

SYNJ1 encodes a polyphosphoinositide phosphatase (Synaptojanin 1) with a prominent role in synaptic vesicle dynamics. Hardies et al. report three families (six patients) with autosomal recessive SYNJ1 variants, who display early-onset refractory seizures and progressive neurological decline. The pathogenic variants entail loss of the dual phosphatase activity of Synaptojanin 1.
Keywords: recessive disorder, early onset epilepsy, neurodegenerative disorder, SYNJ1, SYNJ1 dual phosphatase activity

SYNJ1 encodes a polyphosphoinositide phosphatase (Synaptojanin 1) with a prominent role in synaptic vesicle dynamics. Hardies et al. report three families (six patients) with autosomal recessive SYNJ1 variants, who display early-onset refractory seizures and progressive neurological decline. The pathogenic variants entail loss of the dual phosphatase activity of Synaptojanin 1.
Abstract
SYNJ1 encodes a polyphosphoinositide phosphatase, synaptojanin 1, which contains two consecutive phosphatase domains and plays a prominent role in synaptic vesicle dynamics. Autosomal recessive inherited variants in SYNJ1 have previously been associated with two different neurological diseases: a recurrent homozygous missense variant (p.Arg258Gln) that abolishes Sac1 phosphatase activity was identified in three independent families with early onset parkinsonism, whereas a homozygous nonsense variant (p.Arg136*) causing a severe decrease of mRNA transcript was found in a single patient with intractable epilepsy and tau pathology. We performed whole exome or genome sequencing in three independent sib pairs with early onset refractory seizures and progressive neurological decline, and identified novel segregating recessive SYNJ1 defects. A homozygous missense variant resulting in an amino acid substitution (p.Tyr888Cys) was found to impair, but not abolish, the dual phosphatase activity of SYNJ1, whereas three premature stop variants (homozygote p.Trp843* and compound heterozygote p.Gln647Argfs*6/p.Ser1122Thrfs*3) almost completely abolished mRNA transcript production. A genetic follow-up screening in a large cohort of 543 patients with a wide phenotypical range of epilepsies and intellectual disability revealed no additional pathogenic variants, showing that SYNJ1 deficiency is rare and probably linked to a specific phenotype. While variants leading to early onset parkinsonism selectively abolish Sac1 function, our results provide evidence that a critical reduction of the dual phosphatase activity of SYNJ1 underlies a severe disorder with neonatal refractory epilepsy and a neurodegenerative disease course. These findings further expand the clinical spectrum of synaptic dysregulation in patients with severe epilepsy, and emphasize the importance of this biological pathway in seizure pathophysiology.
Introduction
Synaptic transmission is a highly complex process that mediates rapid propagation of information in the nervous system. Synaptic vesicles play a key role in this process, as they are stored in the presynaptic compartment and release their content by exocytosis in the synaptic cleft. Endocytosis and recycling of synaptic vesicle membranes allow their reuse in multiple cycles of secretion, hence maintain the synapse function during high frequency stimulation (Sudhof, 2004). Synaptic vesicle endocytosis is dynamically regulated and requires transient recruitment to the membrane of a variety of cytosolic factors, which interact with both membrane proteins and phospholipids to control budding and fission (Brodin et al., 2000; Di Paolo and De Camilli, 2006; Saheki and De Camilli, 2012; Posor et al., 2013).
A major pathway of synaptic vesicle recycling involves clathrin-mediated endocytosis. Synaptojanin 1 (SYNJ1), a protein encoded by the SYNJ1 gene on human chromosome 21, is a highly conserved polyphosphoinositide phosphatase that is concentrated at synapses (Takei et al., 1995; Cremona et al., 1999; Mani et al., 2007). In particular, its 145 kDa isoform is enriched on clathrin-coated intermediates (Haffner et al., 1997) where one of its major interactors is endophilin, an adaptor protein that binds the highly curved membrane of endocytotic buds (de Heuvel et al., 1997; Ringstad et al., 1997; Farsad et al., 2001; Gallop et al., 2006; Milosevic et al., 2011). A main function of SYNJ1 is to couple the endocytic reaction to the dephosphorylation of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], a phosphoinositide concentrated at the plasma membrane. Dephosphorylation is critical for shedding of the clathrin coat and other endocytic factors whose recruitment to endocytic sites is mediated by PI(4,5)P2 in the bilayer (Di Paolo and De Camilli, 2006). This function is facilitated by two consecutive phosphatase domains in the SYNJ1 protein: an N-terminal Sac1-like inositol domain (Sac1) and a central 5’-phosphatase domain (5’PP), which enable the removal of a phosphate group from the 4 and 5 position of PI(4,5)P2, respectively. This functional tandem is followed by a C-terminal proline-rich domain (PRD) that primarily functions as a protein-protein interactor and binds SH3 domain-containing proteins such as endophilin and amphiphysin (McPherson et al., 1996; Ramjaun and McPherson, 1996; de Heuvel et al., 1997; Cestra et al., 1999; Cremona et al., 1999; Guo et al., 1999; Gad et al., 2000; Haffner et al., 2000; Harris et al., 2000; Kim et al., 2002; Verstreken et al., 2003; Perera et al., 2006; Mani et al., 2007; McCrea and De Camilli, 2009; Soda et al., 2012; Posor et al., 2013; Dong et al., 2015).
Genetic defects affecting components of the endocytic machinery are well known to cause neurological disorders of variable severity in both humans and model organisms. Animal models with loss-of-function genetic deficits in SYNJ1 homologues display neurological features and early death (Cremona et al., 1999; Harris et al., 2000; Luthi et al., 2001; Kim et al., 2002; Verstreken et al., 2003; Van Epps et al., 2004; Sato et al., 2009). In humans, autosomal recessive inherited variants in SYNJ1 have recently been associated with two different neurological disorders. A recurrent homozygous missense variant located in the Sac1 domain (p.Arg258Gln), which selectively abolishes the phosphatase function of this specific domain, was identified in three independent families with early onset parkinsonism (Krebs et al., 2013; Quadri et al., 2013; Olgiati et al., 2014); whereas a homozygous nonsense variant (p.Arg136*) expected to result in loss of protein formation, was found in a single patient with intractable epilepsy and tau pathology (Dyment et al., 2015a, b). Here, we describe two consanguineous families of Moroccan ethnicity and a non-consanguineous Caucasian family with two affected siblings each presenting early onset refractory seizures and a neurodegenerative disease course. Using next generation sequencing technologies bi-allelic SYNJ1 variants were identified in all three families. We further examined the frequency of SYNJ1 defects in a large epilepsy cohort and studied the effect of the identified variants on SYNJ1 mRNA or phosphatase activity.
