

Abstract
NMDA receptors are ligand-gated ion channels that underlie transmission at excitatory synapses and play an important role in regulating synaptic strength and stability. Functional NMDA receptors require two copies of the GluN1 subunit coassembled with GluN2 (and/or GluN3) subunits into a heteromeric tetramer. A diverse array of allosteric modulators can upregulate or downregulate NMDA receptor activity. These modulators include both synthetic compounds and endogenous modulators, such as cis-unsaturated fatty acids, 24(S)-hydroxycholesterol, and various neurosteroids. To evaluate the structural requirements for the formation and allosteric modulation of NMDA receptor pores, we have replaced portions of the rat GluN1, GluN2A, and GluN2B subunits with homologous segments from the rat GluK2 kainate receptor subunit. Our results with these chimeric constructs show that the NMDA receptor transmembrane domain is sufficient to account for most pore properties, but that regulation by some allosteric modulators requires additional cytoplasmic or extracellular domains.
SIGNIFICANCE STATEMENT Glutamate receptors mediate excitatory synaptic transmission by forming cation channels through the membrane that open upon glutamate binding. Although many compounds have been identified that regulate glutamate receptor activity, in most cases the detailed mechanisms that underlie modulation are poorly understood. To identify what parts of the receptor are essential for pore formation and sensitivity to allosteric modulators, we generated chimeric subunits that combined segments from NMDA and kainate receptors, subtypes with distinct pharmacological profiles. Surprisingly, our results identify separate domain requirements for allosteric potentiation of NMDA receptor pores by pregnenolone sulfate, 24(S)-hydroxycholesterol, and docosahexaenoic acid, three endogenous modulators derived from membrane constituents. Understanding where and how these compounds act on NMDA receptors should aid in designing better therapeutic agents.
Keywords: carboxy terminal domain, docosahexaenoic acid, palmitoylation
Introduction
Vertebrate central neurons express multiple receptors for the excitatory neurotransmitter l-glutamate, including three different families of ionotropic receptors (iGluRs) named for the agonists NMDA, AMPA, and kainate (Traynelis et al., 2010). Distinct sets of subunits, with limited primary sequence identity, contribute to these three iGluR families (Traynelis et al., 2010), but they all share a similar modular organization that includes large extracellular amino-terminal and ligand-binding domains (ATDs and LBDs), a transmembrane domain (TMD) that forms the channel pore (Huettner, 2015), and a cytoplasmic carboxy terminal domain (CTD; Sobolevsky et al., 2009; Karakas and Furukawa, 2014; Lee et al., 2014). Functional iGluRs are tetramers that, in the case of NMDA receptors, require two GluN1 subunits combined in a heterotetramer with GluN2 and/or GluN3 subunits, of which there are four (GluN2A–GluN2D) and two (GluN3A and GluN3B) subtypes, respectively (Paoletti et al., 2013). Additional diversity arises from alternate splicing of GluN1 and post-translational modifications, including phosphorylation and palmitoylation of specific residues in the subunit CTDs (Collingridge et al., 2013).
Conventional NMDA receptors, made up of GluN1 and GluN2 subunits, require coactivation by glutamate, which binds to the GluN2 subunit, and glycine or d-serine, which bind to GluN1 (Traynelis et al., 2010). This subunit combination forms channels that exhibit voltage-dependent block by magnesium but high permeability to monovalent cations and calcium. Because calcium entry through the channels requires both presynaptic transmitter release and postsynaptic depolarization to remove magnesium block, this unique permeation profile underlies the ability of NMDA receptors to regulate plasticity at excitatory synapses by detecting coincidence of presynaptic and postsynaptic activity (Paoletti et al., 2013).
NMDA receptors are subject to regulation by a wide variety of different compounds (Traynelis et al., 2010; Collingridge et al., 2013; Zhu and Paoletti, 2015), including several endogenous modulators derived from constituents of the plasma membrane. Both Ω-6 and Ω-3 cis-unsaturated fatty acids, such as arachidonic acid (AA) and docosahexaenoic acid (DHA), potentiate neuronal and recombinant NMDA receptors (Miller et al., 1992; Nishikawa et al., 1994), whereas lysophospholipids are inhibitory (Casado and Ascher, 1998). In addition, a number of endogenous steroid compounds, including neurosteroids (Gibbs et al., 2006; Korinek et al., 2011) and cholesterol metabolites (Paul et al., 2013; Linsenbardt et al., 2014; Korinek et al., 2015), have been shown to exhibit selective potentiation or inhibition of NMDA receptor-mediated currents. Most of these compounds appear to upregulate or downregulate channel open probability; however, the molecular interactions that underlie modulation remain largely unknown. Indeed, for some of the compounds, it is not clear whether they bind to specific locations on the receptor or exert an indirect influence on channel gating via changes in bulk membrane properties (Casado and Ascher, 1998). Neurosteroids have recently become a focus of drug development and together with oxysterols and cis-unsaturated fatty acids may offer novel targets for therapeutic intervention to reduce excessive NMDA receptor activation.
In the present study we sought to determine which glutamate receptor domains are required for NMDA receptor channel function and allosteric modulation. We generated a series of chimeric subunits that swap domains between kainate receptor subunit GluK2 and NMDA receptor subunits GluN1 and GluN2A or GluN2B (Wilding et al., 2014). Our results show that the TMDs from GluN1 and GluN2, together with the short linkers that connect to their respective LBDs, are sufficient to form functional channels with characteristic features of NMDA receptors, including voltage-dependent block by magnesium and organic pore blockers, permeability to calcium, and positive allosteric modulation by 24(S)-hydroxycholesterol [24(S)-HC; Paul et al., 2013]. In contrast, current potentiation by cis-unsaturated fatty acids required additional membrane proximal portions of the cytoplasmic CTD, whereas potentiation by the neurosteroid pregnenolone sulfate (PS; Wu et al., 1991) was only observed in chimeric receptors that included the LBD and TMD from NMDA receptor subunits.
Materials and Methods
cDNA constructs, cell culture, and transfection.
Rat subunit cDNAs were generously provided by Steve Heinemann, Peter Seeburg, Mark Mayer, Stefano Vicini, and Linda Nowak. Chimeric subunits were produced by a restriction enzyme-free PCR cloning method (Tillett and Neilan, 1999) or by ligation of PCR products with novel restriction sites inserted by silent mutations. All constructs were sequenced by the Washington University Protein Nucleic Acid Chemistry Laboratory. cDNAs were expressed by transient transfection in HEK 293 cells using lipofectamine 2000 (Invitrogen). The cells were propagated in 25 cm2 flasks with MEM plus 10% fetal bovine serum and passaged once each week with protease XXIII (Sigma-Aldrich). Cells used for transfection were seeded onto 12-well plates and transfected the following day. Coexpression of GFP from a second vector was used to identify transfected cells. The day after transfection, cells were plated at low density on 35 mm plates coated with nitrocellulose; recordings were obtained on the following 2 d.
Electrophysiology.
Cultures were bath perfused with Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, pH 7.4 with NaOH. Whole-cell electrodes were pulled from borosilicate tubing (WPI) and filled with an internal solution that contained the following (in mm): 140 Cs-glucuronate, 10 EGTA, 5 CsCl, 5 MgCl2, 5 ATP, 1 GTP, and 10 HEPES, pH adjusted to 7.4 with CsOH. Agonists and antagonists were delivered in control extracellular solution (160 mm NaCl, 2 mm CaCl2, and 10 mm HEPES, pH adjusted to 7.4 with NaOH) by local perfusion from a multibarreled pipette positioned near the recorded cell. Currents were recorded with an Axopatch 200A amplifier controlled by p-Clamp software (Molecular Devices) and are presented as mean ± SEM agonist-evoked current (current during agonist exposure minus holding current recorded in control solution before and/or after agonist superfusion). Chimeras with LBDs from GluK2 were treated with concanavalin A to increase steady-state agonist responses for most experiments (Wilding et al., 2005).
