

Abstract
Brain-derived neurotrophic factor (BDNF) is essential for neuronal differentiation and survival. We know that BDNF levels decline in the brains of patients with Huntington's disease (HD), a neurodegenerative disease caused by the expression of mutant huntingtin protein (mHtt), and furthermore that administration of BDNF in HD mice is protective against HD neuropathology. BDNF is produced in neurons, but astrocytes are also an important source of BDNF in the brain. Nonetheless, whether mHtt affects astrocytic BDNF in the HD brain remains unknown. Here we investigated astrocytes from HD140Q knock-in mice and uncovered evidence that mHtt decreases BDNF secretion from astrocytes, which is mediated by exocytosis in astrocytes. Our results demonstrate that mHtt associates with Rab3a, a small GTPase localized on membranes of dense-core vesicles, and prevents GTP-Rab3a from binding to Rab3-GAP1, disrupting the conversion of GTP-Rab3a into GDP-Rab3a and thus impairing the docking of BDNF vesicles on plasma membranes of astrocytes. Importantly, overexpression of Rab3a rescues impaired BDNF vesicle docking and secretion from HD astrocytes. Moreover, ATP release and the number of ATP-containing dense-core vesicles docking are decreased in HD astrocytes, suggesting that the exocytosis of dense-core vesicles is impaired by mHtt in HD astrocytes. Further, Rab3a overexpression reduces reactive astrocytes in the striatum of HD140Q knock-in mice. Our results indicate that compromised exocytosis of BDNF in HD astrocytes contributes to the decreased BDNF levels in HD brains and underscores the importance of improving glial function in the treatment of HD.
SIGNIFICANCE STATEMENT Huntington's disease (HD) is an inherited neurodegenerative disorder that affects one in every 10,000 Americans. To date, there is no effective treatment for HD, in part because the pathogenic mechanism driving the disease is not fully understood. The dysfunction of astrocytes is known to contribute to the pathogenesis of HD. One important role of astrocytes is to synthesize and release brain-derived neurotrophic factor (BDNF), which is vital for neuronal survival, development, and function. We found that mutant huntingtin protein (mHtt) at the endogenous level decreases BDNF secretion from astrocytes by disrupting the conversion of GTP-Rab3a into GDP-Rab3a and that overexpressing Rab3a can rescue this deficient BDNF release and early neuropathology in HD knock-in mouse brain. Our study suggests that astrocytic Rab3a is a potential therapeutic target for HD treatment.
Keywords: BDNF, exocytosis, glial cell, Huntington's disease, Rab3
Introduction
Huntington's disease (HD) is characterized by selective neurodegeneration, although the mutant HD protein mHtt is expressed in both neuronal and glial cells throughout the brain (Ross and Tabrizi, 2011; Saxena and Caroni, 2011). Despite the lower vulnerability of glial cells in HD brains, emerging evidence shows that mHtt affects the function of astrocytes. For example, mHtt decreases the levels of both EAAT2 (GLT-1) and Kir4.1 in astrocytes, which consequently increases neuronal excitotoxicity (Shin et al., 2005; Bradford et al., 2009; Tong et al., 2014). The consensus has been that dysfunctional astrocytes contribute to the pathogenesis of HD (Chan and Surmeier, 2014).
Brain-derived neurotrophic factor (BDNF), a neurotrophic factor stored in dense-core vesicles, is decreased in the brains of HD patients, as well as animal models (Zuccato et al., 2001; Hermel et al., 2004). The causal relationship between the reduction of BDNF and neuropathology in HD has been proposed (Lu et al., 2005; Zuccato and Cattaneo, 2007) and is supported by the finding that increasing BDNF levels ameliorates the neuropathology and phenotypes of HD animals (Zuccato et al., 2005; Giralt et al., 2011). The decreased BDNF in neuronal cells was found to be due to the effect of mHtt on the transcriptional expression of BDNF (Zuccato et al., 2001) and its axonal transport from cortical neurons to the striatum (Gauthier et al., 2004), implicating neuronal BDNF dysfunction in HD pathogenesis. Astrocytes are also an important source of BDNF (Miyamoto et al., 2015). BDNF is released from astrocytes via exocytosis to support the neighboring neurons and regulate their function (Wu et al., 2008; Parpura and Zorec, 2010; Quesseveur et al., 2013). However, whether mHtt affects astrocytic BDNF production or secretion in the HD brain remains unknown. A previous study found that overexpressing mHtt in cultured astrocytes decreased BDNF levels by sequestering BDNF transcriptional factors into mHtt aggregates (Wang et al., 2012). Considering that very few mHtt aggregates are formed in astrocytes in the HD brains that express mHtt at the endogenous level (Shin et al., 2005), we wanted to investigate whether mHtt at the endogenous level can affect BDNF production or secretion in astrocytes.
We used both full-length HD140Q knock-in (KI) and GFAP-Htt transgenic (TG) mouse models to investigate the effect of mHtt on astrocytic BDNF. In these mouse models, full-length mHtt with 140Q is expressed at the endogenous level in knock-in mice (Menalled et al., 2003), and N-terminal Htt (1–208 aa) with 160Q is expressed at levels similar to endogenous Htt (Bradford et al., 2009). Using primary astrocytes and brain-slice cultures, we found that secreted BDNF from HD astrocytes is reduced, and that this reduction stems from impaired docking of BDNF vesicles attributable to an abnormal association between mHtt and Rab3a in HD astrocytes. More importantly, overexpression of Rab3a improves docking of BDNF-containing vesicles and BDNF release from HD astrocytes. Moreover, overexpression of Rab3a ameliorated reactive astrocytes in the striatum of full-length HD140Q knock-in mice, which is an early HD pathology. Our findings suggest a new mechanism behind the reduced BDNF levels in HD brains and indicate the importance of improving the function of astrocytes in the treatment of HD.
Materials and Methods
Animals.
KI mice were kindly provided by Dr. Michael Levine of the University of California, Los Angeles (Hickey et al., 2008). TG mice with 160Q were generated previously by our laboratory (Bradford et al., 2009). Mice were maintained at the Emory University animal facility. Both male and female pups from these mice were used for primary cultures. Male adult mice of different ages were used for viral injection and brain-slice preparation. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of Emory University (permit number 2002557).
Antibodies and reagents.
Antibodies used were anti-expanded polyQ (1C2; Millipore, MAB1574), anti-Htt (mEM48), anti-Rab3a (BD Biosciences, 610379), anti-GTP-Rab3a (NewEast Biosciences, 26920), anti-Rab3-GAP (Proteintech, 21663-1-AP), anti-V5 (Life Technologies, 46-0705), anti-TrkB (Cell Signaling Technology, 80E3), anti-phospho-TrkB (Epitomics, 2149-1), anti-BDNF (Santa Cruz Biotechnology, sc-546), anti-actin (Sigma-Aldrich, A5060), anti-GFAP (Millipore, MAB360), and anti-NeuN (Millipore, ABN78). Secondary antibodies were HRP-labeled donkey anti-mouse, donkey anti-rabbit, donkey anti-mouse Alexa Fluor 488 or 594, and donkey anti-rabbit Alexa Fluor 488 or 594 from Jackson ImmunoResearch. Proteinase inhibitor mixture, phorbol 12-myristate 13-acetate (PMA), ATP Bioluminescent Assay Kit, and quinacrine dihydrochloride were purchased from Sigma-Aldrich. Rab3-V5 adenovirus was purchased from SignaGen Laboratories (SL174810). The Glutamate Assay Kit was purchased from BioVision.
Primary cell cultures.