Patients and methods
Family collection and clinical description
This study was a combined effort of the EuroEPINOMICS RES consortium working group on recessive epilepsies, a project on presumed mitochondrial diseases from La Pitié-Salpêtrière Hôpital in Paris, and the medical genetics unit of the Children’s Hospitals and Clinics of Minnesota. The study was approved by the Ethical Committees of the local institutes. Parents of each patient signed an informed consent form for participation.
Case report Family A
The two siblings of Family A are the only offspring of a healthy consanguineous couple of Moroccan origin (Fig. 1A). Both were born after a normal pregnancy. The girl, currently 7 years of age, was admitted to the neonatal unit 3 days after birth for poor feeding and hypotonia. Flexion spasms were diagnosed at the age of 10 months, but in retrospect they probably first occurred at the age of 2.5 months. Her younger brother, currently 6 years of age, also presented with hypotonia at birth and was moved to the neonatal unit 2 days after birth for an apparent life-threatening event. Epileptic spasms occurred at the age of 6 months. Neither sibling showed noticeable development after that. Seizure frequency progressively increased despite polytherapy and both patients developed multiple seizures daily, consisting of myoclonic seizures, tonic seizures and eye blinking. EEG showed a modified hypsarrhythmia or multifocal epileptic activity on a slow background, whereas brain MRI was normal. Overall, both children showed a neurodegenerative disease course with a progressive spastic quadriplegia, severe intellectual disability, central visual impairment, and progressive feeding problems for which tube feeding was started around the age of 5 years. The girl recently became oxygen-dependent. Extended clinical information can be found in Supplementary Table 1. This family was included in a project of the EuroEPINOMICS RES consortium that studied five consanguineous and 17 non-consanguineous families with at least two siblings presenting with unexplained early onset seizures and intellectual disability.
Figure 1.

Families with recessive inherited SYNJ1 variants reported here. (A) Pedigrees of Families A–C with an indication of the segregating SYNJ1 variants (NM_203446.2; NP_982271.2). All parents are healthy, affected children are indicated by filled symbols. Double horizontal lines indicate consanguinity between the parents and arrows point towards the patients whose fibroblasts were used in the mRNA expression studies. (B) Visualization of sequencing reads produced by direct Sanger sequencing during variant validation.
Case report Family B
The two sisters of Family B are the only offspring of a healthy Moroccan couple, who are second-degree relatives (Fig. 1A). Both were born after a normal pregnancy, had onset of clonic seizures during the first day of life and subsequently developed myoclonic and tonic-clonic seizures. The oldest sister, currently 5 years of age, had several episodes of convulsive status epilepticus. Although seizure frequency stabilized in the oldest girl, they both developed monthly seizures despite polytherapy. Both have profound intellectual disability, and developed a progressive spastic quadriplegia and feeding problems for which they received a gastrostomy. EEG showed modified hypsarrhythmia or focal spikes on a slow background, whereas brain MRI was normal. Increased serum creatine kinase was suggestive of myopathy. Due to high lactate and a phenotype associating liver disease and myoclonic epilepsy, a mitochondrial origin was suspected. Spectrophotometry of respiratory chain complexes showed a combined deficiency in complex III and IV activity in liver and fibroblasts. Metabolic investigations remained inconclusive. Extended clinical information can be found in Supplementary Table 1. This family was included in a French research study on the genetic aetiology of presumed mitochondrial disorders. Nineteen families including 25 patients were recruited according to strong arguments of mitochondrial diseases (MRI, muscle or liver biopsy, hyperlactataemia), and negative whole mitochondrial DNA screening. The cohort included three consanguineous sib-pairs, 13 sporadic consanguineous cases, and three non-consanguineous sib-pairs.
Case report Family C
Family C consists of a non-consanguineous Caucasian couple with two affected sons and a healthy daughter (Fig. 1A). The eldest brother presented seizures from Day 12, consisting of eye blinking and shoulder movements. Seizures evolved to tonic-clonic and myoclonic seizures and were difficult to treat despite combination therapy with anti-epileptic drug. EEGs were not available for review. MRI at the age of 6 months showed no abnormalities. Whereas he was hypotonic in the neonatal period, he became increasingly hypertonic with tendency to opisthotonus and co-occurring dystonia. He died at the age of 2.5 years after a progressive neurological decline from birth. At that time, he had profound intellectual disability and was tube fed. No autopsy was performed, but he was thought to have died from cardiopulmonary arrest after aspiration in the context of a gastro-intestinal infection. His younger brother had onset of seizures with eye deviation on the first day of life, and subsequently developed refractory generalized tonic-clonic and myoclonic seizures. He developed a progressive spastic quadriparesis and feeding problems for which he received a gastrostomy. He was profoundly impaired, non-verbal and did not present eye contact. He died recently at the age of 8 years, probably from cardiopulmonary impairment in the context of an unspecified infection too. No autopsy was done. EEGs in both sibs showed multifocal epileptic activity on a slow background. MRI was normal in the eldest sib, and showed a thin corpus callosum and limited gliosis and atrophy of the periventricular white matter in the youngest brother. Extended clinical information can be found in Supplementary Table 1. The parents and youngest brother underwent whole exome sequencing as part of the diagnostic workup.
