

Summary
Coffin-Siris syndrome (CSS) (MIM 135900) is characterized by developmental delay, severe speech impairment, distinctive facial features, hypertrichosis, aplasia or hypoplasia of the distal phalanx or nail of the fifth digit and agenesis of the corpus callosum. Recently, it was shown that mutations in the ARID1B gene are the main cause of CSS, accounting for 76% of identified mutations. Here, we report a 15 year-old female patient who was admitted to our clinic with seizures, speech problems, dysmorphic features, bilaterally big, large thumb, café-au-lait (CAL) spots, obesity and hyperinsulinism. First, the patient was thought to have an association of neurofibromatosis and Rubinstein Taybi syndrome. Because of the large size of the NF1 gene for neurofibromatosis and CREBBP gene for Rubinstein Taybi syndrome, whole exome sequence analysis (WES) was conducted and a novel ARID1B mutation was identified. The proband WES test identified a novel heterozygous frameshift mutation c.3394_3395insTA in exon 13 of ARID1B (NM_017519.2) predicting a premature stop codon p.(Tyr1132Leufs*67). Sanger sequencing confirmed the heterozygous c.3394_3395insTA mutation in the proband and that it was not present in her parents indicating de novo mutation. Further investigation and new cases will help to understand this phenomenon better.
Keywords: ARID1B gene, café-au-lait spots, Coffin-Siris syndrome, phenotypic expansion
1. Introduction
Coffin-Siris syndrome (CSS, MIM 135900) is characterized by mild-to-moderate intellectual disability (ID), severe speech impairment, coarse facial features, hypertrichosis, hypoplastic or absent fifth fingernails or toenails and agenesis of the corpus callosum (1,2).
Recently, mutations in the genes ARID1A, ARID1B, SMARCA4, SMARCB1, and SMARCE1, all of them encoding proteins belonging to the BAF complex which is one of the ATP-dependent chromatin remodeling complexes, were determined to be causative of CSS (3,4). It is now clear that mutations in different genes cause the differences in the phenotypes of patients with CSS. Among these genes, it was shown that mutations in ARID1B are the main cause of CSS, accounting for 68%, 83%, and 76% of identified mutations in different studies (3,5). Before the molecular basis of CSS was known, the diagnosis of CSS was based rigorously on clinical findings. After molecular diagnosis of the patients with CSS, the phenotypic spectrum of CSS is becoming wider especially in patients with ARID1B mutations (6).
Here, we report a child with a novel ARID1B mutation identified by whole-exome sequencing (WES) that presented with obesity and hyperinsulinism supporting earlier studies (6). Also she has café-au-lait (CAL) spots expanding the phenotypic spectrum of patients with CSS.
2. Case Report
A 15 year-old female patient was admitted to child neurology outpatient clinic with a speech problem and seizures. She was born to first-cousin parents as a second child after term pregnancy and normal delivery. At 2 months old, she had generalized tonic clonic seizure with fever during lung infection. She was able to sit at 1 year and walk independently at 2.5 years. She could speak single words at 10 years. Now, she can make sentences with two words. She had been evaluated at another clinic and had started a rehabilitation program for 5 years, also Ecocardiography (ECHO) revealed an atrial septal defect at that time. At the age of 13 years, afebrile generalized tonic-clonic seizures had started 3–4 times monthly and oxcarbazepine therapy was added. At that time, her weight was 32 kg (50–75%); height 150 cm (25–50%), head circumference was 54 cm. After the 6 month period, she has been seizure free for three years. At the age of 12 years, stereotypic movements have started. At 14 years old, she was operated for autism spectrum disorder (ASD).
At the age of 15: her weight was 68.5 kg (97%), height 150 cm (3%), BMI: 32.14. Physical examination showed craniofacial abnormalities including coarse face, low frontal hairline, synophrys, thick eyebrows, broad nose, thick, anteverted alae nasi, large mouth, thin upper vermillion, thick lower vermillion, short philtrum, short neck and large ears (Figure 1a). Other dysmorphic features included bilaterally big, large thumbs and short second finger on food, hallux valgus and pes planus. There are 9–10 numbers of CAL lesions on the left gluteal area and internal side of the femur (with sizes between 2–6 centimeters) (Figure 1b). Also she has truncal obesity and excessive body hair especially on the lumbar and sacral region but all of the body. Neurological examination was normal except for mental and motor retardation. There are no other manifestations of NF in the girl and also there are not any other family members with CAL spots and Lisch nodules in the family.