Both male and female brains of postnatal (day 1–3) murine pups were used for culturing cortical astrocytes. Following dissection, the cortex was subjected to 0.3 mg/ml papain digestion. Cell suspension was filtered through 70 μm nylon cell strainers (Fisher Scientific). Microglia and oligodendrocytes were removed from cultures at DIV 14 by shaking. Remaining cells were detached with 0.25% trypsin and plated for the following experiments. For cortical neuron cultures, cortical neurons were prepared from both male and female postnatal day 0 murine pups. The cortex was digested with 0.3 mg/ml papain. Cell suspension was filtered through 40 μm nylon cell strainers (Fisher Scientific) to remove debris. Neurons were plated at a density of 1 × 106 on poly-d-lysine-coated six-well plates and cultured in Neurobasal-A medium supplemented with B27 and glutamine (Invitrogen).
Preparation of astrocyte-conditioned medium.
Primary astrocytes (DIV 25) were cultured at a density of 2 × 106 in six-well plates and treated with 28 mm KCl for 30 min at 37°C. The culture medium was collected for measuring released BDNF via ELISA.
ELISA.
Levels of BDNF were determined by a BDNF ImmunoAssay System (Promega) following the manufacturer's protocol. Briefly, samples or standards (100 μl) were added to 96-well plates, incubated at 4°C overnight, and washed extensively, followed by a 2 h incubation with the anti-BDNF antibody and a 1 h incubation with anti-IgY HRP conjugate plus substrate for signal development. The absorbance was recorded at 450 nm on a plate reader (Microplate Reader, BioTek). The amount of BDNF in each sample was calculated based on the standard curve prepared in the same experiment.
Western blotting.
Primary cultures or brain tissues were homogenized in ice-cold NP-40 buffer containing a protease inhibitor mixture (Thermo Fisher Scientific) and 100 μm PMSF. Samples were boiled for 5 min in SDS/β-mercaptoethanol protein loading dye and run on 4–12% Tris-glycine gels purchased from Invitrogen (catalog #EC60385). Proteins were transferred to a nitrocellulose membrane in Tris-glycine buffer. After blocking, blots were probed with Rab3a or other primary antibodies overnight. The Western blots were developed using the ECL Prime Chemiluminescence kit (GE Healthcare).
Glutamate measurement.
Glutamate release from astrocytes was determined by a Glutamate Assay Kit (BioVision) following the manufacturer's protocol. Briefly, each culture medium was diluted in the assay buffer. Samples or standards (50 μl) were added to 96-well plates. Reaction Mix (100 μl) was added to each well containing the glutamate standard and test samples. Reactions were incubated for 30 min at 37°C and protected from light. Measured OD was at 450 nm in a microplate reader (BioTek). The amount of glutamate in each sample was calculated based on the standard curve prepared in the same experiment.
qRT-PCR.
Total RNA was isolated from WT, TG, and KI astrocytes. Reverse transcription reactions were performed with 1.5 μg of total RNA using the Superscript III First-Strand Synthesis System (Invitrogen). One microliter of cDNA was combined with 10 μl SYBR Select Master Mix (Applied Biosystems) and 1 μl of each primer in a 20 μl reaction. The reaction was performed in a thermal cycler (Eppendorf, RealPlex Mastercycler).
Knockdown assay.
Rab3a siRNA duplexes and negative control siRNA were purchased from OriGene (SR402766). The following combination of oligonucleotides was used to target the Rab3a gene: CGACUAUAUGUUCAAGAUCCUGATC, GGAGUCAUUUAAUGCAGUGCAGGAC, CGCAGUCCUUGACAUUAAAGAGAAT.
Stereotaxic injection of virus.
Male adult mice were used for Rab3-V5 adenoviral and control GFP adenoviral injections (TG, n = 4; WT, n = 4; KI, n = 4). Heads of the animals were placed and fixed in a stereotaxic frame (David Kopf Instruments, Model 1900) equipped with a digital manipulator and a UMP3–1 Ultra pump. The mice were kept deeply anesthetized as assessed by monitoring pinch withdrawal and respiration rate. Animals were injected in the striatum (0.6 mm anterior to bregma, 2.0 mm lateral to the midline, 3.5 mm ventral to dura). The injections were performed at a rate of 0.2 μl/ml. The needle was left in place for 10 min after each injection to minimize the upward flow of viral solution after raising the needle. Rab3a-V5 was allowed to be expressed for 21–30 d in vivo before slicing the brains.
Preparation of brain slices.
Brains from male mice were immersed in chilled artificial CSF (ACSF), and then were cut with the vibratome into 250 μm coronal slices containing striatum (WT, n = 4; TG, n = 4; N171-82Q, n = 4). To stimulate BDNF release from the brain slices, 56 mm KCl was added to the ACSF.
Rab3 GTPase activity assay.
GST-Rab3a was loaded with 20 μCi of [γ-32P]-GTP (5000 Ci/mmol; GE Healthcare Pharmacia Biotech) in 40 μl of loading buffer (20 mm Tris, pH 7.6, 5 mm EDTA, and 0.1 mm DTT) for 15 min at room temperature. The loading reaction was stopped by adding 2 μl of 0.4 m MgCl2. Five microliters of preloaded GTPase was diluted in assay buffer (20 mm Tris, pH 7.6, 1 mm GTP, 1 mg ml−1 BSA, and 0.1 mm DTT) to yield a final volume of 50 μl. The total amount of [γ-32P]-GTP-loaded GTPase was measured by removing 5 μl of the loading sample (t0) into 1 ml of ice-cold dilution buffer (50 mm Tris, pH 7.6, 50 mm NaCl, 5 mm MgCl2). WT or KI astrocyte lysates without Rab3-GAP1 were then added to the assay and incubated with [γ-32P]-GTP-labeled GST-Rab3A for various periods of time at room temperature. Rab3-GAP1 in astrocyte lysate was immunoprecipitated by Rab3-GAP1 antibody. After 30, 60, and 90 min, 5 μl of sample was removed and diluted into 1 ml of ice-cold dilution buffer. The samples were filtered through nitrocellulose filters (Millipore) to trap the γ-32P-labeled GTPase, and the radioactivity retained on the filters was determined by scintillation counting.
Total internal reflection fluorescence microscopy.
A total internal reflection fluorescence (TIRF) microscope (GE Healthcare Life Sciences, GE DeltaVision OMX Blaze) was used to study vesicles docking near the plasma membrane. Astrocytes were plated onto poly-l-lysine-coated coverslips and then transfected with BDNF fused to RFP. After 48 h of transfection, docking of BDNF-RFP vesicles was examined by TIRF microscope, which allows selective imaging of a ∼100 nm region beneath the plasma membrane. Quinacrine dihydrochloride often serves as a fluorescent marker for intracellular ATP-enriched vesicles (Akopova et al., 2012). Living astrocytes were treated with PMA for 15 min at 37°C. Quinacrine staining was performed by incubating living astrocytes with 1 μm quinacrine dihydrochloride for 15 min at room temperature. Quinacrine fluorescence in living astrocytes after stimulation was examined with a Nikon A1R confocal microscope. To examine ATP-containing vesicles, astrocytes were treated with quinacrine dihydrochloride for 15 min at room temperature. Images were acquired by the TIRF microscope every 200 ms. The number of plasma membrane-docked vesicles was counted at the entire cell surface in the evanescent field by Fiji.
Immunoprecipitation.