Next generation sequencing and data analysis
Families A and B were included in two different next generation sequencing research studies, and Family C in a clinical diagnostic workup, with the common goal to identify the underlying genetic defect in the respective siblings. Genomic DNA was isolated from whole blood from patients and all available family members. Differences in the initial study designs led to variability in the number of individuals sequenced (four for Family A, two for Family B, and three for Family C), the choice of sequencing technique (exome or genome sequencing), and analysis platforms. Nevertheless, similar filtering cascades for variant prioritization were used. A schematic overview of the genetic workflow can be found in Supplementary Fig. 1. The most promising candidate variants were confirmed by direct Sanger sequencing.
Whole genome sequencing of Family A
Whole genome sequencing was performed by Complete Genomics on DNA from the two affected children and both healthy parents of Family A (n = 4). Data generation, annotation and variant prioritization was done as previously described (Schubert et al., 2014; Hardies et al., 2015). The pairwise population concordance (Supplementary material) and runs of homozygosity were determined with PLINK v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/) (Purcell et al., 2007).
Whole exome sequencing of Family B
Whole exome sequencing was performed on DNA from the two affected children from Family B (n = 2) by the IGBMC Microarray and Sequencing platform (Strasbourg, France). Exon-capture was performed using the SureSelect XT2 Human all exon V5 enrichment System (Agilent) followed by HiSeq 2500 sequencing (Illumina Inc.) according to the manufacturer’s protocol. Image analysis and base calling were performed using the Real-Time Analysis software from Illumina. DNA sequences were aligned to the reference genome hg19 using Burrows-Wheeler Aligner (BWA) v0.6.1 (Li and Durbin, 2009). Aligned data were then refined with the use of Picard v1.68 (http://picard.sourceforge.net/) to flag duplicate reads, GATK v2.5.2 (DePristo et al., 2011) to perform local realignments and recalibrate base qualities, and Samtools v0.1.18 (Li and Durbin, 2009) to filter out multi-mapped reads. Variant calling was done by combining results of four variant callers i.e. GATK UnifiedGenotyper, GATK HaplotypeCaller, Samtools v0.1.18 mpileup and Samtools 0.1.7 pileup. Variant quality scores were recalibrated using GATK. Variants were annotated using SnpEff v2.0.5, GATK v2.5.2 and SnpSift v3.3c (Cingolani et al., 2012a, b). Finally, VaRank (Geoffroy et al., 2015) was used to rank discovered variants, which were filtered on recessive state in the two affected children.
Whole exome sequencing of Family C
Whole exome sequencing was performed on DNA of the youngest patient of Family C and his parents (n = 3) at Ambry Genetics (Aliso Viejo, CA). Library preparation, sequencing, bioinformatics, and data analysis were performed as previously described (Farwell et al., 2015). Briefly, samples were prepared and sequenced using paired-end, 100 cycle chemistry on the Illumnia HiSeq 2500. Enrichment was performed using the IDT xGen Exome Research Panel V1.0. Data were annotated with the Ambry Variant Analyzer tool.
Genetic follow-up
To investigate the frequency of recessive SYNJ1 variants in a larger epilepsy cohort, a 3-fold genetic follow-up was performed (Supplementary Fig. 2): (i) we re-examined the SYNJ1 region within whole exome sequencing data of 97 patients with epileptic encephalopathies and their healthy parents (previously described in Suls et al., 2013); (ii) we developed a targeted Multiplex Amplification of Specific Targets for Resequencing (MASTR) assay (Multiplicom) and screened all coding regions as well as flanking intronic segments of SYNJ1 in 154 index patients with epilepsy and developmental delay (previously described in Hardies et al., 2015); and (iii) we screened 292 additional patients with a larger gene panel covering both known and candidate genes for neurodevelopmental disorders, including SYNJ1. Ninety of these patients (31%) were suspected to have a recessively inherited genetic defect based on the presence of consanguinity or multiple affected siblings. Target genes were captured with a custom made kit (Agilent) and sequenced on a SOLiD™ 4 System (Applied Biosystems). Reads were aligned with the BWA (Li and Durbin, 2009). Variant calling and analysis was done with a custom pipeline.
Evaluating the effect of missense variants on the enzymatic activity of SYNJ1
To study the effect of identified missense variants in SYNJ1 on protein function, the dephosphorylation by recombinant purified proteins of domain-specific substrates, diC8 PI(4,5)P2 and diC8 PI(3,4,5)P3 for 5’PP, and diC8 phosphatidylinositol 4-phosphate [PI(4)P] for Sac1 was quantified. To obtain wild-type and mutant proteins, the coding sequence of human 145 kD SYNJ1 was cloned into the p3XFLAG-CMV10 vector. Substitution p.Tyr888Cys and p.Met1020Ile/p.Tyr1057Ser were further generated by site-directed mutagenesis from the wild-type constructs (QuikChange® system, Stratagene). Plasmids harbouring wild-type or mutant SYNJ1 were transfected into Expi293F cells for over-expression (Thermo Fisher Scientific). Proteins were first purified by pull down using anti-FLAG M2 beads (Sigma), eluted with the FLAG peptide and further purified by gel filtration on a Superdex™ 200 column (GE Healthcare). The gel filtration buffer contained 20 mM HEPES at pH 8.0, 150 mM NaCl, and 0.5 mM TCEP [tris(2-carboxyethyl)phosphine].
The enzymatic activity of wild-type and mutant proteins was measured by a malachite-green assay as previously described (Maehama et al., 2000). In short, 5 nM SYNJ1 (wild-type or mutant) was incubated with 1 μg of substrate PI(4)P, PI(4,5)P2 or phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] at room temperature for 5 min in a total volume of 25 μl reaction mixture, which also contained 20 mM HEPES at pH 7.4, 100 mM KCl, 2 mM MgCl2, and 1 mM EGTA. After addition of 75 μl of malachite green reagent, phosphate release was measured by absorbance at OD620 (Echelon Biosciences Inc.).