Current–voltage relations (I–Vs) were generated by averaging the currents recorded during a series of five ascending and descending membrane potential ramps (0.75 mV ms−1). I–Vs recorded in the presence of magnesium were fit with a Boltzmann function: I = (a + Vm) * [b + c/(1 + exp[−(Vm − V1/2)/k])] to compare the half-maximal voltage (V1/2) and slope factor (k) for magnesium block. Statistical significance was assigned for p < 0.05.
Molecular modeling.
Homology modeling was performed in Modeler (release 9.7; Eswar et al., 2008) using multiple templates (4PE5: Karakas and Furukawa, 2014; 4TLM: Lee et al., 2014) and twofold symmetry constraint for the A/C and B/D subunit pairs. Predictions for CTD secondary structure were made with JPred3 (Cole et al., 2008) and PEP-FOLD (Thévenet et al., 2012), with subsequent loop-refinement in Modeler.
Results
Chimeric channels with heteromeric NMDA receptor pores
To determine the structural requirements for allosteric modulation of receptors with NMDA receptor pore properties (Traynelis et al., 2010; Huettner, 2015), we generated a number of chimeric subunit constructs that replaced domains of GluN1, GluN2A, and/or GluN2B with the homologous domain from the GluK2 kainate receptor subunit (Fig. 1A,B). All of our initial chimeric constructs contained the full NMDA receptor TMD, including the transmembrane M4 helix and the linkers that connect the TMD and LBD (Fig. 1A,B). Agonist applications failed to evoke significant current in HEK cells transfected with any of the chimeric subunits alone (Fig. 1C), indicating that they were unable to form functional homomeric channels. However, cotransfection of GluN1 and GluN2 TMD-containing subunits together resulted in substantial currents activated by the agonist(s) appropriate for the LBD: kainate for chimeric subunits that included the LBD from GluK2 or NMDA and glycine for subunit combinations that contained the LBDs from GluN1 and GluN2 (Fig. 1C). Each functional combination of homologous constructs displayed voltage-dependent block by magnesium (Fig. 1D; Table 1), a characteristic feature of intact NMDA receptors (Mayer et al., 1984; Nowak et al., 1984) mediated by adjacent asparagine residues near the apex of the GluN2 subunit pore loop (Burnashev et al., 1992; Wollmuth et al., 1998). In addition, I–Vs recorded in normal and elevated calcium (2 and 10 mm) indicated substantial calcium permeability for the chimeric receptors with NMDA receptor pores (Fig. 2A). Based on the change in reversal potential between the two external solutions of 7.5 ± 0.5 mV (n = 72), the permeability ratio for calcium to sodium (PCa/PNa) was estimated at 4.4 ± 0.4, which is consistent with previous work on recombinant wild-type receptors (Jatzke et al., 2002). Finally, chimeric receptors displayed voltage-dependent inhibition by ketamine (Fig. 2B), one of several organic blockers that can be trapped within the closed NMDA receptor channel (Traynelis et al., 2010). Block by 30 μm ketamine was essentially complete at −80 mV (99.8 ± 2.3%, n = 90) but substantially weaker at +40 mV (65.5 ± 4.9%, n = 11). In addition, the potency of ketamine block was similar for wild-type NMDA receptors and constructs in which only the TMD and linkers came from NMDA receptors (Fig. 2C,D). Thus, the results in Figures 1 and 2 indicate that the TMD and linker regions from the GluN1 and GluN2 subunits are together sufficient to specify formation of a functional NMDA receptor pore. These results complement our earlier work (Wilding et al., 2014), which showed that chimeric subunits with the TMD and linkers from GluK2 transplanted into either GluN1 or GluN2B fail to function as homotetramers but form functional heteromeric channels with kainate receptor pore properties upon coexpression. Together, these experiments support the idea that determinants for NMDA receptor subunit heteromerization are distributed across multiple iGluR domains (Ayalon and Stern-Bach, 2001).
Figure 1.
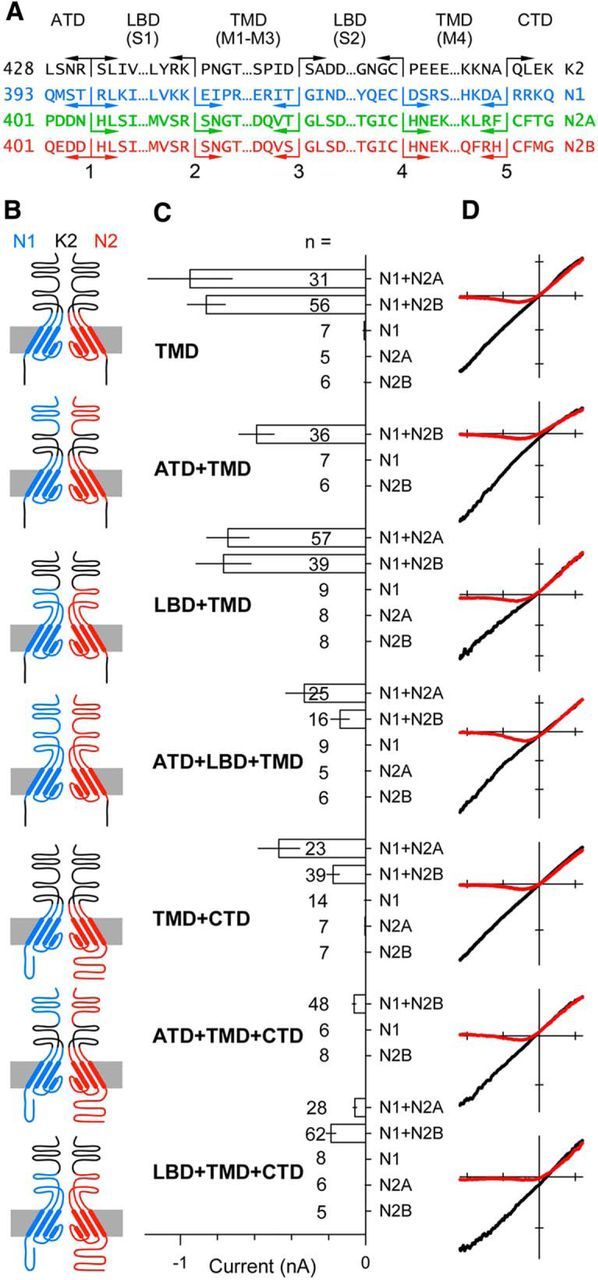
Heteromeric subunit chimeras form functional NMDA receptor pores. A, Protein sequence at the five joints used to construct the chimeric subunits in B. B, Simplified diagram of the four domains in two chimeric subunits of a heteromeric receptor highlighting the pore loop and α helical segments in the TMD derived from the GluN1 (blue) or GluN2 (red) subunit. Non-TMD portions of each chimera derived from the GluK2 kainate receptor subunit are shown in black. Subunits with N1 and N2 extracellular domains are presumed to be adjacent in functional tetramers. For clarity, the subunits are positioned opposite each other in the diagram. C, Mean peak whole-cell agonist-evoked current (± SEM) recorded at −80 mV in HEK 293 cells transiently transfected with chimeric subunit cDNAs and activated with either 10 μm kainate (LBD from GluK2) or 10 μm NMDA and 10 μm glycine (LBDs from NMDA receptor subunits). Agonist-evoked inward currents were significant (i.e., different from zero, t-statistic) for all of the coexpressed construct combinations, but not for any of the chimeric constructs expressed alone. D, I–V for agonist-evoked current recorded as the membrane potential was ramped from −110 to +60 mV at 0.75 mV ms −1 in the presence (red) or absence (black) of 1 mm external magnesium. All recordings from GluN1/2B chimeras. Voltage axis ticks, 50 mV; current axis ticks, 200 pA; except TMD, 500 pA.