Cells were harvested and lysed in ice-cold 0.5% Triton X-100/PBS solution with protease inhibitor mixture and 100 μm PMSF on ice. The lysates were centrifuged at 16,000 × g for 15 min. Protein concentrations were measured with BCA assay (Thermo Fisher Scientific). A total of 300 μg of samples were precleared with protein A agarose beads (Sigma-Aldrich), and huntingtin and Rab3a-V5 proteins were immunoprecipitated with 1C2 and anti-V5 antibodies, respectively, at 4°C overnight. Protein A agarose beads were added to capture the immunoprecipitates for 1 h at 4°C. Ice-cold lysis buffer was used to wash beads three times. Proteins from the immunoprecipitates and inputs were subjected to Western blotting.
Immunofluorescence staining.
Cultured astrocytes were fixed with 4% paraformaldehyde for 8–10 min. Mouse brains were sliced at 10 μm thickness with a cryostat at −20°C, mounted onto gelatin-coated sides, and fixed with 4% paraformaldehyde for 10 min. Fixed samples were blocked with 3% BSA/0.2% Triton X-100 for 30 min at room temperature. Following incubation of fixed samples with primary antibodies at 4°C overnight and washes, fluoroconjugated secondary antibodies and Hoechst nuclear dye were added to the samples for staining. Images were acquired with an Imager Z1 microscope. Quantitative analysis of GFAP staining was performed using the method in our previous studies (Yang et al., 2015). Briefly, ImageJ software was used to measure GFAP immunostaining intensity. Colored images obtained with a 63× objective were converted to eight-bit black-and-white images. The “Threshold” function was used to adjust the background to highlight GFAP-specific staining. The same threshold was applied to all images analyzed. Finally, the “Measure” function was used to quantify GFAP staining intensity in each image. Each group had 7–10 images per section and eight sections per group were examined.
ATP assay.
ATP levels in astrocyte culture medium were analyzed by an ATP bioluminescence assay kit (Sigma-Aldrich) and a luminometer (BioTek) according to the manufacturer's instructions. Briefly, WT, KI, and Rab3a-overexpressed KI astrocytes were treated with PMA for 15 min at 37°C. Each culture medium was harvested for ATP assay. Each sample was run in duplicate and assayed within 5–10 min of collection.
Statistical analyses.
Statistical analyses were performed with unpaired two-tailed Student's t tests. Results are expressed as means ± SEM. P value <0.05 was considered significant. Statistical significance level was set as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
mHtt at endogenous levels impairs BDNF secretion from astrocytes
To examine whether BDNF secretion from astrocytes is affected by mHtt in KI mice, we measured BDNF secretion by cultured astrocytes from WT, KI, and TG mice. TG mice were used because the expression of N-terminal mHtt under the control of the GFAP promoter in these mice is astrocyte-specific (Bradford et al., 2009). Using ELISA to directly detect BDNF in the cell culture medium following 28 mm KCl stimulation, we found that primary astrocytes from either TG (n = 6 independent experiments, p = 1.6381E-05) or KI (n = 4 independent experiments, p = 0.0208) mice release less BDNF into culture medium than do astrocytes from WT mice (Fig. 1A). To verify whether mHtt affects BDNF release in astrocytes in the presence of neuronal cells, we also measured BDNF release from brain slices of TG mice in which mHtt expression is restricted to astrocytes. We found that brain slices from TG mice released less BDNF than slices from WT mice (Fig. 1A; n = 3 independent experiments, p = 0.0449), which also suggests that the expression of mHtt in astrocytes affects BDNF secretion. Nevertheless, BDNF release from brain slices of N171-82Q mice that express N-terminal mHtt primarily in neurons is unchanged compared with WT brain slices, further indicating that mHtt selectively impairs BDNF secretion from astrocytes (Fig. 1A; n = 3 independent experiments, p = 0.1489).
Figure 1.

BDNF secretion from HD astrocytes is reduced. A, ELISA assay showed that cultured primary astrocytes from TG (Student's t test, n = 6 independent experiments, p = 1.6381E-05) and KI (Student's t test, n = 4 independent experiments, p = 0.0208) mice release less BDNF than astrocytes from WT mice. ELISA results also indicated that BDNF secretion is decreased from brain slices of TG mice compared with WT mice (Student's t test, n = 3 independent experiments, p = 0.0449). However, BDNF secretion is unchanged from brain slices of N171-82Q mice compared with WT mice (Student's t test, n = 3 independent experiments, p = 0.1489). B, WT neurons were treated with ACM containing released BDNF from WT or KI astrocytes, and then analyzed via Western blotting to examine p-TrkB levels. p-TrkB is decreased in KI ACM-treated neurons, confirming decreased BDNF in KI ACM. C, Glutamate measurement assay showing no significant reduction in glutamate release in cultured astrocytes from TG mice (Student's t test, n = 5 independent experiments, p = 0.1027) or KI mice (Student's t test, n = 6 independent experiments, p = 0.3647) compared with WT astrocytes. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
To show the functional consequences of reduced BDNF release, we treated WT neurons with either WT or KI astrocyte-conditioned medium (ACM) for 10 min to induce phosphorylation of TrkB, as binding of BDNF to TrkB receptors leads to TrkB phosphorylation (p-TrkB; Duman and Voleti, 2012), so the extent of p-TrkB reflects BDNF levels in the ACM. Consistently, we found that neurons treated with KI ACM showed reduced p-TrkB levels (p-TrkB/actin) compared with those treated with WT ACM (Fig. 1B; n = 4 independent experiments, p = 0.0157). This result further supports our observation that BDNF levels are reduced in KI ACM compared with WT ACM. To verify that mHtt specifically impairs dense-core vesicle release in astrocytes, we also measured glutamate release from astrocytes. This glutamate is stored in small synaptic-like clear-core vesicles. The glutamate measurement assay showed no significantly decreased levels of glutamate in TG astrocyte culture medium (Student's t test, n = 5 independent experiments, p = 0.1027) or in KI astrocyte culture medium (Student's t test, n = 6 independent experiments, p = 0.3647) compared with WT astrocyte culture medium (Fig. 1C). Collectively, our results indicate that mHtt at endogenous levels impairs BDNF secretion from astrocytes.
mHtt does not affect the generation of BDNF in HD astrocytes
To explore the mechanism behind the decreased BDNF secretion from HD astrocytes, we first examined BDNF production in cultured astrocytes, as previous studies suggested that overexpressing mHtt influenced BDNF transcription and translation in astrocytes (Wang et al., 2012). We performed a qRT-PCR assay using BDNF-specific primers and found that levels of different BDNF mRNA isoforms are not significantly changed in TG or KI astrocytes compared with WT astrocytes (Fig. 2A,B). Moreover, using Western blotting and BDNF-specific antibody, we found no significant changes in either mature BDNF (mBDNF) or precursor of BDNF in cultured astrocytes from TG or KI mice (Fig. 2C,D). These results suggest that mHtt at the endogenous level does not affect BDNF mRNA or protein levels.
Figure 2.