Evaluating the effect of premature stop variants on SYNJ1 mRNA formation
Fibroblasts were generated from skin biopsies of the oldest affected sister from Family B and the youngest affected brother from Family C (Fig. 1A). Cells were cultivated at 37°C in either Dulbecco’s modified Eagle medium (DMEM) high glucose (Gibco®) enriched with 10% heat-inactivated foetal calf serum, 1% L-glutamine and 1% penicillin-streptomycin or minimum essential medium (MEM) enriched with 10% heat-inactivated foetal calf serum, 1% penicillin-streptomycin and 100× MEM non-essential amino acid solution (all from Thermo Scientific). To confirm the presence of nonsense-mediated mRNA decay, fibroblasts were split into three subcultures. One subculture remained untreated, whereas the other two were exposed for 4 h at 37°C to 150 µg/ml cycloheximide or an equal volume of solvent (1% dimethyl sulphoxide). After incubation, cells were extensively washed with Dulbecco’s phosphate-buffered saline (DPBS, Thermo Scientific) and harvested by centrifugation.
RNA was extracted with the RNeasy® Mini Kit (Qiagen) from 2 × 106 pelleted fibroblasts, according to the manufacturer’s instructions. The extracted RNA was treated with the TURBO DNA-free™ Kit (Ambion) and 1 µg was converted to cDNA using the SuperScript® III First-Strand Synthesis System (Life Technologies) with both oligo-dT and random hexamer primers. RNA concentrations were measured with the NanoDrop1000 (Thermo Scientific). To visualize SYNJ1 mutant transcripts and confirm cell lineage, direct Sanger sequencing was performed on 50 ng cDNA from cycloheximide treated cells (data not shown). Quantitative polymerase chain reaction (qPCR) for relative quantification of SYNJ1 transcript levels was performed on 10 ng cDNA using a Fast SYBR® Green master mix (Life Technologies) with primer pairs specific for SYNJ1 and five housekeeping genes (primers available on request). Resultant data were analysed with the ViiATM7 PCR system (Life Technologies). Measurements were normalized against the two most stable housekeeping genes determined by geNorm (Vandesompele et al., 2002). Levels of mutant transcript from patient-derived fibroblasts were relatively quantified to the level of wild-type transcript from a control cell line. Unpaired t-testing compared the normalized and relative mean ΔCt values. Statistical analyses were done with GraphPad Prism version 6.05 for Windows (La Jolla California USA, www.graphpad.com).
Results
Next generation sequencing data analysis
Different next generation study strategies were applied in Families A–C and identified potentially causal recessive variants in SYNJ1 (NM_203446.2; NP_982271.2) in all three. Additional variants are listed in Supplementary Table 2.
Whole genome sequencing of Family A
On average, 91.3% of the genome and 93.5% of the exome was covered at 20× or higher for the two siblings and their parents. The common ancestry of Family A was confirmed with a calculated pairwise population concordance value of 1 (Supplementary material) and the identification of 30 homozygous regions covering 160.477 Mb genome wide. Whole genome sequencing of both healthy parents and the two affected children resulted in an average of 4 318 298 variant callings per sample; taken together a total of 6 644 778 different variants were called in at least one family member. Of the latter, only 154 variants were retained after initial prioritization on quality, frequency in control databases, and predicted impact. The only homozygous candidate variant present in both patients was located in SYNJ1 c.2663A>G, which was validated by direct Sanger sequencing (Fig. 1B). The resulting amino acid substitution, p.Tyr888Cys, is conserved through evolution (Supplementary Fig. 3) and was unanimously predicted to be damaging by PolyPhen-2, MutationTaster, and likelihood ratio test. The two siblings did not share any compound heterozygous variants, nor did they share novel heterozygous variants that were absent in both parents (possible germline mosaicism).
Whole exome sequencing of Family B
On average, 93% of the exome was covered at 20× or higher for the two siblings. Whole exome sequencing data analysis of the two affected children of Family B resulted in an average of 66 671 variant callings per sample. Of these, 175 variants were retained after initial prioritization on quality, frequency in control databases, and predicted impact. Seven were homozygous in both sisters and an additional three compound heterozygotes were identified (Supplementary Table 2). Extensive data mining on the remaining genes’ function, disease association, expression and in silico variant predictions, implied a premature stop codon in SYNJ1 (c.2528G>A; p.Trp843*) as best candidate. Using Sanger sequencing (Fig. 1B) this variant was confirmed in the patients and found in a heterozygous state in the parents.
Whole exome sequencing of Family C
On average, 97% of the exome was covered at 20× or higher. Whole exome sequencing of this trio resulted in an average of 127 577 variant callings per sample. Of these 2165 were retained after filtering on quality, frequency in controls and impact on the protein. One homozygous and three compound heterozygous genetic defects were identified in the index patient (Supplementary Table 2). Again, extensive data mining pointed towards two variants located in SYNJ1 (c.1938delT and c.3365-2A>G; p.Gln647Argfs*6 and p.Ser1122Thrfs*6) as best candidates. Recessive segregation was confirmed in the family using Sanger sequencing (Fig. 1B); the proband’s unaffected older sister was found to be a heterozygous carrier of the c.3365-2A>G splice site alteration.
Given the comparable phenotype of the affected siblings in all three families, and the similarities with a previously reported patient (Dyment et al., 2015a, b), the bi-allelic genetic disruption of SYNJ1 was considered likely pathogenic in all three families (Supplementary Fig. 1 and Supplementary Table 2).
Genetic follow-up analyses
Using three different methods, a total of 543 index patients with various forms of epilepsy and/or developmental delay were screened for recessive variants in SYNJ1. Data analysis of the MASTR assay revealed a potentially compound heterozygous SYNJ1 defect in an isolated patient with a severe, early onset fever-sensitive epileptic encephalopathy resembling Dravet syndrome (Supplementary Fig. 2). Sanger sequencing of the patients’ parents confirmed paternal inheritance of variant c.3060G>T (p.Met1020Ile) and maternal inheritance of variant c.3170A>C (p.Tyr1057Ser). Whereas the latter variant was novel, the first has been reported (rs115683257) in a heterozygous state by both the Exome Variant Server (0.48%) and the ExAC Browser (0.07%). In silico prediction programs were inconsistent for both substitutions: predicted tolerated by SIFT, disease causing by MutationTaster and benign to possibly damaging by PolyPhen-2. We did not identify any other potential deleterious variants in SYNJ1.
