

Abstract
Over the past decade, the ability of cancer cells to evade immune destruction has become recognized as one of the hallmarks of cancer. This understanding has paved the way for the development of novel therapeutic agents that can enhance activation of antitumor immune responses or reverse immunosuppressive mechanisms through which tumors escape immune-mediated rejection. The treatment of gynecologic cancers remains a therapeutic challenge, as these malignancies are often diagnosed in advanced stages, and many patients relapse despite appropriate management. Clinical trials have shown efficacy for various immunotherapeutic strategies, especially the use of tumor-targeting antibodies; enhancement of tumor antigen presentation, such as with vaccines and toll-like receptor agonists; and agents targeting immunosuppressive mechanisms, such as checkpoint blockade inhibitors. Emerging data on new and combination approaches currently under investigation provide a strong rationale for these approaches.
Introduction
Each year almost 90,000 women in the United States are diagnosed with gynecologic malignancies, and over 28,000 will die from their disease.[1] Many women with early-stage disease are cured with a combination of surgery, radiation, and chemotherapy. However, especially in the case of ovarian cancer, the malignancies are often diagnosed at advanced stages, and many patients relapse despite appropriate management. The treatment of gynecologic cancers represents a therapeutic challenge, and there is an unmet clinical need for new therapies.
Over the past decades, the field of tumor immunology has gained much attention as the ability of cancer cells to evade immune destruction has become recognized as one of the hallmarks of cancer.[2] Cancer immune surveillance, considered to be an important host protection process for preventing carcinogenesis, relies on components of both the innate and the adaptive immune systems.[3] Recognition of tumor cells by the immune system initially involves the uptake of tumor cell fragments by professional antigen-presenting cells (APCs), such as dendritic cells (DCs). The processing of the tumor fragments involves digestion of the tumor proteins into small peptides, which then get loaded onto major histocompatibility complex (MHC) class I and class II proteins and presented on the surface of the APCs.[4] Some of the displayed peptides represent tumor-associated antigens (TAAs), which are either new peptides resulting from specific mutations (neoantigens) or peptides representing the proteins preferentially expressed in cancer cells over normal tissues (eg, cancer-testis antigens, differentiation antigens).
Activated APCs then migrate to the tumor-draining lymph nodes, where they present the MHC-peptide complexes to naive T cells. Activation of T cells specific for the MHC-peptide complex requires two separate signals: (1) interaction of the MHC-peptide complex with a T-cell receptor (TCR), and (2) interaction of the costimulatory receptor CD28 on the surface of T cells with its ligand (B7-1, B7-2) on the surface of APCs.[5] Following activation, tumor-specific T cells then migrate through the systemic vasculature to the tumor sites, where recognition of TAAs on the surface of tumor cells leads to T-cell–mediated tumor cell lysis.[6]
Recognition of the steps involved in the antitumor immune response has paved the way for the development of novel therapeutic agents that can enhance activation of these responses or reverse immunosuppressive mechanisms that allow tumors to escape from immune-mediated rejection. Various stages of the antitumor immune response can be targeted, and the approaches used to do this can be categorized into three general strategies: (1) augmenting tumor antigen presentation, utilizing agents such as vaccines, toll-like receptor (TLR) agonists, and oncolytic viruses, (2) focusing on enhancement of T-cell activity, either through adoptive cell approaches or through the targeting of activating and inhibitory proteins on T cells; and (3) targeting additional immune inhibitory mechanisms in the tumor microenvironment (Figure).
Figure. Opportunities for Immunotherapy in Gynecologic Malignancies.