Transcription or translation of BDNF is not changed in the HD astrocytes. A, B, qRT-PCR results revealing no significant reduction in BDNF mRNA levels in cultured astrocytes from TG mice (A, Student's t test, n = 5 independent experiments; BDNF transcripts generated by different promoters are indicated: BDNF promoter I, p = 0.5844; BDNF promoter II, p = 0.3669; BDNF promoter III, p = 0.5765; BDNF promoter IV, p = 0.67; BDNF promoter pan, p = 0.2406) or KI mice (B, Student's t test, n ≥ 5 independent experiments; BDNF promoter I, p = 0.0688; BDNF promoter II, p = 0.2283; BDNF promoter III, p = 0.6892; BDNF promoter IV, p = 0.0809; BDNF promoter pan, p = 0.7733) compared with WT astrocytes. C, D, Western blotting revealing similar mature mBDNF and precursor of BDNF (pro-BDNF) levels in cultured astrocytes from TG (C), KI (D), and WT mice. Quantifying ratios of pro-BDNF or mBDNF to actin in TG astrocytes (C, Student's t test; pro-BDNF, n = 8 independent experiments, p = 0.6983; mBDNF, n = 8 independent experiments, p = 0.4087) and KI astrocytes (D, Student's t test; pro-BDNF, n = 8 independent experiments, p = 0.8801; mBDNF, n = 5 independent experiments, p = 0.6349). ns, Not significant.
mHtt binds to Rab3a in astrocytes
Since BDNF synthesis was unchanged, we reasoned that the decreased BDNF protein secretion might be due to decreased exocytosis of BDNF vesicles. Rab3 is a small GTPase comprising four isoforms (Rab3a–Rab3d; Takai et al., 1996). Rab3a, the isoform highly enriched in the brain, is localized on membranes of secretory vesicles, including synaptic vesicles and dense-core vesicles, and mediates the exocytosis of vesicles (Tsuboi and Fukuda, 2006). Although Rab3a is expressed in astrocytes (Maienschein et al., 1999), its function in astrocytes remains elusive. We used Rab3a siRNA to knock down Rab3a expression in cultured WT astrocytes and found that knocking down Rab3a results in the defective release of BDNF and ATP, another cargo in dense-core vesicles, from astrocytes (Fig. 3A,B; BDNF release, n = 3 independent experiments, p = 0.0353; ATP release, n = 5 independent experiments, p = 0.0142). These findings suggest that Rab3a plays an important role in dense-core vesicle-mediated secretion from astrocytes.
Figure 3.
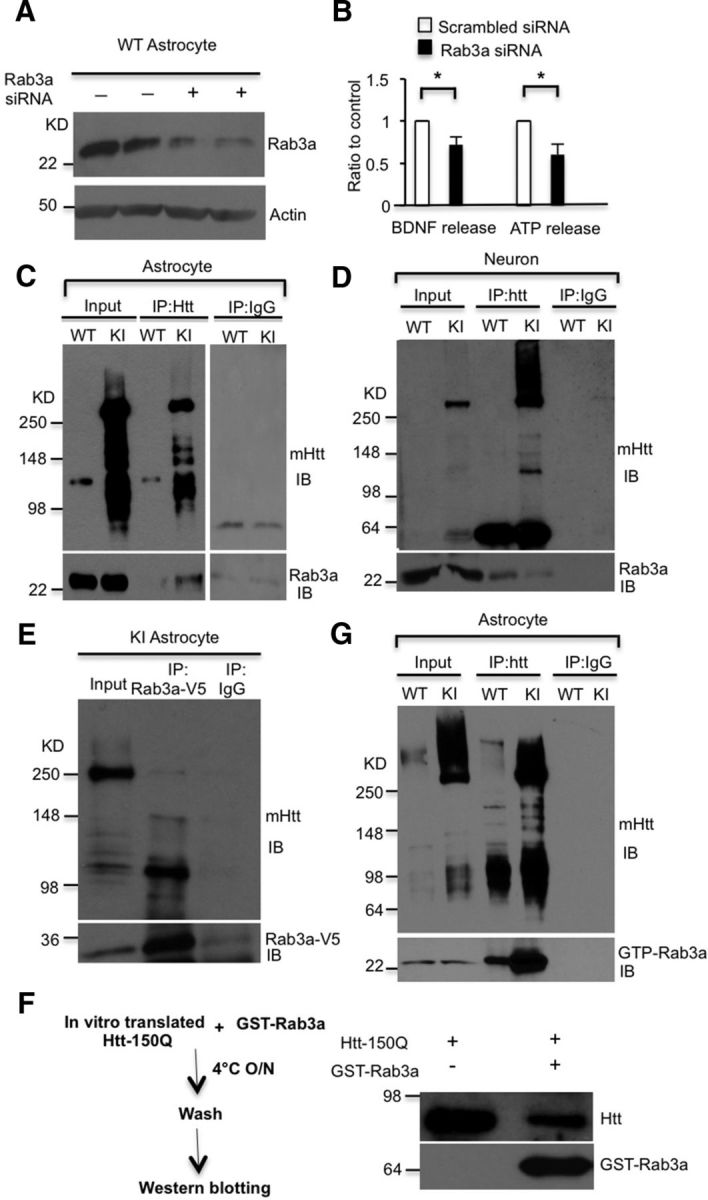
mHtt associates with Rab3a in astrocytes. A, Knocking down Rab3a via siRNA in WT astrocytes. B, ELISA results showing that downregulation of Rab3a inhibits the release of BDNF and ATP from WT astrocytes compared with scrambled siRNA transfected astrocytes (Student's t test: BDNF release, n = 3 independent experiments, p = 0.0353; ATP release, n = 5 independent experiments, p = 0.0142). C, D, Association of mHtt with Rab3a is detected in cultured KI astrocytes (C) but not in cultured KI neurons (D). Endogenous mHtt in KI astrocytes or KI neurons was immunoprecipitated by 1C2 antibody, and the immunoprecipitates were probed with antibody to Rab3a. Immunoprecipitation with IgG served as a control. E, Association of mHtt with Rab3a-V5 was detected in cultured KI astrocytes infected with Rab3a-V5 adenovirus. Rab3a was immunoprecipitated by anti-V5 antibody, and the immunoprecipitates were probed with antibodies to 1C2 to detect mHtt. Immunoprecipitation with IgG served as a control. F, In vitro binding assay showed that in vitro translated Htt (1–212 aa) with 150Q binds to purified GST-Rab3a. G, Binding of mHtt to GTP-Rab3a was found in cultured HD KI astrocytes. To immunoprecipitate endogenous mHtt in HD KI astrocytes, 1C2 antibody was used, and the immunoprecipitates were probed with the antibody specific to GTP-Rab3a. *p < 0.05.
Given that mHtt binds a variety of proteins to affect intracellular trafficking (Li and Li, 2004), we wanted to know whether mHtt binds Rab3a and affects its mediated BDNF secretion from astrocytes. To test this hypothesis, we performed a coimmunoprecipitation assay using an antibody (1C2) to selectively precipitate mHtt and found that mHtt could coprecipitate with Rab3a from cultured KI astrocyte lysates (Fig. 3C), but not from cultured KI neuronal lysates (Fig. 3D). These results indicate that mHtt specifically binds to Rab3a in astrocytes. Using an antibody to precipitate Rab3a, we noticed that more degraded mHtt than full-length mHtt was coprecipitated with Rab3a (Fig. 3E). This result suggests that N-terminal mHtt is likely to affect the function of Rab3a. To confirm the interaction of N-terminal mHtt with Rab3a, we in vitro synthesized N-terminal mHtt (1–212 aa) with 150Q and then incubate the product with purified GST-Rab3a. This in vitro binding assay also showed an association between mHtt and Rab3a (Fig. 3F). Since GTP-Rab3a is an active form of Rab3a for exocytosis, we immunoprecipitated mHtt from KI astrocytes and found it also interacts with GTP-Rab3a (Fig. 3G). Together, our results suggest that Rab3a plays a critical role in BDNF secretion in astrocytes, and the association between mHtt and Rab3a may contribute to the impaired BDNF release from HD astrocytes.