Enzymatic activity is reduced for the p.Tyr888Cys variant (Family A)
We tested the effect of the identified substitutions (p.Tyr888Cys and p.Met1020Ile/p.Tyr1057Ser) on the enzymatic activity of SYNJ1 in vitro, using purified proteins from transfected Expi293F cells (Fig. 2). A clear reduction (P < 0.0001–0.0003) towards all tested substrates was found for the p.Tyr888Cys substitution located in the 5’PP domain of the protein (Fig. 3). We did not find any difference in enzymatic activity for aberrant proteins carrying the p.Met1020Ile/p.Tyr1057Ser substitution identified during our genetic follow-up study. These findings are consistent with (i) the localization of these two amino acids in a predicted unfolded region of the protein outside the phosphatase domains (Fig. 3); (ii) the non-conservation of this residue between SYNJ1 and SYNJ2, two enzymes with similar catalytic activities (Supplementary Fig. 3); (iii) the difference in phenotype compared to other patients; and (iv) the relative high frequency of one of the variants in population databases (rs115683257; 0.07–0.48%).
Figure 2.

Phosphatase activity assay of the identified SYNJ1 missense variants. The effect on enzymatic activity of SYNJ1 was tested for mutants resulting from missense variants. Wild-type or mutant FLAG-tagged SYNJ1 (NM_203446.2) was expressed in Expi293F cells, purified and tested for enzymatic activity against short chain phosphoinositides using a malachite-based assay (Maehama et al., 2000). The dephosphorylation of domain specific substrates was investigated: PI(4)P for the Sac1 domain, and PI(4,5)P2 and PI(3,4,5)P3 for the 5’PP domain. Because dephosphorylation of PI(4,5)P2 by the 5’PP domain results in PI4P, which can then be dephosphorylated by the Sac1 domain, the phosphate released when using PI(4,5)P2 as substrate reflects the action of both phosphatase domains. When PI(3,4,5)P3 was used as a substrate, selective action of the 5’PP domain could be assessed. Graphics depict mean values of released inorganic free phosphate from three independent experiments (bars reflect standard error of mean). A clear reduction, but not a complete loss, of phosphate activity towards all tested substrates was found for the p.Tyr888Cys substitution identified in Family A. Compared to wild-type expressing cells, no differences in enzymatic activity was found for cells expressing the p.Met1020Ile/p.Tyr1057Ser substitutions. Unpaired t-test was used to analyse the difference between wild-type and mutants (***P = 0.0002–0.0003, ****P < 0.0001). ns = non-significant.
Figure 3.

Protein structure of human SYNJ1. Domains of the protein are shown in different colours. The positions of newly identified (above) and previously reported (below) pathogenic variants are indicated (NP_982271.2). Variants identified in patients with early onset refractory epilepsy and progressive neurological decline are marked in red, the recurrent substitution identified in three families with early onset parkinsonism in blue, and the compound heterozygous substitutions that are likely to be polymorphisms (p.Met1020Ile/p.Tyr1057Ser), in green. Sac = Sac1 domain; 5PPtase = 5-phosphatase domain; RRM = RNA recognition motif; PRD = proline rich domain.
Messenger RNA level is diminished by the premature stop variants (Families B and C)
The p.Trp843* variant identified in Family B is located in the 5’PP domain and is predicted to result in a truncated protein, and thus a misfolded/inactive 5’PP domain. Premature stop variants can, however, induce mRNA degradation via nonsense-mediated mRNA decay. Fibroblasts carrying the p.Trp843* variant indeed contained only 15% (mutant) SYNJ1 mRNA transcript (P < 0.0001) compared to wild-type expressing cells (Fig. 4). Transcript levels could be increased after treatment with the translation inhibitor cycloheximide that blocks nonsense-mediated mRNA decay (Lejeune et al., 2003) (data not shown). Likewise, the p.Gln647Argfs*6/p.Ser1122Thrfs*3 genetic defect identified in Family C resulted in only 5% of mRNA transcript (P < 0.0001; Fig. 4).
Figure 4.
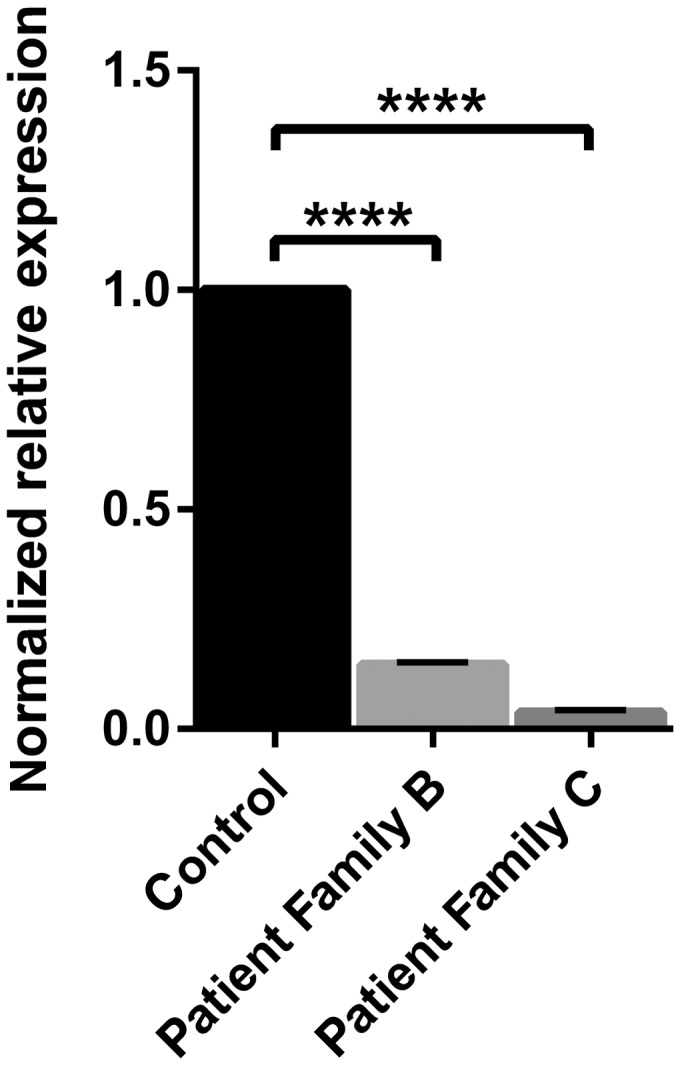
Levels of SYNJ1 mRNA in patient and control fibroblasts. The effect of the premature stop variants identified in Families B and C was evaluated by the amount of mutant mRNA transcript in patient fibroblasts. Messenger RNA was extracted from human fibroblasts expressing either homozygous wild-type (control) or mutant SYNJ1: patient Family B homozygous for p.Trp843* and patient Family C compound heterozygous for p.Gln647Argfs*6/p.Ser1122Thrfs*3. Quantitative PCR was performed on the converted cDNA. Graphics depict mean values of normalized ΔCt, relatively compared to wild-type mRNA levels, from at least two independent experiments (bars reflect standard error of mean). P-values were determined by comparing the normalized ΔCt (unpaired t-test, ****P < 0.0001). A significant reduction in SYNJ1 transcript expression was found in both patients’ fibroblasts compared to control cells.