Table 1.
Boltzmann parameters for voltage-dependent magnesium block
| Construct | Slope | V1/2 | # |
|---|---|---|---|
| GluN2A | |||
| Wild type | 14.7 ± 1.2 | −31.3 ± 4.3 | 8 |
| TMD | 16.7 ± 1.3 | −20.2 ± 2.6 | 4 |
| LBD + TMD | 15.3 ± 0.4 | −25.9 ± 3.0 | 7 |
| ATD + LBD + TMD | 14.6 ± 0.3 | −25.9 ± 6.8 | 7 |
| TMD + CTD | 17.1 ± 1.2 | −18.5 ± 4.9 | 6 |
| LBD + TMD + CTD | 13.7 ± 0.3 | −6.7 ± 1.6* | 14 |
| GluN2B | |||
| Wild type | 12.1 ± 0.5 | −16.8 ± 1.5 | 16 |
| TMD | 13.7 ± 0.5 | −32.1 ± 4.9 | 14 |
| ATD + TMD | 15.0 ± 1.0 | −18.1 ± 5.2 | 6 |
| LBD + TMD | 15.0 ± 0.4 | −15.8 ± 3.9 | 15 |
| ATD + LBD + TMD | 14.9 ± 0.6 | −18.5 ± 1.9 | 3 |
| TMD + CTD | 14.4 ± 0.9 | −12.1 ± 3.0 | 3 |
| ATD + TMD + CTD | 12.8 ± 1.2 | −21.6 ± 4.0 | 3 |
| LBD + TMD + CTD | 13.8 ± 0.2 | −6.8 ± 1.8* | 14 |
Slope factors (k) and V1/2 values were obtained from best fits of a Boltzmann function: I = (a + Vm) ∗ [b + c / (1 + exp[−(Vm − V1/2)/k])] to I–V in the presence of 1 mm magnesium. Each GluN2 construct was coexpressed with the homologous GluN1.1 construct. * denotes significant difference from wild type (ANOVA on ranks with post-hoc Dunn's test).
Figure 2.
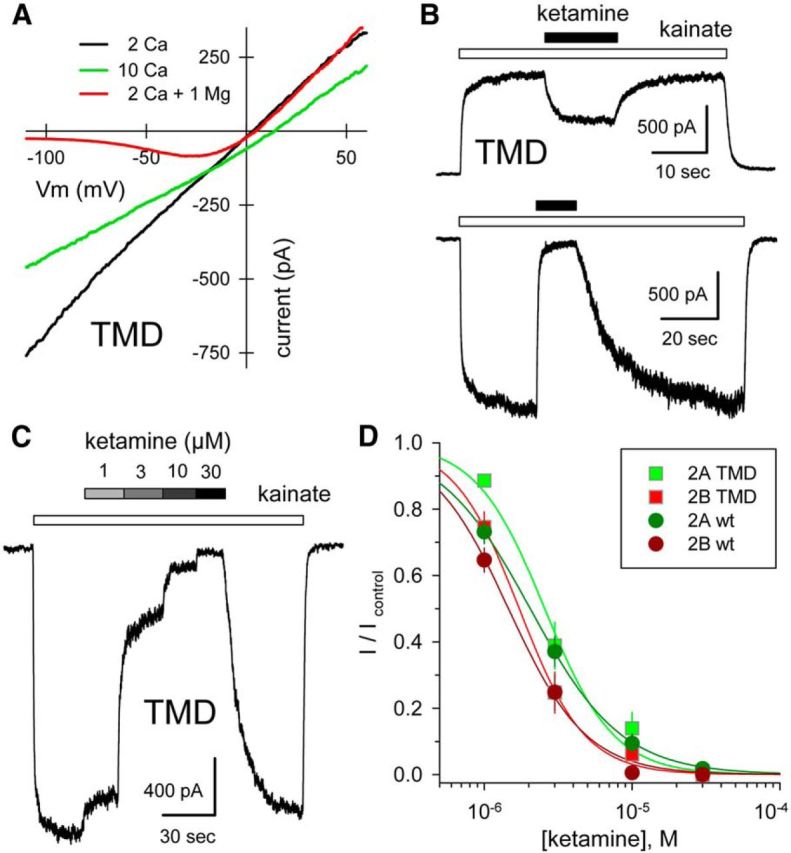
Calcium permeability and ketamine block of chimeric channels. A, Whole-cell current evoked by 10 μm NMDA plus 10 μm glycine as a function of voltage as membrane potential was ramped from −110 to +60 mV at 0.75 mV ms −1 in 160 mm NaCl, 10 mm HEPES with 2 mm CaCl2 (black), or with 10 mm CaCl2 (green), or with 2 mm CaCl2 and 1 mm MgCl2 (red). Note the rightward shift in reversal potential in elevated calcium. GluN1/2A TMD. B, Exposure to 30 μm ketamine (black bars) blocks current evoked by 10 μm kainate (open bars) at −80 mV, and to a lesser extent at +40 mV. GluN1/2A TMD. C, Whole-cell current evoked by 10 μm kainate is blocked progressively by 1, 3, 10, and 30 μm katamine. GluN1/2A TMD. D, Current in the presence of ketamine as a fraction of control (mean ± SEM) is plotted as a function of ketamine concentration. Smooth curves are best fits of 1/(1 + [(ketamine)/IC50]n) where IC50 is the concentration producing half-maximal inhibition and n is the slope factor. Values for chimeric constructs (N1/2A: IC50 = 2.6 ± 0.2, n = 1.9 ± 0.3, 7 cells; N1/2B: IC50 = 1.7 ± 0.1, n = 2.0 ± 0.3, 9 cells) were not significantly different from those of the respective wild-type subunits (N1/2A: IC50 = 2.0 ± 0.1 μm, n = 1.4 ± 0.2, 13 cells; N1/2B: IC50 = 1.6 ± 0.2, n = 1.7 ± 0.2, 10 cells) by t test.
Domain requirements for positive allosteric modulation
NMDA receptor activity can be potentiated by exposure to a number of different membrane-derived modulatory compounds including AA and DHA (Miller et al., 1992; Nishikawa et al., 1994), 24(S)-HC (Paul et al., 2013), and PS (Wu et al., 1991). To evaluate the structural basis for this modulation, we recorded from HEK cells transfected with chimeric subunits and compared the amplitude of agonist-evoked currents before and immediately after exposure to 24(S)-HC (Fig. 3), PS (Fig. 4), or DHA (Fig. 5). As shown in Figure 3, agonist-evoked whole-cell currents increased following exposure to 24(S)-HC in heteromeric chimeric combinations that included the NMDA receptor TMD, whereas treatment with 24(S)-HC had no effect on edited or unedited homomeric wild-type GluK2 receptors or on heteromeric chimeric receptors with the extracellular ATD and LBD from NMDA receptors fused to the TMD and CTD from GluK2 (Wilding et al., 2014). Together, these results strongly suggest that the NMDA receptor TMD alone is necessary and sufficient to determine channel modulation by 24(S)-HC.
Figure 3.