mHtt binds GTP-Rab3a to prevent its association with Rab3-GAP in astrocytes
The ability to associate/dissociate GTP and GDP is essential for the function of Rab3a in dense-core vesicle docking (van Weering et al., 2007). Given the increased binding of mHtt to GTP-Rab3a, we wanted to know whether mHtt disrupts the conversion of GTP-Rab3a into GDP-Rab3a (GTP/GDPRab3a exchange), which could result in altered levels of GTP-Rab3a. Using Western blotting with a GTP-Rab3a-specific antibody, we found that GTP-Rab3a protein levels were indeed increased in KI astrocytes (Fig. 4A), supporting the idea that mHtt inhibits the conversion of GTP-Rab3a. As Rab3-GAP binds GTP-Rab3a and then converts the active GTP bound form to the inactive GDP bound form (Burstein et al., 1993), we examined whether mHtt could reduce the association of GTP-Rab3a with Rab3-GAP1, the catalytic subunit of Rab3-GAP. Indeed, we found that the association between GTP-Rab3a and Rab3-GAP1 is significantly reduced in astrocytes from KI mice compared with WT astrocytes (Fig. 4B,C; n = 4 independent experiments, p = 0.0041). In addition, we observed the same result in protein lysates from the KI corpus callosum, a brain region enriched in glial cells (Fig. 4B,C; n = 4 independent experiments, p = 0.0106). Western blotting using a Rab3-GAP1-specific antibody showed that there is no difference in Rab3-GAP1 levels between WT and KI astrocytes, ruling out the possibility that the increased GTP-Rab3a is caused by a reduction of Rab3-GAP1 (Fig. 4D). We also used an in vitro assay to measure GTP-Rab3a activity. In this assay, Rab3-GAP1 was removed from WT and KI astrocyte lysates by immunodepletion to examine the direct effect of mHtt in astrocyte lysates on purified Rab3a for its GTPase activity to incorporate [γ-32P]-GTP (Fig. 4E). The result revealed no difference between WT and KI lysates, suggesting that mHtt in KI lysates does not directly affect the GTPase activity of Rab3a, but disrupts GTP/GDP-Rab3a exchange by interfering with the association between GTP-Rab3a and Rab3-GAP1.
Figure 4.

Reduced association between Rab3-GAP1 and Rab3a by mHtt results in increased GTP-Rab3a in HD astrocytes. A, GTP-Rab3a protein levels were increased in cultured KI astrocytes (Student's t test, n = 3 independent experiments, p = 0.0402). B, C, Association between GTP-Rab3a with Rab3-GAP1 is decreased in both cultured KI astrocytes (C, Student's t test, n = 4 independent experiments, p = 0.0041) and in the corpus callosum of KI mice (C, Student's t test, n = 4 independent experiments, p = 0.0106). D, Rab3-GAP1 levels did not change in KI astrocytes. E, GTPase activity of purified GST-Rab3a was not affected by binding to mHtt (Student's t test, n = 4 independent experiments; 30 min, p = 0.4807; 60 min, p = 0.9353; 90 min, p = 0.4071). *p < 0.05, **p < 0.01; ns, not significant.
Overexpression of Rab3a in HD astrocytes rescued deficits of BDNF secretion
If mHtt binds Rab3a to inhibit GTP/GDP-Rab3a exchange, overexpression of Rab3a should antagonize this inhibition and the related decrease in BDNF release. We therefore generated adenoviral vector to overexpress Rab3a-V5 in cultured astrocytes from KI mice. Immunofluorescent staining and Western blotting of these cells verified the expression of Rab3a-V5 (Fig. 5A,B). Using stereotaxic techniques, we injected Rab3a-V5 adenovirus into the striatum of TG mice and confirmed the expression of Rab3a-V5 via Western blotting (Fig. 5C). It is important to see whether overexpressed Rab3a could increase BDNF release in HD astrocytes, so we isolated mouse brain slices 30 d after stereotaxic injection. TG mouse brain slices containing the striatum area were prepared and incubated in ACSF, immediately followed by stimulation with 56 mm KCl for 30 min to trigger BDNF release, and the ACSF in the culture dish was harvested and used for ELISA (Fig. 5D). Similar to cultured KI astrocytes (n = 3 independent experiments, p = 0.0056), the BDNF secretion from TG brain slices is also increased after viral Rab3a-V5 injection (n = 3 independent experiments, p = 0.0464) compared with control virus-infected TG mice (Fig. 5E). Moreover, this increased BDNF release is further supported by the increased levels of p-TrkB found in cultured WT neurons incubated with ACM from Rab3a-overexpressing KI astrocytes (Fig. 5F; n = 5 independent experiments, p = 0.0292). These results demonstrated that defective BDNF secretion from HD astrocytes could be rescued by overexpressing Rab3a.
Figure 5.

Overexpression of Rab3a rescues defective release of BDNF from HD astrocytes. A, B, Overexpression of Rab3a in KI astrocytes by adenovirus infection was confirmed by immunostaining (A) and Western blotting (B). C, Rab3a was overexpressed in the striatum of TG mice by injecting Rab3a-V5 adenovirus into the striatum. D, E, ELISA results indicated that BDNF secretion was increased from cultured KI astrocytes infected with Rab3a-V5 adenovirus (Student's t test, n = 3 independent experiments, p = 0.0056) or from Rab3a-V5 adenovirus-injected brain slices containing striatum of TG mice (Student's t test, n = 3 independent experiments, p = 0.0464). F, WT neurons were treated with ACM containing released BDNF from KI or Rab3a-overexpressed KI astrocytes. p-TrkB levels are increased in neurons treated with Rab3a-overexpressed KI ACM relative to those treated with KI ACM (Student's t test, n = 5 independent experiments, p = 0.0292). *p < 0.05, **p < 0.01; ns, not significant.
Overexpression of Rab3a rescued the defective release of ATP from HD astrocytes
Since BDNF is not the only cargo in dense-core vesicles, we also examined whether mHtt also impairs the release of ATP, which is stored in dense-core vesicles. Quinacrine often serves as a fluorescent marker for intracellular ATP-enriched vesicles. After PMA treatment, quinacrine staining was performed by incubating living cultured astrocytes with quinacrine for 15 min at room temperature. Quinacrine staining results showed that ATP levels are higher in KI astrocytes compared with WT astrocytes after stimulation, perhaps because the reduced release of ATP increased the intracellular accumulation of ATP. Importantly, these increased ATP levels are attenuated by overexpression of Rab3a in KI astrocytes (Fig. 6A,B; WT vs KI: WT, n = 12 cells; KI, n = 14 cells, p = 0.0204; KI vs KI+Rab3a-V5: KI, n = 14 cells; KI+Rab3a-V5, n = 15 cells, p = 0.0068). We next measured the released ATP in culture medium after stimulation. ATP release was decreased in KI astrocyte culture medium, and overexpression of Rab3a could restore the release of ATP from KI astrocytes (Fig. 6C; n = 5 independent experiments; WT vs KI, p = 0.00006; KI vs KI+Rab3a-V5, p = 0.0125). These results indicated that overexpressing Rab3a could also rescue the defective ATP release from HD astrocytes.
Figure 6.

Overexpression of Rab3a rescues the deficient release of ATP from HD astrocytes. A, Quinacrine staining was performed by incubating living cultured WT, KI, and Rab3a-overexpressed KI astrocytes for 15 min after treatment of PMA. B, The quantitative results showed that ATP levels are higher in KI astrocytes than in WT astrocytes after stimulation, and overexpression of Rab3a reduces ATP levels in astrocytes compared with KI astrocytes (Student's t test; WT vs KI: WT, n = 12 cells; KI, n = 14 cells, p = 0.0204; KI vs KI+Rab3a-V5: KI, n = 14 cells; KI+Rab3a-V5, n = 15 cells, p = 0.0068). C, Bioluminescence assay showed that ATP secretion was rescued from cultured KI astrocytes infected with Rab3a-V5 adenovirus (Student's t test, n = 5 independent experiments, WT vs KI, p = 0.00006; KI vs KI+Rab3a-V5, p = 0.0125). *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar, 10 μm.