Figure 1.
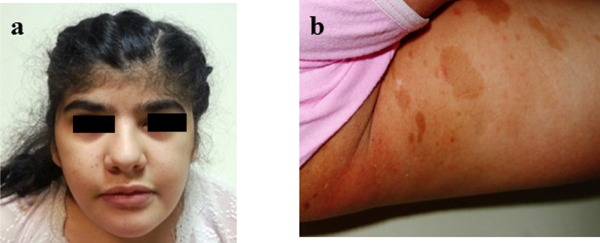
(a) Facial view of the patient (b) multiple café-au-lait spots on the left gluteal area.
Laboratory investigation including hemogram, biochemical and metabolic investigations including lactic acid, ammonia, urine and blood amino acids, and tandem MS were normal except for high TSH (6.46 IU/mL), insulin (29,4 mIU/mL, N: 2.6–24.9) levels and PTH levels (122.2 pg/mL, N:15–65) and low Vitamin D (5.1microgr/L). Cranial MRI showed small cerebellum, thin corpus callosum, and right lateral temporal periventricular (subependymal) nodular heterotopia. One year later, cranial MRI was repeated for evaluation of the cerebellum. There was no change in size. So, it was accepted as cerebellar hypoplasia. The Spinal MRI showed flat cervical lordosis. Renal doppler USG revealed bilateral Grade I increased echo. Pelvic ultrasonography was normal. Ophthalmological examination was first made at another center and Lisch nodules were reported on the left eye. According to these findings patient's prediagnosis was thought to be neurofibromatosis and Rubinstein Taybi syndrome. Because of the big size of the NF1 gene for neurofibromatosis and CREBBP gene for Rubinstein Taybi syndrome, we decided to perform a whole exome sequence analyses.
3. Genetics
Cytogenetic analyses showed 46,XX. There is no translocation or chromosomal rearrangement that involves both genes. After karyotype analyses, we decided to perform a WES analyses on this patient. We especially paid attention to NF1 and CREBBP gene sequences during WES data analysis in terms of mutation. However we found nothing in these genes. After variant prioritization in the WES data, we identified the ARID1B (ARID-containing protein 1B [MIM 614556]) mutation, which was located on chromosome 6, involving a TA insertion (c.3394_3395insTA [NM_017519.2]) leading to p.(Tyr1132Leufs*67) (RefSeq NP_059989). Our case was heterozygous for this mutation, and the parents were normal indicating de novo mutation (Figure 2). This mutation caused the frameshift mutation which possibly caused the truncated protein formation. ARID1B mutation in this study was not annotated in databases of human variation [Exome Variant Server (http://evs.gs.washington.edu/EVS/), 1000 genomes (http://www.1000genomes.org/), dbSNP (http://www.ncbi.nlm. nih.gov/projects/SNP/)]. Additionally, the mutation was not seen in the IGBAM in-house exome database (n = 777). To determine the predicted consequences of the mutations on ARID1B function, we used an in silico prediction method. Mutation Taster indicated that the mutation would be harmful. The ARID1B variant c.3394_3395insTA identified in this study was submitted to the Leiden Open Variation Database (LOVD) at www.lovd.nl/ARID1B. The mutation was confirmed by Sanger sequencing. We also sequenced the mutation site for both parents and determined that mutation to be de novo (Figure 3).
Figure 2.
