

Summary
Allan-Herndon-Dudley syndrome (AHDS) is an X-linked disorder caused by impaired thyroid hormone transporter. Patients with AHDS usually exhibit severe motor developmental delay, delayed myelination of the brain white matter, and elevated T3 levels in thyroid tests. Neurological examination of two patients with neurodevelopmental delay revealed generalized hypotonia, and not paresis, as the main neurological finding. Nystagmus and dyskinesia were not observed. Brain magnetic resonance imaging demonstrated delayed myelination in early childhood in both patients. Nevertheless, matured myelination was observed at 6 years of age in one patient. Although the key finding for AHDS is elevated free T3, one of the patients showed a normal T3 level in childhood, misleading the diagnosis of AHDS. Genetic analysis revealed two novel SLC16A2 mutations, p.(Gly122Val) and p.(Gly221Ser), confirming the AHDS diagnosis. These results indicate that AHDS diagnosis is sometimes challenging owing to clinical variability among patients.
Keywords: Thyroid function, delayed myelination, monocarboxylate transporter 8 (MCT8)
1. Introduction
Allan-Herndon-Dudley syndrome (AHDS; MIM #300523) is an X-linked disorder caused by impaired thyroid hormone transporters (monocarboxylate transporter 8: MCT8) (1). Patients with this syndrome exhibit severe motor developmental delay and radiological examination using magnetic resonance imaging (MRI) reveals delayed maturation of brain white matter (2,3). An abnormal thyroid hormone profile is another key finding for clinical diagnosis of AHDS (1). However, thyroid hormone abnormalities are not common among AHDS patients (4). The severity of delayed myelination in patients is also variable (5). Therefore, clinical diagnosis of patients is sometimes challenging, and the disorder is often under-diagnosed. A final and complete diagnosis requires detection of a mutation in the solute carrier family 16 member 2 gene (SLC16A2) (5). This requirement also contributes to under-diagnosis.
Previously, we identified some SLC16A2 mutations (6,7). In this study, we identified a new SLC16A2 mutation in additional patients.
2. Patients and Methods
2.1. Patients
Case report 1 A 19-year-old male patient is a third-born child and had a birth weight of 2,940 g (25–50th centile). His elder brother (II-2) showed developmental delay from infancy, and required a tracheotomy at 12 years of age due to respiratory dysfunction (Figure 1A). He died suddenly at 14 years of age owing to bleeding from a trachea brachiocephalic artery fistula. The patient's elder sister is healthy and has a healthy son. The paternal aunt and the maternal uncle died in childhood from unknown etiology.
Figure 1.
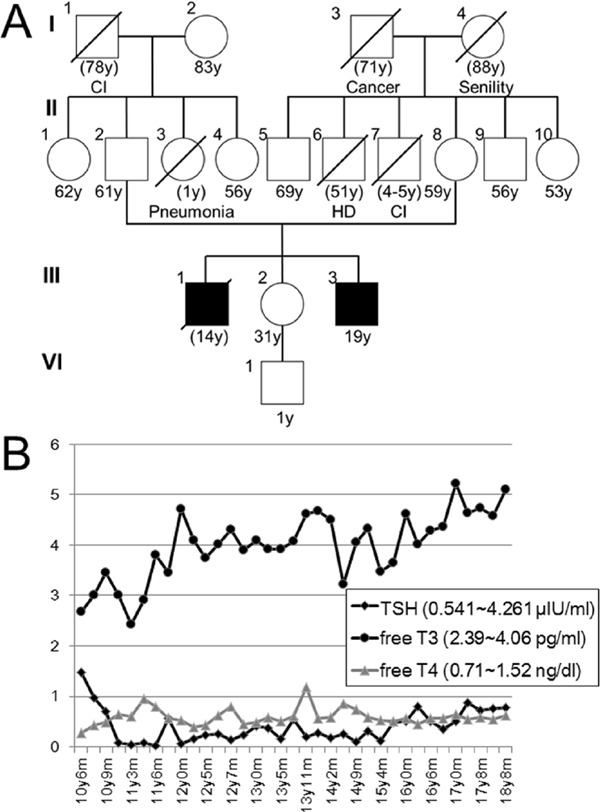
Clinical information for patient 1. (A) Family tree of patient 1. CI; cerebral infarction, HD; heart disease. (B) Serial results of thyroid hormone examinations in patient 1. Normal ranges are shown on the left. Free T3 gradually increased.