Potentiation by 24(S)-HC only requires the NMDA receptor TMD. A, Exposure to 10 μm 24(S)-HC (shaded bar) potentiated whole-cell current evoked by 10 μm kainate (open bars) in an HEK cell transfected with the N1/N2B TMD chimera. B, Minimal effect of 24(S)-HC on the N1/N2B ATD+LBD chimera activated by 10 μm NMDA plus 10 μm glycine. C, Plot of current (mean ± SEM) evoked after exposure to 24(S)-HC as a fraction of control current before 24(S)-HC treatment. *, Significant difference from wild-type GluN1/2B was observed for homomeric GluK2 (Q or R) and for the N1/N2B ATD+LBD chimera, which has an unedited (Q) kainate receptor TMD but not for chimeric constructs with the NMDA receptor LBD+TMD or TMD alone (p < 0.001, ANOVA on ranks with post hoc Dunn's test).
Figure 4.

PS potentiates chimeric channels that include the NMDA receptor LBD and TMD. A, B, Exposure to 100 μm PS (shaded bar) potentiates whole-cell current evoked by 10 μm NMDA plus 10 μm glycine in an HEK cell transfected with the N1/N2B LBD+TMD chimeric subunits (A), but not in a cell transfected with the N1/N2B TMD-alone chimera (B). C, Plot of current (mean ± SEM) evoked during exposure to PS as a fraction of control current before PS treatment. *, Significant difference from wild-type GluN1/2B was observed for homomeric GluK2 (Q or R), for heteromeric chimeras with the N1/2B TMD and linkers without the LBD and for the N1/N2B ATD+LBD chimera, which has an unedited (Q) kainate receptor TMD; however, potentiation for the chimeric construct with the NMDA receptor LBD+TMD was not significantly different from N1/2B. (p < 0.001, ANOVA on ranks with post hoc Dunn's test).
Figure 5.
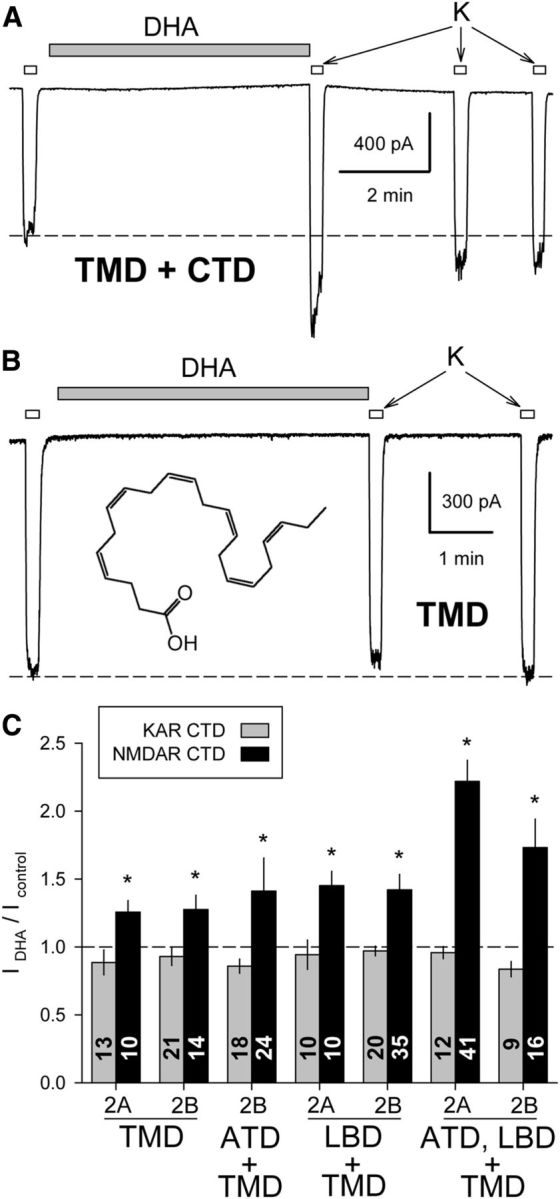
DHA potentiates chimeric channels that include the NMDA receptor TMD and CTD. A, Exposure to 15 μm DHA (shaded bar) potentiated whole-cell current evoked by 10 μm kainate (open bars) in an HEK cell transfected with the N1/N2B TMD+CTD chimera. B, Lack of effect of DHA on the N1/N2B TMD chimera. C, Plot of current (mean ± SEM) evoked immediately after exposure to DHA as a fraction of control current before DHA treatment. *, Significant potentiation for chimeric constructs with the NMDA receptor TMD+CTD, but not for chimeras that included the NMDA receptor TMD and linkers without the CTD (p < 0.05, Mann–Whitney rank-sum tests).
In contrast to 24(S)-HC, which potentiated constructs with an NMDA receptor TMD (Fig. 3), PS and DHA affected distinct subsets of the chimeric receptors depending upon the presence of additional NMDA receptor domains. Consistent with previous evidence that potentiation by PS involves interaction with the NMDA receptor LBD (Jang et al., 2004; Horak et al., 2006), we observed substantial potentiation by PS of chimeric receptors that included both the TMD and LBD from NMDA receptor subunits (Fig. 4), but significantly less effect on chimeric constructs that lacked the NMDA receptor LBD. In addition, PS did not potentiate currents mediated by edited or unedited GluK2 receptors. Importantly, exposure to PS did not cause significant potentiation of currents mediated by the chimeric construct with NMDA receptor ATD and LBD fused to the TMD and CTD from GluK2. Collectively, these results confirm earlier evidence that interaction with the LBD is required for allosteric potentiation of NMDA receptor channels by PS. Additional portions of the linkers and M4 helix may also be essential (Korinek et al., 2011; Kostakis et al., 2011) as chimeric receptors that included the kainate receptor TMD and linkers were not potentiated by PS even when activated by NMDA and glycine binding to the NMDA receptor LBD.
Unlike PS and 24(S)-HC, exposure to AA and DHA modulates activity of both NMDA and kainate receptors, potentiating NMDA-evoked currents in neurons (Miller et al., 1992) and inhibiting currents mediated by neuronal kainate receptors (Wilding et al., 1998). In addition, work on recombinant wild-type and mutant or chimeric subunits indicates that the kainate receptor TMD is sufficient to determine the effect of DHA exposure on pore operation (Wilding et al., 2014). Strong inhibition of GluK2 by DHA requires the presence of arginine at the glutamine/arginine (Q/R) editing site of all four subunits in the tetramer, both for wild-type full-length homomeric GluK2 (Wilding et al., 2005) and for chimeric receptors with the GluK2 TMD (Wilding et al., 2014). Moreover, an M3 helix Leu to Ala substitution at the level of the central cavity converts inhibition of edited (R) channels to potentiation, both for full-length GluK2(R) (Lopez et al., 2013) and for chimeric channels that include the TMD and linker segments from GluK2 combined with the ATD and LBD from NMDA receptor subunits (Wilding et al., 2014). In sharp contrast to the evidence that DHA modulation targets the kainate receptor TMD, potentiation of NMDA receptor channels by DHA was only observed for chimeric constructs that included both the TMD and CTD from NMDA receptors. As shown in Figure 5, exposure to DHA had no significant effect on constructs in which the CTDs of both the GluN1 and GluN2 subunits were replaced with the GluK2 CTD but potentiated currents mediated by constructs that included both the TMD and CTD from NMDA receptors.