Perturbed GTP/GDP-Rab3a exchange leads to impaired docking of dense-core vesicles in KI astrocytes
Disruption of GTP/GDP-Rab3a exchange is reported to cause defective docking of dense-core vesicles in mammalian chromaffin cells (van Weering et al., 2007). To explore whether the disrupted GTP/GDP-Rab3a exchange by mHtt influences docking of BDNF-containing dense-core vesicles in HD astrocytes, we used TIRF microscopy (TIRFM) to examine the vesicle docking in cultured astrocytes. We transfected BDNF-RFP into cultured WT and KI astrocytes and quantified BDNF-containing vesicles docking on astrocytic plasma membranes. The numbers of BDNF docking vesicles are significantly decreased in KI astrocytes (n = 16 cells, p = 0.0043) compared with WT astrocytes (n = 11 cells). Moreover, the impaired docking could be rescued by overexpressing Rab3a in KI astrocytes (n = 8 cells, p = 0.0275) compared with KI astrocytes (n = 16 cells; Fig. 7A,C). It is well known that Rab3a is a membrane protein of dense-core vesicles that carry not only BDNF but also other kinds of cargoes, such as ATP in astrocytes (Pangrsic et al., 2007). The disruption of BDNF vesicle docking implies that mHtt probably perturbs the docking of all dense-core vesicles in KI astrocytes. We performed fluorescent labeling of endogenous ATP with quinacrine dihydrochloride (Pangrsic et al., 2007); indeed, fewer docked ATP-containing vesicles were observed in KI (n = 13 cells, p = 0.0435) than in WT (n = 16 cells) astrocytes (Fig. 7B,D). Moreover, overexpressing Rab3a (n = 15 cells, p = 0.0226) could rescue the defective docking compared with KI astrocytes (n = 13 cells) without overexpression of Rab3a (Fig. 7B,D).
Figure 7.

Defective docking of BDNF-containing and ATP-containing vesicles are rescued by Rab3a overexpression in HD astrocytes. A–D, TIRFM revealing that the docking of BDNF-containing vesicles (A, C; Student's t test; WT vs KI: WT, n = 11 cells, KI, n = 16 cells, p = 0.0043; KI vs KI+Rab3a-V5: KI, n = 16 cells; KI+Rab3a-V5, n = 8 cells, p = 0.0275) and ATP-containing vesicles (B, D; Student's t test; WT vs KI: WT, n = 16 cells; KI, n = 13 cells, p = 0.0435; KI vs KI+Rab3a-V5: KI, n = 13 cells; KI+Rab3a-V5, n = 15 cells, p = 0.0226) are compromised in KI astrocytes, which can be improved by overexpressing Rab3a. *p < 0.05, **p < 0.01. Scale bar, 10 μm.
Overexpression of Rab3a reduces reactive astrocytes in the striatum of HD140Q knock-in mice
In HD KI mouse brains, the elevated staining of GFAP, an astrocyte marker, reflects reactive astrocytes in the absence of neuronal loss (Yu et al., 2003), which represents an early HD pathological event. To examine whether overexpression of Rab3a would decrease HD-related pathology in vivo, we injected Rab3a-V5 adenovirus into the striatum of HD140Q KI mice. This Rab3a-V5 adenovirus expresses Rab3a under the control of the CMV promoter. After 21 d, we performed immunofluorescent staining and observed the preferential transduction of Rab3a-V5 adenovirus into astrocytes (Fig. 8A), which is in agreement with previous findings that adenovirus preferentially infects glia in vivo (Iino et al., 2001; Yue et al., 2005). We then performed stereotaxic injection of Rab3a and GFP adenovirus into different sides of the striatum in the same animal (Fig. 8B). Compared with GFP adenovirus injection, immunofluorescent staining showed that GFAP expression level is significantly decreased in the Rab3a-V5 injection side (Fig. 8C). We also examined NeuN-positive neuronal cells and mutant Htt aggregates, but did not see differences between adenoviral Rab3a-V5 and GFP injection sides (Fig. 8C,D). Thus, overexpression of Rab3a could selectively decrease reactive astrocytes in the HD KI mouse model. Based on these findings, we propose that mHtt binds Rab3a to affect its GTP/GDP exchange and impairs the docking of dense-core vesicles in HD astrocytes, resulting in decreased release of BDNF (Fig. 9).
Figure 8.
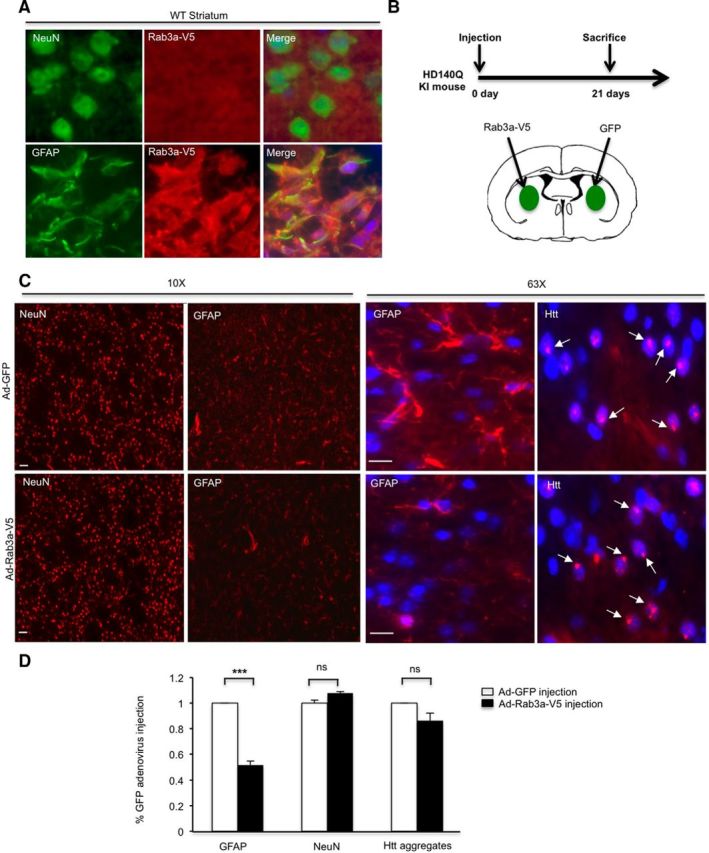
Overexpression of Rab3a reduces reactive astrocytes in the striatum of HD140Q KI mice A, Five-month-old WT mice were injected with Rab3a-V5 adenovirus into the left side of the striatum. After 21 d, immunofluorescent staining showed that Rab3a-V5 preferentially infected astrocytes in the striatum. B, Stereotaxic injection of 9-month-old HD140Q KI mice. C, Low-magnification (10×) micrographs showing NeuN and GFAP staining in adenoviral GFP and Rab3a-V5-injected striatum. High-magnification (63×) micrographs showing GFAP (red) or mutant Htt aggregates (arrows, red) staining in the merged images in which the nuclei are stained by Hoechst (blue). Scale bars, 10 μm. D, Quantitative analysis of the GFAP immunofluorescent density showing GFAP staining is significantly decreased in the Rab3a-V5-injection side compared with GFP-injection side (Student's t test, randomly selected 7–10 images per section, n = 8 sections per group, p = 7.51767E-13). The percentage of NeuN-positive cells and the number of mutant Htt aggregates per image are unchanged (Student's t test, n = 600 cells per group, NeuN-positive cells, p = 0.1016; aggregates number, p = 0.1511). ***p < 0.001; ns, not significant.