At 5 months of age, developmental delay was suggested due to head lagging. Subsequently, he showed failure to thrive as evidenced by his weight, which was consistently under the 3rd centile (7,150 g at 12 months old, 7,950 g at 3 years old, 8,420 g at 6 years old, 8,435 g at 9 years old, 10.2 kg at 12 years old, and 12.0 kg at 19 years old). Gross motor development was severely delayed and he was bedridden. His maximum motor ability was rolling over to the right lateral decubitus position. Developmental deterioration was not noted. At the age of 9 years, tube feeding was initiated owing to insufficient water intake. At 19 years old, he received a tracheostomy owing to recurrent respiratory infections. At night, he used a respirator. At this age, a gastric fistula was constructed.
At 10 years, a low free T4 level of 0.28 ng/dL (normal range: 0.71–1.52 ng/dL) was noted. Thyroid stimulating hormone (TSH) of 1.478 µIU/mL (normal range: 0.541–4.261 µIU/mL) and free T3 of 2.67 pg/mL (normal range: 2.39–4.06 pg/mL) were within normal limits. Then, thyroid hormone was prescribed and TSH level was decreased. Free T3 level was gradually increased from 12 years old (Figure 1B). Brain MRIs were regularly examined (Figures 2A–F). At 2 years and 6 months of age, subcortical white matter showed a high T2 signal (Figures 2A and 2B), indicating delayed myelination. Since then, dilatation of the lateral ventricles and extracerebral spaces were observed (Figure 2E and 2F).
Figure 2.
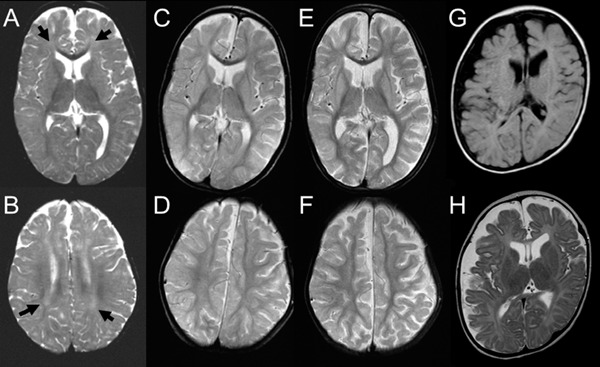
Brain magnetic resonance imaging. (A–F) Serial T2-weighted axial images for patient 1. Delayed myelination is demonstrated at 2 years and 6 months for the deep white matter (A, B; arrows). At 6 years and 1 month, myelination is mature (C, D). Diffuse brain atrophy is exhibited at 7 years and 8 months (E, F). (G, H) Axial images for patient 2 examined at 6 months. T1(G)- and T2(H) axial images demonstrate hypomyelination and brain volume loss.
At present, his height is 129 cm (< 3rd centile) and his weight is 12 kg (< 3rd centile). He shows emotional reactions, characterized by various expressions, to environmental stimuli, but does not convey meaningful words or gestures. He occasionally nods in question and vocalizes during songs. He still shows head lagging. Neurological examination indicates generalized hypotonia. His upper and lower extremities show flexion and extension with scoliosis and knee contractures.