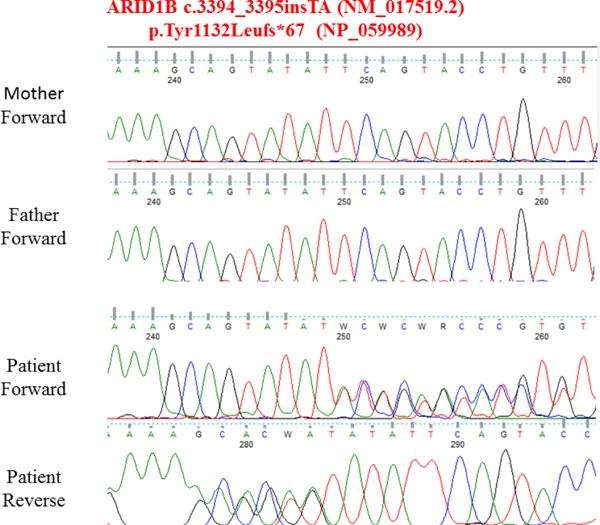
Electropherogram of the ARID1B mutation identified in our case compared with her parents electropherogram.
Figure 3.
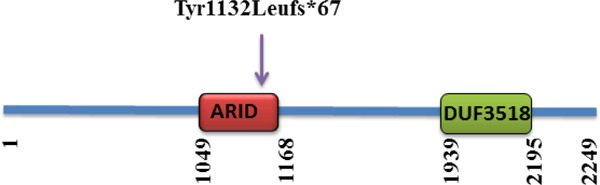
The protein domain organization of ARID1B and the localization of ARID1B mutation associated with Coffin-Siris syndrome.
We wanted to confirm Lisch nodules after molecular diagnosis in our hospital (ARID1B mutation) and it revealed that the patient has no Lisch nodules on her eyes. So it is especially important that molecular diagnosis changed from the first diagnosis. Also, so we believe that it is a phenotypic expansion rather than the probability of co-occurrence.
4. Discussion
CSS is characterized by developmental delay, severe speech impairment, distinctive facial features, hypertrichosis, aplasia or hypoplasia of the distal phalanx or nail of the fifth digit and agenesis of the corpus callosum (2). This definition was based completely on clinical findings and was made before the molecular basis of CSS was well known. With the recent detection of truncated heterozygous mutations in the BAF complex in some individuals with CSS, it is an inevitable requirement that diagnostic criteria and definition for CSS must evolve.
In line with this view, most common features of CSS patients with an ARID1B mutation are determined to have intellectual disability, speech delay, coarse facial features, and hypertrichosis in a recent study. Other findings, present in a smaller subgroup of CSS patients are determined to have small 5th finger or toe nails, short fifth finger, feeding difficulties, agenesis of the corpus callosum, seizures, myopia, and growth delay in the same study (7).
Our case has no fifth digit involvement similar to some earlier CSS cases with ARID1B mutations (3,8,9). Although microcephaly has been mentioned as a feature of CSS (10), we did not observe it in our patient compared to some larger CSS cohort studies (11).
Our patient presented with obesity and hyperinsulinism supporting Vals et al. They suggest that these features may be associated with ARID1B gene mutations, further broadening the phenotypic spectrum of CSS, and they may be added to the list of clinical features of ARID1B mutations (6). These results reinforce the view that reevaluation of individuals with a broader phenotype is needed to determine the frequency of this finding in persons with molecularly confirmed CSS.
To the best of our knowledge this is the first report of a patient with CSS who also has CAL spots. CALs are present as well-circumscribed, evenly pigmented macules and patches that range in size from 1 to 2 mm to greater than 20 cm in greatest diameter. CAL macules are common in children. Although most CAL is present as 1 or 2 spots in an otherwise healthy child, the presence of multiple CAL, large segmental CAL, associated facial dysmorphism, other cutaneous anomalies, or unusual findings on physical examination should suggest the possibility of an associated syndrome. Neurofibromatosis type 1 is the most common syndrome seen in children with multiple CAL. Other associated multisystemic genetic disorders include McCune Albright Syndrome, Tuberous sclerosis, Fanconi anemia, Bloom syndrome, Ataxia telangiectasia, Russell-Silver Syndrome, Multiple endocrine neoplasia type 2b, Bannayan-Riley- Ruvalcaba syndrome, and Multiple lentigines (LEOPARD) syndrome (12). Also the presence of multiple CAL has been described in patients with ring chromosome syndromes involving chromosomes 7, 11, 12, 15, and 17 (13).