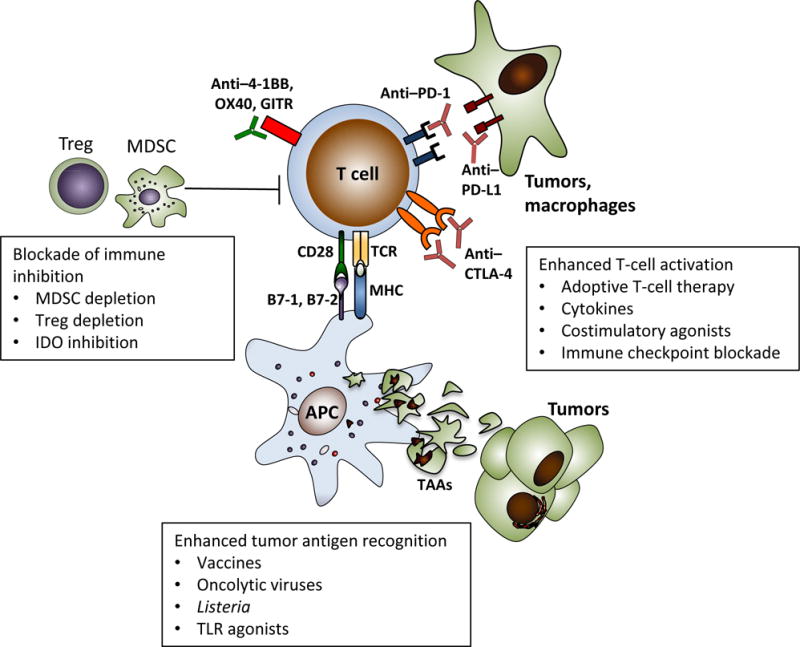
Successful immunotherapy for gynecologic cancers will require multimodal approaches that target different aspects of the antitumor immune response, including tumor antigen recognition, T-cell activation, and blockade of immune inhibitory mechanisms. Strategies currently in development are presented in boxes. APC = antigen-presenting cell; CTLA-4 = cytotoxic T-lymphocyte–associated antigen 4; GITR = glucocorticoid-induced tumor necrosis factor receptor–related protein; IDO = indoleamine 2,3-dioxygenase; MDSC = myeloid-derived suppressor cell; MHC = major histocompatibility complex; TAAs = tumor-associated antigens; TCR = T-cell receptor; TLR = toll-like receptor; Treg = regulatory T cell.
A number of immunotherapeutic approaches have been tested in gynecologic malignancies. In this review, we will summarize clinical trials that have used various immunotherapeutic strategies, with a particular focus on recently emerging data for new agents and combinations.
Ovarian Cancer
In 2015, there will be an estimated 21,290 new cases of epithelial ovarian cancer (EOC) in the United States, with 14,180 deaths, representing 2.4% of all US cancer deaths.[7] EOC is the fifth leading cause of cancer death among women, accounting for more deaths than any other cancer of the female reproductive system. Unfortunately, the majority of patients with EOC relapse despite appropriate treatment, and ultimately they die from their disease.
While it was originally felt that EOC would not respond well to immunotherapy, research has, in fact, demonstrated a key role for the immune system in the control of EOC cell growth. This is supported by the observation that increased levels of tumor-infiltrating lymphocytes (TILs) are associated with improved prognosis.[8] Several immunotherapeutic strategies have been explored in EOC.
Therapies to enhance tumor antigen recognition
Strategies that aim to enhance tumor recognition by the immune system can be collectively grouped into vaccines and innate immune activators; included in the second group are TLR agonists, type I interferon (IFN), and oncolytic viruses.
Vaccines
The identification of unique differentiation proteins expressed in EOC has led to the exploration of various vaccination approaches, including simple vaccine preparations consisting of specific peptides and proteins, as well as more complex strategies, such as engineered cellular vaccines, DC vaccines, virus-vectored vaccines, and oncolytic viruses. The majority of studies explored the cancer-testis antigens (eg, NY-ESO-1) and proteins known to be overexpressed in EOC (eg, p53, survivin, and MUC1); a comprehensive review of vaccination strategies that have been explored in EOC is published elsewhere.[9] Although many studies have demonstrated induction of an immune response to the vaccines, very few have demonstrated clinical benefit. It is likely that these strategies are insufficient to overcome immune tolerance to self-antigens and to result in efficient activation of antigen-specific T cells, although they may prove to be valuable in combination with other therapies.