Requirement for membrane proximal portions of the GluN2 CTD
To rule out the possibility that elements within the GluK2 CTD might be incompatible with NMDA receptor channel potentiation by DHA, we generated deletion constructs with stop codons immediately after M4 corresponding to position K841 in GluN1 and R846 or R847 in GluN2A or GluN2B, respectively. As shown in Figure 6A,C, currents evoked by NMDA and glycine in cells cotransfected with both deletion constructs, or either deletion construct together with a full-length partner, were unaffected by exposure to DHA, suggesting a requirement for both NMDA receptor CTDs. However, we also tested combinations in which only the GluN1 CTD or only the GluN2 CTD was replaced with the kainate receptor CTD from GluK2 (Fig. 6B,C). DHA potentiated receptors that included full-length GluN2A or GluN2B together with chimeric GluN1 bearing the CTD from GluK2, albeit to a lesser extent than completely wild-type receptors. In contrast, DHA had no effect on receptors comprising full-length GluN1 paired with chimeric GluN2A or GluN2B subunits bearing the GluK2 CTD (Fig. 6B,C). Together, these results indicate that fatty-acid potentiation requires the GluN2 CTD, but on the GluN1 subunit either the native wild-type CTD or the GluK2 CTD will suffice.
Figure 6.
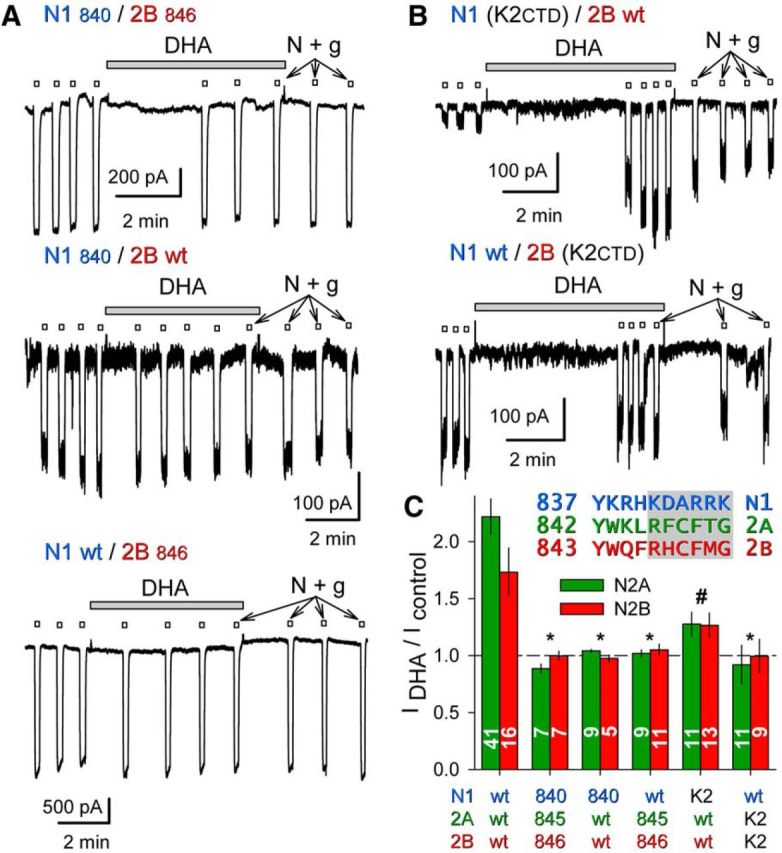
DHA potentiation requires the GluN2 CTD. A, Exposure to 15 μm DHA (shaded bars) did not potentiate whole-cell currents evoked by 10 μm NMDA plus 10 μm glycine (open bars) in HEK cells cotransfected with truncated GluN1840 and GluN2B846 (top trace), or for cotransfection of a wild-type and a truncated subunit: GluN1840/GluN2Bwt (middle trace), GluN1wt/GluN2B846 (bottom trace). B, Exposure to 15 μm DHA (shaded bar) potentiated whole-cell current evoked by 10 μm NMDA plus 10 μm glycine (open bars) in an HEK cell transfected with a GluN1 chimera bearing a GluK2 CTD and wild-type GluN2B (top); lack of effect of DHA on wild-type GluN1 with the GluN2B chimera bearing a GluK2 CTD (bottom). C, Plot of current (mean ± SEM) evoked immediately after exposure to DHA as a fraction of control current before DHA treatment for wild-type and truncated or chimeric NMDA receptor subunits. *, Significant difference from wild-type (p < 0.0001, ANOVA on ranks with post hoc Dunn's test) and not significantly different from no effect (i.e., IDHA/Icontrol = 1, t-statistic). # denotes that for the N1 (K2CTD)/N2 wild-type combinations IDHA/Icontrol was significantly >1 but also significantly less than that for wild-type receptors. Inset, Proximal CTD sequences of GluN1, GluN2A, and GluN2B. Shading denotes truncation after N1 H840, 2A L845, and 2B F846.
Additional deletion mutants were created to determine which portion of the GluN2 CTD was essential. Potentiation was maintained with truncation after position 931 in either GluN2A or GluN2B, resulting in deletion of the C-terminal 526 or 544 aa, respectively (Fig. 7A–C). Weaker potentiation was also observed for more proximal truncation after residue 877 of GluN2A or 878 of GluN2B, but not for several additional truncations closer to M4 (Fig. 7A–C; data not shown). These results suggest that the proximal GluN2 CTD is necessary for NMDA receptor potentiation by DHA. Our previous work (Wilding et al., 2014) indicated that kainate receptor modulation by DHA was similar for wild-type GluK2 and for chimeric receptors with a TMD from GluK2, heteromeric ATD and LBD from NMDA receptors, and either the GluK2 CTD or heteromeric NMDA receptor CTDs. In the present study we also tested whether the proximal GluN2 CTD alone was sufficient to modify the effect of DHA on homomeric edited (R) or unedited (Q) GluK2. As shown in Figure 7D, DHA inhibition of GluK2(R) was unchanged by substitution of the GluN2B CTD. Wild-type GluK2(Q) is unaffected by DHA, whereas receptors substituted with the GluN2B CTD were weakly inhibited. Collectively, these results suggest that the GluN2 proximal CTD renders the NMDA receptor TMD susceptible to potentiation by DHA, but has minimal effect on the GluK2 TMD.
Figure 7.