Discussion
We describe the clinical and molecular findings of three families with four different autosomal recessive inherited SYNJ1 variants and a devastating phenotype with early onset treatment-resistant seizures and progressive neurological decline. The homozygous p.Tyr888Cys substitution identified in Family A and localized in the 5’PP domain, surprisingly showed an impairment of the dephosphorylation activity of both the 5’PP and the Sac1 domain. The unexpected impact on Sac1 domain function suggests the occurrence of an intramolecular regulation. We further showed a significant loss of mutant SYNJ1 transcript for the premature stop variants in SYNJ1, namely a homozygous p.Trp843* variant in Family B and the compound heterozygous variant p.Gln647Argfs*6/p.Ser1122Thrfs*3 in Family C. A similar reduction in SYNJ1 transcript was shown for the previously reported premature stop variant p.Arg136*, identified in a patient with a comparable phenotype (Dyment et al., 2015a, b). A large genetic follow-up study (n = 543) identified one additional patient who was compound heterozygous for two SYNJ1 substitutions (p.Met1020Ile/p.Tyr1057Ser). Phosphatase activity of purified proteins carrying these latter variants, located in the C-terminal of the protein and downstream of the catalytic domains, did not differ from wild-type proteins. Although it remains possible that these variants disrupt unknown binding sites and subsequently disable protein-protein interactions or mistarget SYNJ1 (Haffner et al., 1997), we concluded that there is currently no evidence that these variants are pathogenic. Our follow-up screening indicates that bi-allelic defects in SYNJ1 are not a frequent cause of severe epilepsy, even though the disease variant yield might be higher in a more selected cohort of patients with early onset epilepsy and progressive neurological decline.
Bi-allelic genetic disruption of SYNJ1 in animal models results in impaired synaptic vesicle recycling dynamics and accumulation of clathrin-coated vesicles (Cremona et al., 1999; Harris et al., 2000; Kim et al., 2002; Verstreken et al., 2003; Van Epps et al., 2004). Knock-out mice that survive longer than 24 h after birth (maximum up to 15 days) progressively develop a variety of neurological manifestations, including ataxia and generalized seizures (Cremona et al., 1999). Zebrafish larvae with synaptojanin mutations present with peripheral blindness and abnormal swimming behaviour (Van Epps et al., 2004; Jia et al., 2014). In humans, recessive variants in SYNJ1 have been associated with two different neurological disorders, which seem to be related to the variant type and their effect on protein function. A first disease entity consists of refractory epileptic seizures with neonatal or early infancy onset, little or no development after birth, and a progressive neurodegenerative disease course leading to spastic quadriplegia and loss of feeding capacities, as seen in the affected children reported here and in a previously reported patient (Supplementary Table 1; Dyment et al., 2015a, b). Seizure types vary and include clonic, myoclonic, tonic and/or tonic-clonic seizures. The progressive neurological decline in the presence of myoclonic seizures even led to the suspicion of a progressive myoclonic epilepsy of potential mitochondrial origin in one family (Family B). Three children died between the ages of 2.5 and 8 years, probably due to cardiorespiratory arrest, whereas the oldest living child reported here is currently 7 years old and recently became oxygen-dependent. The SYNJ1 variants associated with this severe phenotype include two homozygous nonsense variants [the novel p.Trp843*, Family B; and the previously reported p.Arg136* (Dyment et al., 2015a, b)] and two premature stop variants in a compound heterozygous state (p.Gln647Argfs*6/p.Ser1122Thrfs*3, Family C) leading to loss of functional SYNJ1 expression, plus a homozygous missense variant (p.Tyr888Cys, Family A) located in the 5’PP domain leading to a decreased dephosphorylation capacity towards all substrates. A second disease entity was recently described in three independent families, and consists of parkinsonism with onset in the third decade of life. Half of the patients with parkinsonism (3/6) also had infrequent (or even a single) generalized tonic-clonic seizures with onset during infancy or adolescence (Krebs et al., 2013; Quadri et al., 2013; Olgiati et al., 2014). All three families carried the same homozygous missense variant (p.Arg258Gln) located in the Sac1 domain. This missense variant specifically abolishes the capacity of the protein to dephosphorylate Sac1-specific substrates, whereas the 5’PP domain activity remains unchanged (Krebs et al., 2013). Hence, the phenotypic presentation associated with autosomal recessive SYNJ1 variants in humans seems to correlate with the residual SYNJ1 phosphatase activity: variants resulting in a (nearly) complete loss of SYNJ1 dual phosphatase activity, either by loss of protein formation or protein function, lead to a devastating phenotype with early onset refractory seizures, progressive neurological decline and early mortality; whereas loss of dephosphorylation activity limited to the Sac1 domain leads to parkinsonism with onset in early adulthood and an apparent mild increase in seizure susceptibility. The identification of additional patients will help addressing full phenotype–genotype correlations.
