries K (1985). Changes in bronchial hyperreactivity induced by 4 weeks of treatment with antiasthmatic drugs in patients with allergic asthma: a comparison between budesonide and terbutaline. J Allergy Clin Immunol 76: 628–636. [DOI] [PubMed] [Google Scholar]
- Lan B, Deng L, Donovan GM, Chin LY, Syyong HT, Wang L et al. (2015). Force maintenance and myosin filament assembly regulated by Rho‐kinase in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 308: L1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ et al. (2011). CAL‐101, a p110delta selective phosphatidylinositol‐3‐kinase inhibitor for the treatment of B‐cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117: 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte JC, Moore PE, Baraldo S, Jouvin MH, Church TL, Schwartzman IN et al. (2001). Direct effects of interleukin‐13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am J Respir Crit Care Med 164: 141–148. [DOI] [PubMed] [Google Scholar]
- Lee KS, Lee HK, Hayflick JS, Lee YC, Puri KD (2006). Inhibition of phosphoinositide 3‐kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J 20: 455–465. [DOI] [PubMed] [Google Scholar]
- Leguillette R, Lauzon AM (2008). Molecular mechanics of smooth muscle contractile proteins in airway hyperresponsiveness and asthma. Proc Am Thorac Soc 5: 40–46. [DOI] [PubMed] [Google Scholar]
- Ma X, Cheng Z, Kong H, Wang Y, Unruh H, Stephens NL et al. (2002). Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol Lung Cell Mol Physiol 283: L1181–L1189. [DOI] [PubMed] [Google Scholar]
- McCrea KE, Hill SJ (1993). Salmeterol, a long‐acting beta 2‐adrenoceptor agonist mediating cyclic AMP accumulation in a neuronal cell line. Br J Pharmacol 110: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashed BF, Zhang T, Al‐Alwan M, Srinivasan G, Halayko AJ, Okkenhaug K et al. (2007). Role of the phosphoinositide 3‐kinase p110delta in generation of type 2 cytokine responses and allergic airway inflammation. Eur J Immunol 37: 416–424. [DOI] [PubMed] [Google Scholar]
- Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI (1989a). A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol 256: C329–C335. [DOI] [PubMed] [Google Scholar]
- Panettieri RA Jr, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI (1989b). A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol Cell Physiol 256: C329–C335. [DOI] [PubMed] [Google Scholar]
- Ringdal N, Derom E, Wahlin‐Boll E, Pauwels R (1998). Onset and duration of action of single doses of formoterol inhaled via Turbuhaler. Respir Med 92: 1017–1021. [DOI] [PubMed] [Google Scholar]
- Robinett KS, Koziol‐White CJ, Akoluk A, An SS, Panettieri RA Jr, Liggett SB (2014). Bitter taste receptor function in asthmatic and nonasthmatic human airway smooth muscle cells. Am J Respir Cell Mol Biol 50: 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Johnson PR, Borger P, Bihl MP, Rudiger JJ, King GG et al. (2004). Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth‐muscle cells. N Engl J Med 351: 560–574. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016) The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44:D1054‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Smolock EM, Marcel KN, Moreland RS (2004). Phosphatidylinositol 3‐kinase modulates vascular smooth muscle contraction by calcium and myosin light chain phosphorylation‐independent and ‐dependent pathways. Am J Physiol Heart Circ Physiol 286: H657–H666. [DOI] [PubMed] [Google Scholar]
- Sward K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP (2000). Inhibition of Rho‐associated kinase blocks agonist‐induced Ca2+ sensitization of myosin phosphorylation and force in guinea‐pig ileum. J Physiol 522 (Pt 1): 33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Ito W, Tanabe M, Ueki S, Kato H, Kihara J et al. (2009). Allergic airway hyperresponsiveness, inflammation, and remodeling do not develop in phosphoinositide 3‐kinase gamma‐deficient mice. J Allergy Clin Immunol 123: 805–812. [DOI] [PubMed] [Google Scholar]