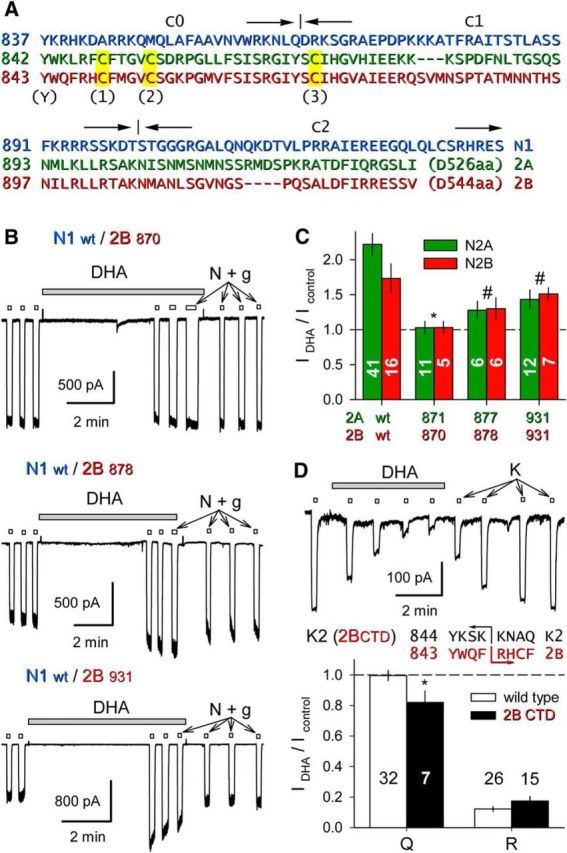
The GluN2 proximal CTD supports NMDA receptor TMD potentiation. A, Sequence of the GluN1.1 CTD (blue) noting joints for the C0, C1, and C2 splice cassettes; alignment of the membrane-proximal portions of GluN2A (green) and GluN2B (red) noting location of conserved Tyr (Y) and Cys (C1, 2, 3) residues. B, Exposure to 15 μm DHA (shaded bar) did not potentiate whole-cell current evoked by 10 μm NMDA plus 10 μm glycine (open bars) in HEK cells cotransfected with wild-type GluN1.1 and GluN2B truncated after S870 (top trace), but did potentiate for N2B truncation after E878 (middle trace) or V931 (bottom trace). C, Plot of current (mean ± SEM) evoked immediately after exposure to DHA as a fraction of control current before DHA treatment for wild-type and GluN2 truncated NMDA receptors. *, Significant difference from wild-type (p < 0.0001, ANOVA on ranks with post hoc Dunn's test) and not significant difference from no effect (IDHA/Icontrol = 1, t-statistic). # denotes that for the other N2 truncations IDHA/I control was significantly >1 but also significantly less than for wild-type receptors. D, Exposure to 15 μm DHA (shaded bar) inhibited whole-cell current evoked by 10 μm kainate (open bars) in an HEK cell transfected with chimeric GluK2(R) bearing a CTD from GluN2B. Plot of current (mean ± SEM) evoked at the end of DHA exposure as a fraction of control current before DHA treatment. *, DHA produced weak but significant (p = 0.030) inhibition of unedited (Q) chimeric GluK2 subunits and strong inhibition of edited (R) chimeric receptors that was not different from wild-type GluK2(R) (p = 0.148). Inset, Proximal CTD sequences of GluK2 and GluN2B indicating the position of the K2/2B joint.
Cysteine replacement in the proximal CTD reduces potentiation by DHA
Previous biochemical studies have identified a number of post-translational modification sites within the minimal essential region of the GluN2 CTD identified in Figure 7, including a cluster of cysteine residues that can undergo palmitoylation (Hayashi et al., 2009; Mattison et al., 2012) and a conserved tyrosine that may be phosphorylated by Src kinase (Vissel et al., 2001). As an initial test for whether any of these potential modification sites are required for allosteric modulation by DHA, we generated mutant versions of GluN2A and GluN2B with Y-to-F substitution at the conserved tyrosine and C-to-A substitution at all three of the proximal cysteines. An additional cysteine (C838) located near the cytoplasmic end of M4 in GluN2B was substituted with Trp, the residue at the homologous position in GluN2A. As shown in Figure 8, full-length receptors bearing all of these substitutions were unaffected by exposure to DHA. Additional mutant full-length GluN2A constructs revealed that potentiation by DHA was reduced relative to wild-type for all of the single, double, or triple substitutions in the proximal cysteine cluster, but was least affected for the Y842F substitution, which exhibited significant increases in agonist-gated current after DHA exposure (Fig. 8A,B). Together, these results strengthen the evidence for involvement of the proximal CTD in allosteric modulation by DHA and raise the possibility that cysteine modification could be a contributory factor.
Figure 8.

Proximal GluN2 CTD substitutions prevent DHA potentiation. A, Plot of current (mean ± SEM) evoked immediately after exposure to DHA as a fraction of control current before DHA treatment for NMDA receptors with wild-type GluN1 and the indicated substitutions to full-length GluN2. Residues highlighted in yellow are wild-type. *, Significant difference from wild-type (p < 0.0001, ANOVA on ranks with post hoc Dunn's test) and not significant difference from no effect (IDHA/Icontrol = 1, t-statistic). # denotes that for the N1 wt/N2A Y842F combination IDHA/Icontrol was significantly >1 but also significantly less than for wild-type receptors. B, Traces show the quadruple FAAA mutant lacked potentiation by DHA (top trace). Potentiation was preserved for the Y842F substitution, in some cases reaching the level observed for wild-type receptors (bottom trace).
Although there is no x-ray crystal structure available for any of the iGluR CTDs, algorithms that predict secondary structure suggest that there may be several α helical segments within the proximal CTD of each iGluR subunit (Ryan et al., 2008). The minimal GluN2 cytoplasmic domain essential for DHA potentiation includes a predicted helical region between C2 and C3 that encompasses a conserved SRGIYSC sequence (Fig. 7A) that is present in all four GluN2 subunits. Although the helical properties of this proximal CTD segment require structural confirmation, together our results suggest a hypothetical model for the GluN2 proximal CTD shown in Figure 9.
Figure 9.

NMDA receptor model with GluN2A proximal CTD. Homology model for GluN1/GluN2A generated by Modeler (Eswar et al., 2008) using PDB files 4PE5 (Karakas and Furukawa, 2014) and 4TLM (Lee et al., 2014) as templates and with twofold symmetry constraint for the A/C and B/D subunits. Prediction for the proximal CTD generated by PEP-FOLD (Thévenet et al., 2012) with subsequent loop-refinement in Modeler. Palmitoylation at C1, 2, and 3 is expected to tether the proximal CTD to the membrane.
Discussion
Our results support several conclusions about iGluR structure, operation, and regulation by positive allosteric modulators. First, we show that GluN1 and GluN2 subunit TMDs and linkers include sufficient constraints for the assembly of functional heteromeric tetramers. Chimeric subunits that included GluN1 or GluN2 TMDs and linkers failed to generate functional channels when expressed alone, but heteromeric coexpression gave rise to robust agonist-gated currents that displayed signature features of NMDA receptor pores, including permeability to calcium and voltage-dependent block by magnesium and ketamine (Traynelis et al., 2010). Second, we demonstrate distinct domain requirements for positive allosteric modulation by three membrane-derived regulators. The cholesterol metabolite 24(S)-HC had no effect on homomeric GluK2 but strongly potentiated chimeric channels that included the TMD and linkers from NMDA receptor subunits. Potentiation by the neurosteroid PS required both the LBD and TMD from NMDA receptors, confirming earlier mutation studies (Jang et al., 2004; Horak et al., 2006). In contrast, potentiation by DHA did not require the NMDA receptor LBD but was only observed for chimeric channels that included a proximal portion of the GluN2 subunit CTD.
Functional NMDA/kainate receptor subunit chimeras
Although much remains to be learned about specification of specific subunit combinations, there is growing evidence that several distinct domain interactions contribute to production of heteromeric iGluRs (Traynelis et al., 2010; Herguedas et al., 2013). For NMDA receptors, assembly as obligate heterotetramers is thought to involve initial association controlled by the extracellular ATDs (Hansen et al., 2010). Indeed, our earlier work showed that chimeric subunits with the extracellular domains from NMDA receptors and a transmembrane and cytoplasmic portion from GluK2 kainate receptors fail to function as homotetramers but only form functional channels when coexpressed as N1/K2+N2B/K2 heteromers (Wilding et al., 2014). Thus, NMDA receptor extracellular domains are sufficient to impose a requirement for heteromerization onto the kainate receptor pore (Wilding et al., 2014), despite the fact that native full-length GluK2 forms functional homotetramers (Egebjerg et al., 1991).