Figure 9.

Proposed model for the decreased dense-core vesicles released from HD astrocytes. GTP/GDP-Rab3 exchange is essential for docking of dense-core vesicles in astrocytes. In HD, the binding of mHtt to GTP-Rab3a keeps GTP-Rab3a from associating with Rab3-GAP1, disrupts GTP/GDP-Rab3 exchange, inhibits docking of dense-core vesicles, and results in the decreased release of secreted molecules, such as BDNF.
Discussion
Decreased BDNF levels in HD brains are believed to play a critical role in HD pathogenesis (Zuccato and Cattaneo, 2007). While previous studies have focused on the effect of mHtt on neuronal BDNF, our findings demonstrate that mHtt in astrocytes can also affect BDNF release, yielding new insights into the decreased BDNF levels in the HD brain.
Given the fact that >70% of cells in the striatum are astrocytes, the role of astrocytes in the striatum is vitally important. Astrocytes have a wide range of functions to support neuronal cells, including the reuptake of neurotransmitters and release of growth factors. mHtt was found to reduce the transcription of an important neurotransmitter transporter, glutamate transporter (GLT-1; Bradford et al., 2009) in astrocytes, which may contribute to the increased glutamate levels and excitotoxic neuronal death in the brains of HD mice (Espey et al., 1998; Liévens et al., 2001; Behrens et al., 2002; Hassel et al., 2008; Estrada-Sánchez et al., 2009; Bradford et al., 2010; Wolfram-Aduan et al., 2014). Our findings indicate that mHtt can also affect the secretion of neurotrophic factors of astrocytes. Astrocytic secretion of neurotrophic factors in supporting neuronal function and survival is a well studied subject (Quesseveur et al., 2013). BDNF is reported to play an important role in neuronal development and survival, and decreased BDNF levels in neurodegenerative diseases, including HD, Parkinson's disease, and Alzheimer's disease, have been proposed as a major player in neuronal death (Howells et al., 2000; Lee et al., 2005; Zuccato and Cattaneo, 2007). Elucidating the mechanism behind the decreased BDNF in these diseases could help us find ways to restore BDNF levels in diseased brains, providing new therapeutic targets for HD, as well as other neurodegenerative diseases.
In the present study, using primary cultures and mouse brain slice models from HD mice, we found that mHtt at the endogenous level was able to compromise BDNF secretion from HD astrocytes but not from HD neurons. This idea is supported by the fact that brain slices from the N171-82Q mouse model, in which N-terminal Htt with 82Q is primarily expressed in neuronal cells (Schilling et al., 1999), do not show defective BDNF release. Additionally, cultured astrocytes and brain slices from TG mice that express mHtt selectively in astrocytes show defective release of BDNF. This selectivity is likely due to the specific binding of mHtt to Rab3a in astrocytes, but not in neurons. Although it remains to be investigated how mHtt selectively binds Rab3a in astrocytes, we can speculate that astrocyte-specific molecules, proteins, and post-translational modifications may contribute to this selective binding, given that mHtt can associate with a large number of proteins and its function is regulated by complex post-translational modifications. Our studies also indicate that N-terminal mHtt fragments can affect the secretion of BDNF. This is because immunoprecipitation results show that N-terminal mHtt binds Rab3a and that TG mice expressing N-terminal mHtt show defective BDNF release from their astrocytes.
On the other hand, we found no changes in BDNF mRNA and protein levels in cultured astrocytes from astrocyte-specific transgenic HD or HD knock-in mice. This finding differs from an earlier report that showed that BDNF mRNA in cultured astrocytes was decreased by overexpressed N-terminal mHtt via adenoviral infection (Wang et al., 2012). This discrepancy may be due to different expression levels of mHtt. In our studies, we used astrocytes that express endogenous full-length mHtt or N-terminal mHtt at a level similar to endogenous Htt.
Rab3 undergoes functional conversion between active and inactive forms, which is essential for vesicle docking and exocytosis in cells (Darchen and Goud, 2000; Schlüter et al., 2004; Tsuboi and Fukuda, 2006; van Weering et al., 2007). It has been reported that Rab3 is located on the membrane of synaptic vesicles and dense-core vesicles regulating vesicle exocytosis in neurons; however, the location and function of Rab3 in astrocytes remains unknown. Although electron microscopy studies have shown that astrocytes contain both dense-core vesicles and synaptic-like clear-core vesicles, whether the exocytosis of these vesicles is mediated by different mechanisms is currently unknown (Parpura and Zorec, 2010). Despite this, we know that BDNF is packed in dense-core vesicles in astrocytes in a similar manner as in neurons, which requires GTP/GDP-Rab3a exchange (Parpura and Zorec, 2010). We found that the association of mHtt with Rab3a perturbs this exchange. First, we detected an increased level of GTP-Rab3a in KI astrocytes, which may be the result of the impaired GTP/GDP-Rab3a exchange. Second, our results demonstrate that the binding of mHtt to GTP-Rab3a inhibits the association of GTP-Rab3a with Rab3-GAP1. Previous studies showed that Rab27 and Rab3 sequentially regulate dense-core vesicle exocytosis in human sperm cells (Bustos et al., 2012). However, we did not find the association of mHtt with Rab27 by immunoprecipitation (data not shown). These results suggest that the disruption of the GTP/GDP-Rab3a exchange causes the deficient release of BDNF from HD astrocytes by binding mHtt to Rab3a. More importantly, we found that overexpressing Rab3a significantly improved vesicle docking and BDNF release from HD astrocytes.
Previous studies also showed that overexpression of Rab3a increases vesicle docking (Martelli et al., 2000), and conversely, disruption of GTP/GDP-Rab3 exchange inhibits vesicle docking (van Weering et al., 2007). Using TIRFM, we found a reduced number of BDNF-containing docking vesicles underneath the plasma membranes of KI astrocytes, which was increased through Rab3a overexpression. This result also supports the notion that disrupting conversion of Rab3-GTP to Rab3-GDP suppresses vesicle docking. Because BDNF is not a unique cargo for dense-core vesicles (Parpura and Zorec, 2010), we examined ATP release and the docking of ATP-containing dense-core vesicles. Consistently, we found defective ATP secretion and less docking of ATP-containing dense-core vesicles, which are also rescued by Rab3a overexpression. Therefore, our results indicate that mHtt impairs the docking of dense-core vesicles by intervening in the conversion of GTP/GDP-Rab3a in astrocytes. The defective docking of dense-core vesicles in astrocytes could impair the release of BDNF, ATP, and other molecules, which can synergistically contribute to HD pathology. In support of this idea, overexpressing Rab3a in the striatum in HD140Q knock-in mice can reduce reactive astrocytes, an early neuropathology event in HD KI mouse brains. We did not observe that overexpressed Rab3a influences mHtt aggregates, also suggesting that the rescue effect is due to the antagonistic effect on the abnormal association of mHtt with Rab3a rather than a direct effect on the expression of mutant Htt.
Substances like BDNF released from astrocytic dense-core vesicles are well known to play broad roles in neuronal survival and excitability, as well as astrocyte–neuron communication. Our findings provide new insight into the decreased secretion of BDNF from HD astrocytes in which mHtt is expressed at endogenous levels. Our findings also suggest that mHtt could affect other functions of astrocytes or the secretion of different molecules via a similar mechanism. Furthermore, our finding that Rab3a overexpression can rescue the defective docking, increase BDNF secretion, and ameliorate reactive astrocytes in vivo suggests that improving the exocytosis function of astrocytes could be beneficial to the treatment of HD.