- Townley RG (2007). Interleukin 13 and the beta‐adrenergic blockade theory of asthma revisited 40 years later. Ann Allergy Asthma Immunol 99: 215–224. [DOI] [PubMed] [Google Scholar]
- Trian T, Burgess JK, Niimi K, Moir LM, Ge Q, Berger P et al. (2011). beta2‐Agonist induced cAMP is decreased in asthmatic airway smooth muscle due to increased PDE4D. PLoS One 6: e20000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng P, Pushkarsky I, Di Carlo D (2014). Metallization and biopatterning on ultra‐flexible substrates via dextran sacrificial layers. PLoS One 9: e106091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijverberg SJ, Hilvering B, Raaijmakers JA, Lammers JW, Maitland‐van der Zee AH, Koenderman L (2013). Clinical utility of asthma biomarkers: from bench to bedside. Biologics: Targets & Therapy 7: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF (1994). A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 269: 5241–5248. [PubMed] [Google Scholar]
- Wang IY, Bai Y, Sanderson MJ, Sneyd J (2010). A mathematical analysis of agonist‐ and KCl‐induced Ca(2+) oscillations in mouse airway smooth muscle cells. Biophys J 98: 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yoshioka K, Azam MA, Takuwa N, Sakurada S, Kayaba Y et al. (2006). Class II phosphoinositide 3‐kinase alpha‐isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem J 394: 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Sugimoto N, Takuwa N, Takuwa Y (2007). Essential role for class II phosphoinositide 3‐kinase alpha‐isoform in Ca2 + −induced, Rho‐ and Rho kinase‐dependent regulation of myosin phosphatase and contraction in isolated vascular smooth muscle cells. Mol Pharmacol 71: 912–920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Figure S1 Time course of airway dilation to CAL‐101 and Y27632. Airways in lung slices were preconstricted to carbachol, then dilated to CAL‐101 or Y27632 (60 μM), 0–30 min. Data is representative of mean ± SEM for 4–15 slices/condition.
Figure S2 PI3K p110δ (A) and γ (B) mRNA expression in airway smooth muscle assessed by RNAseq. PI3K inhibition has little effect on expression of total protein levels (C) of MYPT1 (D), Akt (E), and MLC (F). Data is representative of n = 5–8 (A and B) or n = 2 (C‐F) HASM cell lines.
Figure S3 PI3K inhibition has little effect on agonist‐induced calcium mobilization. HASM were stimulated with 10 μM carbachol following a 30 min pretreatment with vehicle (DMSO), CAL‐101 or LY294002 (1 μM) and single cell calcium transients measured using Fluo‐8 calcium sensing dye. (A) Time course tracings of calcium flux. Both peak calcium (B) and AUC (C) for each treatment were plotted as mean ± SD.
Figure S4 siRNA knockdown of PI3K p110γ had little effect on carbachol‐induced (10 min, 10 μM) phosphorylation of MLC (pMLC), MYPT1, or Akt in HASM cells. (A) Immunoblot analyses of HASM cells transfected with PI3K p110γ siRNA or scrambled siRNA. 3 (B) Slight inhibition of carbachol‐induced phosphorylation of Akt (pAkt) and MYPT1 (pMYPT1) in HASM cells by PI3K p110δ siRNA. Data are representative of five independent experiments (n = 5, mean + SEM); *P < 0.05.
Figure S5 PI3K inhibition has little effect alone to induce or augment cAMP levels. Data are representative of 3 donors and each condition in triplicate, with bars representing mean ± SEM with a P value of <0.05 considered significant.
Table S1 Donor demographics of asthma and non‐asthma HASM cell donors.
Table S2 Donor demographics of non‐asthma hPCLS donors.
Table S3 Pharmacologic properties of the inhibitors utilized in this study. In vitro kinase assay results, IC50 values in cell based assays, and the corresponding literature citations are shown for CAL‐101, LY294002, and Y27632.
Movie S1 Formoterol‐induced relaxation of bradykinin contracted HASM. Link: https://youtu.be/51oJeQ73LEA.
Movie S2 CAL‐101‐induced relaxation of bradykinin contracted HASM. Link: https://youtu.be/2H3DKMqI‐GU.