In this study we found that interactions promoting heteromerization are also present within the NMDA receptor TMD and linkers (Ayalon and Stern-Bach, 2001). Previous work (Villmann et al., 1999; Liu and Nowak, 2000) showed that chimeric subunits in which only the M1–M3 pore domain from GluN1 was transplanted into GluK2 formed functional homomeric channels that were activated by kainate, suggesting the homomeric extracellular domains from GluK2 enabled assembly of a chimeric pore. However, similar constructs using M1–M3 from GluN2B failed to function as homomeric channels or when coexpressed with GluN1 pore-domain chimeras (Villmann et al., 2008). Moreover, chimeric GluN1 M1–M3 homomers lacked characteristic NMDA receptor features (Villmann et al., 1999), including voltage-dependent magnesium and MK-801 block, properties preserved in our constructs that swap the entire TMD including linkers to the LBD and the M4 helix. Previous studies have highlighted the importance of the M4 helix for iGluR assembly and function (Schorge and Colquhoun, 2003; Terhag et al., 2010; Salussolia et al., 2013). In addition, recent NMDA receptor crystal structures (Karakas and Furukawa, 2014; Lee et al., 2014) confirm extensive contact between GluN1 and GluN2 involving the M4 helix of each subunit with M1 and M3 helices of the adjacent heteromeric subunit. Our results suggest that this M4-mediated contact is sufficient to allow for heteromeric association of the M1–M3 pore domain into functional channels sensitive to magnesium and ketamine as expected for native NMDA receptors.
Domain requirements for positive allosteric modulation
In addition to competitive antagonists and channel blockers, a diverse assortment of allosteric regulators have been shown to modify the operation of NMDA receptors, including endogenous molecules, such as polyamines, zinc, fatty acids, lysophospholipids, and cholesterol metabolites, as well as a growing repertoire of synthetic compounds (Collingridge et al., 2013; Zhu and Paoletti, 2015). Binding sites and mechanisms of modulation have been proposed for a number of these allosteric modulators (Zhu and Paoletti, 2015), but in many cases the basis for regulation remains incompletely understood. Our results using chimeric subunits to evaluate three positive allosteric modulators derived from components of the lipid bilayer revealed three distinct domain requirements: PS (Jang et al., 2004; Horak et al., 2006) required the NMDA receptor LBD and TMD, whereas 24(S)-HC potentiated chimeras that only included the TMD and linkers from NMDA receptors combined with the ATD, LBD, and CTD from GluK2. Recent work suggests that 24(S)-HC (Paul et al., 2013) acts by a different mechanism than either PS or cis-unsaturated fatty acids as potentiation by 24(S)-HC does not occlude modulation by PS or AA. In addition, the 25-hydroxy derivative of cholesterol noncompetitively reduces potentiation by 24(S)-HC but does not prevent potentiation by PS or AA (Linsenbardt et al., 2014). Thus, our results suggesting distinct structural requirements for allosteric modulation are broadly consistent with recent analysis of oxysterol pharmacology.
The finding that potentiation by fatty acid exposure requires a proximal segment of the GluN2 CTD was not expected. Our recent work on chimeric receptors with kainate receptor pores revealed that DHA produced similar modulation of the GluK2 TMD and linkers regardless whether the CTD came from GluK2 or from GluN1/2B (Wilding et al., 2014). In addition, earlier work by Casado and Ascher (1998) had suggested that NMDA receptor modulation by AA and lysophospholipids, as well as mechanosensitivity (Paoletti and Ascher, 1994), did not involve the CTD. They showed that channel modulation by mechanical deformation of the membrane was preserved in truncated mutants lacking either the GluN1 CTD or the distal CTD of GluN2A (terminated at residue 1082 or 1438). Channel inhibition by lysophosphatidyl inositol persisted in receptors with proximal GluN2A CTD truncation at position 844; however, the effect on mechanosensitivity or potentiation by AA was not reported (Casado and Ascher, 1998). In addition, modulation by lysophospholipids was only observed with extracellular application. Several groups have reported consistent potentiation of NMDA receptors by AA or DHA applied to outside-out patches (Miller et al., 1992; Nishikawa et al., 1994), but also in at least one experiment on inside-out patches with exposure from the cytoplasmic side (Casado and Ascher, 1998). We have not had success with inside-out recordings from chimeric channels with NMDA receptor CTDs, possibly because of their relatively low expression (Fig. 1C), nor have we attempted to elicit modulation in whole-cell recordings by adding DHA to the internal solution, as lipophilic molecules often appear inactive when delivered by intracellular dialysis from the electrode (Jo and Bean, 2014). Our results in the present study do confirm earlier evidence that anchorage to cytoplasmic scaffolding proteins by the GluN2 distal C terminus is not essential for modulation (Casado and Ascher, 1998), but they also reveal an absolute requirement for the GluN2 proximal CTD in potentiation by DHA. Further work will be needed to determine the exact relationship between mechanosensitivity and allosteric regulation by fatty acids, lysophospholipids, oxysterols, or other lipophilic modulators.
Previous work identified several post-translational modifications within the proximal CTD of GluN2A and GluN2B, including tyrosine phosphorylation near the cytoplasmic end of M4 (Vissel et al., 2001) and palmitoylation of three conserved cysteine residues within the first 30–40 CTD amino acids (Hayashi et al., 2009). All of these modifications have been implicated in the regulation of receptor trafficking: the tyrosine residue is part of consensus binding motif for the clathrin adaptor protein AP-2 present in all four GluN2 subunits (Scott et al., 2004). Phosphorylation of this Tyr stabilizes receptors at the surface but is suppressed by serine substitution at the three proximal Cys residues of either GluN2A or GluN2B. Thus, modification of proximal cysteines by palmitate thioesterification has been suggested to reduce NMDA receptor internalization by enhancing phosphotyrosine levels (Hayashi et al., 2009). Because robust potentiation by AA (Casado and Ascher, 1998) or DHA (Nishikawa et al., 1994) persists in outside-out patches, it is likely to involve a direct allosteric effect rather than a change in receptor trafficking to and from the surface membrane (Linsenbardt et al., 2014). Indeed, our results in the present study indicate that allosteric potentiation by cis-unsaturated fatty acids depends on the three proximal cysteine residues but does not require the conserved tyrosine that directly regulates internalization.
In summary, our study demonstrates formation of functional heteromeric NMDA receptor pores by chimeric subunits constructed by replacing intracellular and extracellular portions of GluN1, GluN2A, and GluN2B with homologous segments from kainate receptor subunit GluK2. In addition, we demonstrate that distinct NMDA receptor domains underlie positive allosteric modulation by PS, 24(S)-hydroxycholesterol, and DHA. Although further work will be needed to understand the detailed mechanisms that underlie channel regulation, one possible interpretation is that our results implicate regions where each of the modulators directly interacts with the receptor. Alternately, these domains may be required for sensing specific changes in membrane mechanical properties or contacting other structural elements necessary for indirect modulation. Our results with DHA identify an unexpected requirement for three conserved cysteine residues in the proximal cytoplasmic tail, suggesting that palmitoylation of these cysteines may be important for fatty acid modulation. Establishing where and how endogenous allosteric modulators act should facilitate ongoing efforts (Collingridge et al., 2013; Zhu and Paoletti, 2015) to identify more potent and selective drugs capable of upregulating or downregulating specific iGluR subpopulations (Paoletti et al., 2013).
Footnotes
This work was supported by the National Institutes of Health (NS30888, J.E.H.; GM083914, M.N.L.). We thank Steve Mennerick for providing 24(S)-hydroxycholesterol and for critical reading of the manuscript; Linda Nowak for advice and cDNA constructs; and Steve Heinemann, Peter Seeburg, Mark Mayer, and Stefano Vicini for additional cDNA constructs.
The authors declare no competing financial interests.