Our results add new evidence to the concept that dysregulation of endocytic recycling of synaptic vesicles in neurons is an important biological pathway in the aetiology of epilepsy with disturbance of neurodevelopment. Consistent with these findings, mice knock-out studies have shown that loss of function variants in endophilin and amphiphysin, two SYNJ1 binding proteins, also lead to severe epilepsy (Di Paolo et al., 2002; Milosevic et al., 2011), and de novo mutations in DNM1, which impairs synaptic vesicle endocytosis, were shown to cause a severe epileptic encephalopathy in humans (EuroEPINOMICS RES Consortium et al., 2014). Increased understanding of the different pathways leading to neurodevelopmental disorders will guide future development of novel treatments for these severely affected patients.
Supplementary Material
Acknowledgements
We thank the families and patients for their participation to this study. The primary collection of samples was done within the autosomal recessive working group of the EuroEPINOMICS RES consortium, therefore we acknowledge the work of: Zaid Afawi, Stéphanie Baulac, Nina Barisic, Hande Caglayan, Dana Craiu, Carolien G. De Kovel, Rosa Guerrero Lopez, Renzo Guerrini, Helle Hjalgrim, Holger Lerche, Johanna Jahn, Karl Martin Klein, Bobby C. Koeleman, Eric Leguern, Johannes Lemke, Carla Marini, Hiltrud Muhle, Felix Rosenow, José M. Serratosa, Katalin Štěrbová, Rikke S. Møller, Pasquale Striano, Yvonne Weber, Federico Zara. The Family Genomics group at the Institute for Systems Biology deserves thanks for their support and project management of the whole genome sequencing data. We thank the IGBMC Microarray and Sequencing platform, member of the ‘France Génomique’ consortium (ANR-10-INBS-0009), for whole exome sequencing of Family B. We acknowledge the contribution of Peter De Rijk for computational support and the VIB Genetic Service Facility for the genetic follow-up analyses (http://www.vibgeneticservicefacility.be).
Glossary
Abbreviations
- 5’PP
5’-phosphatase domain
- PI(4,5)P2
phosphatidylinositol 4,5-bisphosphate
Appendix 1
The AR working group of the EuroEPINOMICS RES Consortium consists of the following consortium members:
Zaid Afawi; Stéphanie Baulac; Nina Barisic; Hande Caglayan; Dana Craiu; Carolien G. De Kovel; Rosa Guerrero Lopez; Renzo Guerrini; Helle Hjalgrim; Holger Lerche; Johanna Jahn; Karl Martin Klein; Bobby C. Koeleman; Eric Leguern; Johannes Lemke; Carla Marini; Hiltrud Muhle; Felix Rosenow; José M. Serratosa; Katalin Štěrbová; Rikke S. Møller; Aarno Palotie; Pasquale Striano; Yvonne Weber; Federico Zara
Contributor Information
Collaborators: Zaid Afawi, Stéphanie Baulac, Nina Barisic, Hande Caglayan, Dana Craiu, Carolien G. De Kovel, Rosa Guerrero Lopez, Renzo Guerrini, Helle Hjalgrim, Holger Lerche, Johanna Jahn, Karl Martin Klein, Bobby C. Koeleman, Eric Leguern, Johannes Lemke, Carla Marini, Hiltrud Muhle, Felix Rosenow, José M. Serratosa, Katalin S Štěrbová, Rikke S. Møller, Aarno Palotie, Pasquale Striano, Yvonne Weber, and Federico Zara
Funding
P.D.J. (G.A.136.11.N and FWO/ESF-ECRP) and I.H. (HE5415/3-1) received financial support from the Eurocores program of the European Science Foundation. C.J. received funding from the Fondation Maladies Rares ‘high throughput sequencing and rare diseases’ (CJ call 2013-2) for the project ‘Towards the identification of novel genes involved in mitochondrial functions in genetically and biochemically informative patients’ enabling whole exome sequencing of Family B. P.D.C. was supported by NIH grants (R37NS036251 and DA018343), and a grant from The Michael J. Fox Foundation for Parkinson’s research (MJFF; 11353). This work was further supported by the University of Antwerp and the Fund for Scientific Research Flanders (FWO), International Coordination Action (ICA) grant G0E8614N. M.C. has a fellowship from the Parkinson disease Foundation. A.S. was a postdoctoral fellow of the FWO and is now supported by the University of Antwerp. K.H. and T.D. were PhD fellows of the Institute for Science and Technology (IWT)-Flanders. P.M. is supported as an ISB/LCSB fellow by ‘le plan Technologies de la Santé par le Government du Grand-Duché de Luxembourg’ through the Luxembourg Centre for Systems Biomedicine (LCSB) at the University of Luxembourg.
Supplementary material
Supplementary material is available at Brain online.
Conflict of interest
Since mid-October 2015, K. Hardies is under employment of UCB Pharma (Braine-l'Alleud, Belgium). The company had no part in this study, all the data was generated and analysed before enrolment. K. L. Helbig and S.A. Sajan are employed by Ambry Genetics, a company that provides exome sequencing among its commercially available tests (here used for Family C). None of the other authors have disclosures.