Case report 2 A 6-month-old male baby is the first born to healthy parents by emergency caesarian section at 41 weeks of gestation, with a birth weight of 3,820 g (> 97th centile). Apgar score was 8/8 at 1/5 minutes. His mother had a past history of spontaneous abortion. Family history was not remarkable. In early infancy, poor sucking was noted. At 6 months, owing to uncontrolled head, he was referred to the hospital. At that time, his weight was 7.75 kg (25–50th centile), length was 66.2 cm (10–25th centile), and occipitofrontal circumference was 44.2 cm (50–75th centile). Neurological examination showed generalized hypotonia. There was no nystagmus. Blood examination showed elevated TSH (4.720 µIU/mL) and free T3 (10.74 pg/mL) and a low level of free T4 (0.59 ng/dL). Conventional G-banding showed normal males karyotype of 46,XY. Brain MRI showed remarkable hypomyelination and reduced volume of the cerebrum (Figures 2G, H). Auditory brainstem response showed normal latency time. Due to poor sucking, tube feeding was initiated.
2.2. Genetic examination
A genetic study was performed in accordance with the Declaration of Helsinki and approval by the Ethics Committee at the Tokyo Women's Medical University. Based on clinical and radiological examinations, we suspected AHDS as a candidate diagnosis for both patients. After obtaining written informed consent from the patient's family, Sanger sequencing of the SLC16A2 coding exons was performed and single nucleotide alterations were identified; for patient 1 in exon 1: NM_006517.4 (SLC16A2_v001):c.365G>T [p.(Gly122Val)] and for patient 2 in exon 2: NM_006517(SLC16A2_v001):c.661G>A [p.(Gly221Ser)]. Both variants were not reported in the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB). The affected amino acids were conserved among species (Figures 3A and 3B). Functional effects of these variants were predicted using Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/www/SIFT_help.html), CADD (http://cadd.gs.washington.edu/info), and MutationTaster (http://www.mutationtaster.org/). All scores predicted a damaging effect. Because the identified two mutations were not reported previously, those were considered as novel mutations (8).
Figure 3.
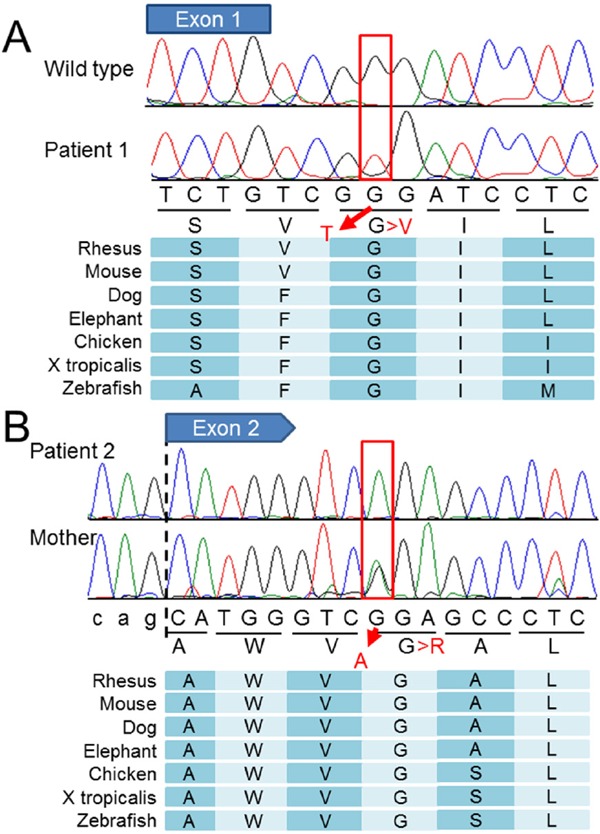
Results of Sanger sequencing and conservation among species. (A) Electropherogram demonstrates hemizygous G>T nucleotide change in patient 1. The affected codon is conserved among species (below). (B) Electropherogram demonstrates hemizygous G>A mutation in patient 2. His mother is heterozygous for the mutation, indicating an obligate carrier. The affected codon is conserved among species (bottom).
The mother of patient 2 was heterozygous for the mutation, indicating an obligate carrier. The mother of patient 1 declined to be genotyped.