References
- Ayalon G, Stern-Bach Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron. 2001;31:103–113. doi: 10.1016/S0896-6273(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Schoepfer R, Monyer H, Ruppersberg JP, Günther W, Seeburg PH, Sakmann B. Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science. 1992;257:1415–1419. doi: 10.1126/science.1382314. [DOI] [PubMed] [Google Scholar]
- Casado M, Ascher P. Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. J Physiol. 1998;513:317–330. doi: 10.1111/j.1469-7793.1998.317bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, Tidball P, Fang G, Irvine MW, Costa BM, Monaghan DT, Bortolotto ZA, Molnár E, Lodge D, Jane DE. The NMDA receptor as a target for cognitive enhancement. Neuropharmacology. 2013;64:13–26. doi: 10.1016/j.neuropharm.2012.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egebjerg J, Bettler B, Hermans-Borgmeyer I, Heinemann S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature. 1991;351:745–748. doi: 10.1038/351745a0. [DOI] [PubMed] [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Gibbs TT, Russek SJ, Farb DH. Sulfated steroids as endogenous neuromodulators. Pharmacol Biochem Behav. 2006;84:555–567. doi: 10.1016/j.pbb.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol. 2010;78:535–549. doi: 10.1124/mol.110.067157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Thomas GM, Huganir RL. Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron. 2009;64:213–226. doi: 10.1016/j.neuron.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herguedas B, Krieger J, Greger IH. Receptor heteromeric assembly—how it works and why it matters: the case of ionotropic glutamate receptors. Prog Mol Biol Transl Sci. 2013;117:361–386. doi: 10.1016/B978-0-12-386931-9.00013-1. [DOI] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Chodounska H, Vyklicky L., Jr Subtype-dependence of N-methyl-d-aspartate receptor modulation by pregnenolone sulfate. Neuroscience. 2006;137:93–102. doi: 10.1016/j.neuroscience.2005.08.058. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Glutamate receptor pores. J Physiol. 2015;593:49–59. doi: 10.1113/jphysiol.2014.272724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mierke DF, Russek SJ, Farb DH. A steroid modulatory domain on NR2B controls N-methyl-d-aspartate receptor proton sensitivity. Proc Natl Acad Sci U S A. 2004;101:8198–8203. doi: 10.1073/pnas.0401838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jatzke C, Watanabe J, Wollmuth LP. Voltage and concentration dependence of Ca(2+) permeability in recombinant glutamate receptor subtypes. J Physiol. 2002;538:25–39. doi: 10.1113/jphysiol.2001.012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Bean BP. Sidedness of carbamazepine accessibility to voltage-gated sodium channels. Mol Pharmacol. 2014;85:381–387. doi: 10.1124/mol.113.090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek M, Kapras V, Vyklicky V, Adamusova E, Borovska J, Vales K, Stuchlik A, Horak M, Chodounska H, Vyklicky L., Jr Neurosteroid modulation of N-methyl-d-aspartate receptors: molecular mechanism and behavioral effects. Steroids. 2011;76:1409–1418. doi: 10.1016/j.steroids.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Korinek M, Vyklicky V, Borovska J, Lichnerova K, Kaniakova M, Krausova B, Krusek J, Balik A, Smejkalova T, Horak M, Vyklicky L. Cholesterol modulates open probability and desensitization of NMDA receptors. J Physiol. 2015;593:2279–2293. doi: 10.1113/jphysiol.2014.288209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostakis E, Jang MK, Russek SJ, Gibbs TT, Farb DH. A steroid modulatory domain in NR2A collaborates with NR1 exon-5 to control NMDAR modulation by pregnenolone sulfate and protons. J Neurochem. 2011;119:486–496. doi: 10.1111/j.1471-4159.2011.07442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenbardt AJ, Taylor A, Emnett CM, Doherty JJ, Krishnan K, Covey DF, Paul SM, Zorumski CF, Mennerick S. Different oxysterols have opposing actions at N-methyl-d-aspartate receptors. Neuropharmacology. 2014;85:232–242. doi: 10.1016/j.neuropharm.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Nowak LM. Large conductance ligand-gated channels formed by homomeric expression of a GluR6-NMDA NR1 chimera subunit. Soc Neurosci Abstr. 2000;26:527.13. [Google Scholar]
- Lopez MN, Wilding TJ, Huettner JE. Q/R site interactions with the M3 helix in GluK2 kainate receptor channels revealed by thermodynamic mutant cycles. J Gen Physiol. 2013;142:225–239. doi: 10.1085/jgp.201311000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison HA, Hayashi T, Barria A. Palmitoylation at two cysteine clusters on the C-terminus of GluN2A and GluN2B differentially control synaptic targeting of NMDA receptors. PLoS One. 2012;7:e49089. doi: 10.1371/journal.pone.0049089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Kimura S, Akaike N. Facilitatory effect of docosahexaenoic acid on N-methyl-d-aspartate response in pyramidal neurones of rat cerebral cortex. J Physiol. 1994;475:83–93. doi: 10.1113/jphysiol.1994.sp020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P. Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron. 1994;13:645–655. doi: 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, Crawford DC, Linsenbardt AJ, Shu HJ, Izumi Y, Mennerick SJ, Zorumski CF. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-d-aspartate receptors. J Neurosci. 2013;33:17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TJ, Emes RD, Grant SG, Komiyama NH. Evolution of NMDA receptor cytoplasmic interaction domains: implications for organisation of synaptic signalling complexes. BMC Neurosci. 2008;9:6. doi: 10.1186/1471-2202-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salussolia CL, Gan Q, Kazi R, Singh P, Allopenna J, Furukawa H, Wollmuth LP. A eukaryotic specific transmembrane segment is required for tetramerization in AMPA receptors. J Neurosci. 2013;33:9840–9845. doi: 10.1523/JNEUROSCI.2626-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorge S, Colquhoun D. Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J Neurosci. 2003;23:1151–1158. doi: 10.1523/JNEUROSCI.23-04-01151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhag J, Gottschling K, Hollmann M. The transmembrane domain C of AMPA receptors is critically involved in receptor function and modulation. Front Mol Neurosci. 2010;3:117. doi: 10.3389/fnmol.2010.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40:W288–W293. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillett D, Neilan BA. Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 1999;27:e26. doi: 10.1093/nar/27.19.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villmann C, Strutz N, Morth T, Hollmann M. Investigation by ion channel domain transplantation of rat glutamate receptor subunits, orphan receptors and a putative NMDA receptor subunit. Eur J Neurosci. 1999;11:1765–1778. doi: 10.1046/j.1460-9568.1999.00594.x. [DOI] [PubMed] [Google Scholar]
- Villmann C, Hoffmann J, Werner M, Kott S, Strutz-Seebohm N, Nilsson T, Hollmann M. Different structural requirements for functional ion pore transplantation suggest different gating mechanisms of NMDA and kainate receptors. J Neurochem. 2008;107:453–465. doi: 10.1111/j.1471-4159.2008.05623.x. [DOI] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Chai YH, Huettner JE. Inhibition of rat neuronal kainate receptors by cis-unsaturated fatty acids. J Physiol. 1998;513:331–339. doi: 10.1111/j.1469-7793.1998.331bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding TJ, Zhou Y, Huettner JE. Q/R site editing controls kainate receptor inhibition by membrane fatty acids. J Neurosci. 2005;25:9470–9478. doi: 10.1523/JNEUROSCI.2826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding TJ, Lopez MN, Huettner JE. Radial symmetry in a chimeric glutamate receptor pore. Nat Commun. 2014;5:3349. doi: 10.1038/ncomms4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Sakmann B. Adjacent asparagines in the NR2-subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg2+ J Physiol. 1998;506:13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-d-aspartate receptor. Mol Pharmacol. 1991;40:333–336. [PubMed] [Google Scholar]
- Zhu S, Paoletti P. Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr Opin Pharmacol. 2015;20:14–23. doi: 10.1016/j.coph.2014.10.009. [DOI] [PubMed] [Google Scholar]