References
- Brodin L, Low P, Shupliakov O. Sequential steps in clathrin-mediated synaptic vesicle endocytosis. Curr Opin Neurobiol 2000; 10: 312–20. [DOI] [PubMed] [Google Scholar]
- Cestra G, Castagnoli L, Dente L, Minenkova O, Petrelli A, Migone N, et al. The SH3 domains of endophilin and amphiphysin bind to the proline-rich region of synaptojanin 1 at distinct sites that display an unconventional binding specificity. J Biol Chem 1999; 274: 32001–7. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet 2012a; 3: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang lL, Nguyen T, Wang L, Land SJ, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012b; 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Kim WT, Takei K, Daniell L, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 1999; 99: 179–88. [DOI] [PubMed] [Google Scholar]
- de Heuvel E, Bell AW, Ramjaun AR, Wong K, Sossin WS, McPherson PS. Identification of the major synaptojanin-binding proteins in brain. J Biol Chem 1997; 272: 8710–16. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006; 443: 651–7. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, et al. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 2002; 33: 789–804. [DOI] [PubMed] [Google Scholar]
- Dong Y, Gou Y, Li Y, Liu Y, Bai J. Synaptojanin cooperates in vivo with endophilin through an unexpected mechanism. Elife 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyment DA Smith AC Humphreys P Schwartzentruber J Beaulieu CL FORGE Canada Consortium, et al. Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol Aging 2015a; 36: 1222–5. [DOI] [PubMed] [Google Scholar]
- Dyment DA, Tetreault M, Beaulieu CL, Hartley T, Ferreira P, Chardon JW, et al. Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet 2015b; 88: 34–40. [DOI] [PubMed] [Google Scholar]
- EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014; 95: 360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol 2001; 155: 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015; 17: 578–86. [DOI] [PubMed] [Google Scholar]
- Gad H, Ringstad N, Low P, Kjaerulff O, Gustafsson J, Wenk M, et al. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron 2000; 27: 301–12. [DOI] [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, et al. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J 2006; 25: 2898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy V, Pizot C, Redin C, Piton A, Vasli N, Stoetzel C, et al. VaRank: a simple and powerful tool for ranking genetic variants. PeerJ 2015; 3: e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Stolz LE, Lemrow SM, York JD. SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem 1999; 274: 12990–5. [DOI] [PubMed] [Google Scholar]
- Haffner C, Di Paolo G, Rosenthal JA, De Camilli P. Direct interaction of the 170 kDa isoform of synaptojanin 1 with clathrin and with the clathrin adaptor AP-2. Curr Biol 2000; 10: 471–4. [DOI] [PubMed] [Google Scholar]
- Haffner C, Takei K, Chen H, Ringstad N, Hudson A, Butler MH, et al. Synaptojanin 1: localization on coated endocytic intermediates in nerve terminals and interaction of its 170 kDa isoform with Eps15. FEBS Lett 1997; 419: 175–80. [DOI] [PubMed] [Google Scholar]
- Hardies K, May P, Djemie T, Tarta-Arsene O, Deconinck T, Craiu D, et al. Recessive loss-of-function mutations in AP4S1 cause mild fever-sensitive seizures, developmental delay and spastic paraplegia through loss of AP-4 complex assembly. Hum Mol Genet 2015; 24: 2218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Hartwieg E, Horvitz HR, Jorgensen EM. Mutations in synaptojanin disrupt synaptic vesicle recycling. J Cell Biol 2000; 150: 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Muto A, Orisme W, Henson HE, Parupalli C, Ju B, et al. Zebrafish Cacna1fa is required for cone photoreceptor function and synaptic ribbon formation. Hum Mol Genet 2014; 23: 2981–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WT, Chang S, Daniell L, Cremona O, Di Paolo G, De Camilli P. Delayed reentry of recycling vesicles into the fusion-competent synaptic vesicle pool in synaptojanin 1 knockout mice. Proc Natl Acad Sci USA 2002; 99: 17143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive parkinsonism with generalized seizures. Hum Mutat 2013; 34: 1200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Li X, Maquat LE. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 2003; 12: 675–87. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi A, Di Paolo G, Cremona O, Daniell L, De Camilli P, McCormick DA. Synaptojanin 1 contributes to maintaining the stability of GABAergic transmission in primary cultures of cortical neurons. J Neurosci 2001; 21: 9101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Slama JT, Dixon JE. A sensitive assay for phosphoinositide phosphatases. Anal Biochem 2000; 279: 248–50. [DOI] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, et al. The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 2007; 56: 1004–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea HJ, De Camilli P. Mutations in phosphoinositide metabolizing enzymes and human disease. Physiology (Bethesda) 2009; 24: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, et al. A presynaptic inositol-5-phosphatase. Nature 1996; 379: 353–7. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Collesi C, Shen H, Paradise S, et al. Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 2011; 72: 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgiati S, De Rosa A, Quadri M, Criscuolo C, Breedveld GJ, Picillo M, et al. PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 2014; 15: 183–8. [DOI] [PubMed] [Google Scholar]
- Perera RM, Zoncu R, Lucast L, De Camilli P, Toomre D. Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. Proc Natl Acad Sci USA 2006; 103: 19332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posor Y, Eichhorn-Gruenig M, Puchkov D, Schöneberg J, Ullrich A, Lampe A, et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013; 499: 233–7. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 2013; 34: 1208–15. [DOI] [PubMed] [Google Scholar]
- Ramjaun AR, McPherson PS. Tissue-specific alternative splicing generates two synaptojanin isoforms with differential membrane binding properties. J Biol Chem 1996; 271: 24856–61. [DOI] [PubMed] [Google Scholar]
- Ringstad N, Nemoto Y, De Camilli P. The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci USA 1997; 94: 8569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P. Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol 2012; 4: a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Ernstrom GG, Watanabe S, Weimer RM, Chen CH, Sato M, et al. Differential requirements for clathrin in receptor-mediated endocytosis and maintenance of synaptic vesicle pools. Proc Natl Acad Sci USA 2009; 106: 1139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 2014; 46: 1327–32. [DOI] [PubMed] [Google Scholar]
- Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, et al. Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest 2012; 122: 4401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci 2004; 27: 509–47. [DOI] [PubMed] [Google Scholar]
- Suls A, Jaehn JA, Kecskes A, Weber Y, Weckhuysen S, Craiu DC, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013; 93: 967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K, McPherson PS, Schmid SL, De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature 1995; 374: 186–90. [DOI] [PubMed] [Google Scholar]
- Van Epps HA, Hayashi M, Lucast L, Stearns GW, Hurley JB, De Camilli P, et al. The Zebrafish nrc mutant reveals a role for the polyphosphoinositide phosphatase synaptojanin 1 in cone photoreceptor ribbon anchoring. J Neurosci 2004; 24: 8641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, et al. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 2003; 40: 733–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.