3. Results and Discussion
In this study, neurological findings of both patients were mainly generalized hypotonia, not paresis. Nystagmus and dyskinesia were not observed. Delayed myelination was observed during childhood. The difference between the two patients was the result of thyroid functions. The key finding for AHDS is an elevated free T3 level (1). Patient 2 showed the typical free T3 elevation in infancy. Thus, AHDS was easily suspected as a tentative diagnosis. On the other hand, patient 1 did not show free T3 elevation in childhood. Rather, free T3 was gradually elevated in adolescence.
Familial occurrence in patient 1 suggested an X-linked recessive disorder; however, there are many X-linked disorders associated with intellectual disability. Thus, we could not list AHDS as a candidate diagnosis for this patient. A final diagnosis of AHDS was eventually suspected for this patient based on gradually elevated free T3 levels and retrospective MRI evaluations suggesting transient delay of white matter maturation. Finally, a novel mutation in SLC16A2 was identified at 19 years of age.
Tsurusaki et al. reported a patient with white matter abnormalities (9). Because an elevated T3 level was not detected in the patient, they did not suspect AHDS as a candidate diagnosis. The exome sequencing identified an SLC16A2 mutation in this case, and a final diagnosis of AHDS was made (9). This finding supports our inference that AHDS diagnosis is sometime challenging. Patterns of thyroid dysfunction vary among patients with AHDS (4). Although an elevated T3 level is listed as a necessary finding (8), we should consider AHDS as a candidate diagnosis when patients show generalized hypotonia and developmental delay, irrespective of thyroid function abnormalities.
Generally, growth impairment is not observed in early childhood, but later in patients with AHDS (1). Only a few reported children with AHDS showed growth impairment in early childhood (6,10). In this study, severe growth impairment and psychomotor developmental delay were the main symptoms in patient 1.
Acknowledgements
We would like to express our gratitude to the patients and their families for their cooperation. This work was supported by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and development (AMED), a Grant-in-Aid for Scientific Research from Health Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare, Japan, and JSPS KAKENHI Grant Number 15K09631.
References
- 1. Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005; 77:41-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tonduti D, Vanderver A, Berardinelli A, Schmidt JL, Collins CD, Novara F, Genni AD, Mita A, Triulzi F, Brunstrom-Hernandez JE, Zuffardi O, Balottin U, Orcesi S. MCT8 deficiency: Extrapyramidal symptoms and delayed myelination as prominent features. J Child Neurol. 2013; 28:795-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matheus MG, Lehman RK, Bonilha L, Holden KR. Redefining the pediatric phenotype of X-linked monocarboxylate transporter 8 (MCT8) deficiency: Implications for diagnosis and therapies. J Child Neurol. 2015; 30:1664-1668. [DOI] [PubMed] [Google Scholar]
- 4. Boccone L, Dessi V, Meloni A, Loudianos G. Allan-Herndon-Dudley syndrome (AHDS) in two consecutive generations caused by a missense MCT8 gene mutation. Phenotypic variability with the presence of normal serum T3 levels. Eur J Med Genet. 2013; 56:207-210. [DOI] [PubMed] [Google Scholar]
- 5. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004; 74:168-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kobayashi S, Onuma A, Inui T, Wakusawa K, Tanaka S, Shimojima K, Yamamoto T, Haginoya K. Clinical course and images of four familial cases of Allan-Herndon-Dudley syndrome with a novel monocarboxylate transporter 8 gene mutation. Pediatr Neurol. 2014; 51:414-416. [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto T, Shimojima K, Umemura A, Uematsu M, Nakayama T, Inoue K. SLC16A2 mutations in two Japanese patients with Allan-Herndon-Dudley syndrome. Hum Genome Var. 2014; 1:14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friesema EC, Visser WE, Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol. 2010; 322:107-113. [DOI] [PubMed] [Google Scholar]
- 9. Tsurusaki Y, Osaka H, Hamanoue H, Shimbo H, Tsuji M, Doi H, Saitsu H, Matsumoto N, Miyake N. Rapid detection of a mutation causing X-linked leucoencephalopathy by exome sequencing. J Med Genet. 2011; 48:606-609. [DOI] [PubMed] [Google Scholar]
- 10. Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005; 153:359-366. [DOI] [PubMed] [Google Scholar]