As can be seen from the above discussion and earlier studies, there is a big variation in phenotype between patients with CSS. The underlying mechanism of this variation has yet to be answered. Different functional effects on the BAF complex consists of over 25 core and interchangeable protein subunits by ARID1B haploinsufficiency and SMARCB1/SMARCE1 missense mutations could give rise to broad variation in the clinical phenotypes (14). Also, the set of genes transcriptionally changed due to mutations in the BAF complex genes like ARID1B could modify the phenotype and determine how severely affected a patient with ARID1B haploinsufficiency will be.
Also our data supports the view by Dixon-Salazar et al. that not only is whole-exome sequencing a useful tool for identifying disease-causing genes, but it is also able to redefine or modify the diagnosis for some patients (15).
5. Conclusions
We report a patient with diagnosed CSS with WES. Clinically, in addition to classic features of CSS, the patient presented with CAL spots, obesity and hyperinsulinism. To the best of our knowledge this is the first report of a patient with CSS who has CAL spots. Further investigations and new cases will help to understand this phenomenon better.
Acknowledgements
This work was supported by the Republic of Turkey Ministry of Development Infrastructure Grant (no: 2011K120020) and BILGEM-TUBITAK (The Scientific and Technological Research Council of Turkey) (grant no: 100132). We thank the proband and her family for participating in this study.
Note: We obtained a written informed consent from the families of the indexed individuals for participation in this study and accompanying images. The study was performed according to the Declaration of Helsinki protocols.
References
- 1. Coffin GS, Siris E. Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child. 1970; 119:433-439. [DOI] [PubMed] [Google Scholar]
- 2. Fleck BJ, Pandya A, Vanner L, Kerkering K, Bodurtha J. Coffin-Siris syndrome: Review and presentation of new cases from a questionnaire study. Am J Med Genet. 2001; 99:1-7. [DOI] [PubMed] [Google Scholar]
- 3. Santen GW, Aten E, Sun Y, et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012; 44:379-380. [DOI] [PubMed] [Google Scholar]
- 4. Tsurusaki Y, Okamoto N, Ohashi H, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012; 44:376-378. [DOI] [PubMed] [Google Scholar]
- 5. Tsurusaki Y, Okamoto N, Ohashi H, et al. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2014; 85:548-554. [DOI] [PubMed] [Google Scholar]
- 6. Vals MA, Oiglane-Shlik E, Noukas M, Shor R, Peet A, Kals M, Kivistik PA, Metspalu A, Ounap K. Coffin-Siris Syndrome with obesity, macrocephaly, hepatomegaly and hyperinsulinism caused by a mutation in the ARID1B gene. Eur J Hum Genet. 2014; 22:1327-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Santen GW, Clayton-Smith J, ARID1B-CSS consortium. The ARID1B phenotype: What we have learned so far. Am J Med Genet C Semin Med Genet. 2014; 166C:276-289. [DOI] [PubMed] [Google Scholar]
- 8. Halgren C, Kjaergaard S, Bak M, et al. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet. 2012; 82:248-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoyer J, Ekici AB, Endele S, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012; 90:565-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MA, D'Avanzo M G, Lynch SA, Musio A, Nyazov DM, Sanchez-Lara PA, Shalev SA, Deardorff MA. The Coffin-Siris syndrome: A proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet A. 2012; 158A:1865-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santen GW, Aten E, Vulto-van Silfhout AT, et al. Coffin-Siris syndrome and the BAF complex: Genotype-phenotype study in 63 patients. Hum Mutat. 2013; 34:1519-1528. [DOI] [PubMed] [Google Scholar]
- 12. Tekin M, Bodurtha JN, Riccardi VM. Cafe au lait spots: The pediatrician's perspective. Pediatr Rev. 2001; 22:82-90. [DOI] [PubMed] [Google Scholar]
- 13. Shah KN. The diagnostic and clinical significance of cafe-au-lait macules. Pediatr Clin North Am. 2010; 57:1131-1153. [DOI] [PubMed] [Google Scholar]
- 14. Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011; 21:396-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dixon-Salazar TJ, Silhavy JL, Udpa N, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012; 4:138ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]