Footnotes
This work was supported by the National Institutes of Health (AG19206 and NS041449 to X.-J.L.; AG031153 and NS045016 to S.L.). We thank the Integrated Cellular Imaging Core at Emory University for the use of imaging facilities and Cheryl Strauss for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Akopova I, Tatur S, Grygorczyk M, Luchowski R, Gryczynski I, Gryczynski Z, Borejdo J, Grygorczyk R. Imaging exocytosis of ATP-containing vesicles with TIRF microscopy in lung epithelial A549 cells. Purinergic Signal. 2012;8:59–70. doi: 10.1007/s11302-011-9259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, Li S, Li XJ. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem. 2010;285:10653–10661. doi: 10.1074/jbc.M109.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein ES, Brondyk WH, Macara IG, Kaibuchi K, Takai Y. Regulation of the GTPase cycle of the neuronally expressed Ras-like GTP-binding protein Rab3A. J Biol Chem. 1993;268:22247–22250. [PubMed] [Google Scholar]
- Bustos MA, Lucchesi O, Ruete MC, Mayorga LS, Tomes CN. Rab27 and Rab3 sequentially regulate human sperm dense-core granule exocytosis. Proc Natl Acad Sci U S A. 2012;109:E2057–E2066. doi: 10.1073/pnas.1121173109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Surmeier DJ. Astrocytes go awry in Huntington's disease. Nat Neurosci. 2014;17:641–642. doi: 10.1038/nn.3705. [DOI] [PubMed] [Google Scholar]
- Darchen F, Goud B. Multiple aspects of Rab protein action in the secretory pathway: focus on Rab3 and Rab6. Biochimie. 2000;82:375–384. doi: 10.1016/S0300-9084(00)00219-4. [DOI] [PubMed] [Google Scholar]
- Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35:47–56. doi: 10.1016/j.tins.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey MG, Kustova Y, Sei Y, Basile AS. Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. J Neurochem. 1998;71:2079–2087. doi: 10.1046/j.1471-4159.1998.71052079.x. [DOI] [PubMed] [Google Scholar]
- Estrada-Sánchez AM, Montiel T, Segovia J, Massieu L. Glutamate toxicity in the striatum of the R6/2 Huntington's disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol Dis. 2009;34:78–86. doi: 10.1016/j.nbd.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Giralt A, Carretón O, Lao-Peregrin C, Martín ED, Alberch J. Conditional BDNF release under pathological conditions improves Huntington's disease pathology by delaying neuronal dysfunction. Mol Neurodegener. 2011;6:71. doi: 10.1186/1750-1326-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Tessler S, Faull RL, Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington's disease. Neurochem Res. 2008;33:232–237. doi: 10.1007/s11064-007-9463-1. [DOI] [PubMed] [Google Scholar]
- Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE, Hackam AS, Logvinova AV, Peel AL, Chen SF, Hook V, Singaraja R, Krajewski S, Goldsmith PC, Ellerby HM, Hayden MR, Bredesen DE, Ellerby LM. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 2004;11:424–438. doi: 10.1038/sj.cdd.4401358. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, Chesselet MF. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington's disease mice. Neuroscience. 2008;157:280–295. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- Lee J, Fukumoto H, Orne J, Klucken J, Raju S, Vanderburg CR, Irizarry MC, Hyman BT, Ingelsson M. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp Neurol. 2005;194:91–96. doi: 10.1016/j.expneurol.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Liévens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Maienschein V, Marxen M, Volknandt W, Zimmermann H. A plethora of presynaptic proteins associated with ATP-storing organelles in cultured astrocytes. Glia. 1999;26:233–244. doi: 10.1002/(SICI)1098-1136(199905)26:3<233::AID-GLIA5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Martelli AM, Baldini G, Tabellini G, Koticha D, Bareggi R, Baldini G. Rab3A and Rab3D control the total granule number and the fraction of granules docked at the plasma membrane in PC12 cells. Traffic. 2000;1:976–986. doi: 10.1111/j.1600-0854.2000.11207.x. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Shindo A, Liang AC, Maeda M, Egawa N, Itoh K, Lo EK, Lok J, Ihara M, Arai K. Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor. J Neurosci. 2015;35:14002–14008. doi: 10.1523/JNEUROSCI.1592-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangrsic T, Potokar M, Stenovec M, Kreft M, Fabbretti E, Nistri A, Pryazhnikov E, Khiroug L, Giniatullin R, Zorec R. Exocytotic release of ATP from cultured astrocytes. J Biol Chem. 2007;282:28749–28758. doi: 10.1074/jbc.M700290200. [DOI] [PubMed] [Google Scholar]
- Parpura V, Zorec R. Gliotransmission: exocytotic release from astrocytes. Brain Res Rev. 2010;63:83–92. doi: 10.1016/j.brainresrev.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesseveur G, David DJ, Gaillard MC, Pla P, Wu MV, Nguyen HT, Nicolas V, Auregan G, David I, Dranovsky A, Hantraye P, Hen R, Gardier AM, Déglon N, Guiard BP. BDNF overexpression in mouse hippocampal astrocytes promotes local neurogenesis and elicits anxiolytic-like activities. Transl Psychiatry. 2013;3:e253. doi: 10.1038/tp.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Schmitz F, Jahn R, Rosenmund C, Südhof TC. A complete genetic analysis of neuronal Rab3 function. J Neurosci. 2004;24:6629–6637. doi: 10.1523/JNEUROSCI.1610-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes toneuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Shirataki H, Nakanishi H. Rab3A small GTP-binding protein in Ca2+-dependent exocytosis. Genes Cells. 1996;1:615–632. doi: 10.1046/j.1365-2443.1996.00257.x. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Fukuda M. Rab3A and Rab27A cooperatively regulate the docking step of dense-core vesicle exocytosis in PC12 cells. J Cell Sci. 2006;119:2196–2203. doi: 10.1242/jcs.02962. [DOI] [PubMed] [Google Scholar]
- van Weering JR, Toonen RF, Verhage M. The role of Rab3a in secretory vesicle docking requires association/dissociation of guanidine phosphates and munc18-1. PLoS One. 2007;2:e616. doi: 10.1371/journal.pone.0000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lin F, Wang J, Wu J, Han R, Zhu L, Zhang G, DiFiglia M, Qin Z. Truncated N-terminal huntingtin fragment with expanded-polyglutamine (htt552-100Q) suppresses brain-derived neurotrophic factor transcription in astrocytes. Acta Biochim Biophys Sin. 2012;44:249–258. doi: 10.1093/abbs/gmr125. [DOI] [PubMed] [Google Scholar]
- Wolfram-Aduan A, Altemus M, Wickwire JH, Sandstrom MI. Presymptomatic glutamate levels in prefrontal cortex in the Hdh(CAG150) mouse model of Huntington's disease. J Huntingtons Dis. 2014;3:387–399. doi: 10.3233/JHD-140114. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Wang G, Wang CE, Guo X, Yin P, Gao J, Tu Z, Wang Z, Wu J, Hu X, Li S, Li XJ. Mutant alpha-synuclein causes age-dependent neuropathology in monkey brain. J Neurosci. 2015;35:8345–8358. doi: 10.1523/JNEUROSCI.0772-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X. PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development. 2005;132:3281–3291. doi: 10.1242/dev.01891. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]