Innate immune activators
Another strategy for enhancing tumor antigen presentation by APCs involves agents that target the innate immune response. Antigen processing and presentation by APCs requires activation signals, which is accomplished through activation of pattern-recognition receptors (PRRs) such as TLRs.[10] TLRs recognize signature molecules that are broadly shared by various pathogens and, in addition, sense “danger signals” in the tumor microenvironment, which consist of endogenous molecules produced by dying cells. A phase I study of VTX-2337 (motolimod), a small-molecule agonist of TLR8, in combination with liposomal doxorubicin in patients with advanced EOC, demonstrated safety and evidence of immune activation and clinical benefit.[11] A phase II study evaluating motolimod in combination with liposomal doxorubicin (ClinicalTrials.gov identifier: NCT01666444) is ongoing.
Activated APCs produce type I IFN, which plays a role in the antiviral immune response; it has also been demonstrated to be indispensible for tumor antigen presentation by APCs.[12] Although type I IFN has been evaluated in various cancer types and is approved for use as adjuvant therapy in patients with resected melanoma, in a study by Alberts et al, systemic or intraperitoneal administration of IFNα had limited activity in patients with EOC and was associated with frequent toxicities.[13]
Oncolytic viruses have inherent properties that allow them to replicate in cancer cells while sparing normal tissues. While serving as tumor-debulking agents, oncolytic viruses also activate the innate immune response on multiple levels through release of tumor antigens, PRR ligands and danger signals, and production of type I IFN. Several trials using oncolytic viruses in patients with EOC have demonstrated safety and durable clinical benefit in some patients.[14]
Overall, strategies to enhance tumor antigen presentation by the innate immune system have been demonstrated to be safe, but to date, their efficacy has been marginal. The future of drugs that enhance tumor antigen presentation in patients with EOC probably will be seen in combination therapies in which the T-cell response primed with a vaccine or an innate immune activator is further driven through therapies targeting T-cell activation and adaptive immune responses.
Therapies to enhance T-cell activation
The survival, proliferation, and activation of T cells are controlled by a variety of factors, including cytokines and a range of immunostimulatory and inhibitory receptors. Several studies have explored agents targeting T cells as immunotherapy in EOC, including drugs that target pathways of T-cell activation as well as adoptive T cell strategies.
Cytokines
The cytokines interleukein (IL)-2 and IL-12 are potent activators of T-cell proliferation and cytotoxicity. Their use as anticancer agents has been explored in multiple types of cancer, including ovarian. The use of both agents, administered systemically, is limited by toxicity. A phase I/II study of intraperitoneal IL-2 in patients with persistent or recurrent EOC showed an overall response rate of 25.7%, although the regimen was associated with significant toxicity.[15] A different strategy for delivery of IL-12—the use of IL-12–expressing plasmids—has been explored. In a recent study, 22 patients with recurrent EOC receiving intraperitoneal EGEN-001, an IL-12 plasmid formulated with lipopolymer, demonstrated a 35% stable disease rate.[16]
Immune checkpoint blockade
Identification of the costimulatory and coinhibitory receptors that regulate T-cell activation led to the development of antibodies that target these receptors.[5] Targeting such receptors, an approach termed “immune checkpoint blockade,” has demonstrated significant activity in preclinical cancer models and in clinical trials.[17] In particular, antibodies targeting the inhibitory receptors cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) and programmed death 1 (PD-1), as well as the PD-1 ligand (PD-L1), are the agents of this type that are most advanced in clinical development, with the CTLA-4–targeting agent ipilimumab approved for use in treating metastatic melanoma and the PD-1–targeting agents nivolumab and pembrolizumab approved for use in treating melanoma and non–small-cell lung cancer.
Based on these findings, therapy with immune checkpoint blockade has been evaluated in trials in patients with EOC. Despite its activity in metastatic melanoma, the efficacy of the CTLA-4–targeting antibody in EOC as a single agent has so far been limited. In 11 patients with EOC who received GVAX, an autologous tumor cell vaccine expressing granulocyte-macrophage colony-stimulating factor, treatment with ipilimumab led to an objective response in 1 patient that was durable for over 4 years.[18] In comparison, emerging clinical data indicate that targeting of PD-1 and PD-L1 may be a promising strategy in EOC. In a phase I study of an anti–PD-L1 antibody in patients with advanced cancer, 22% of the 17 patients with EOC had evidence of objective response or stable disease lasting at least 24 weeks.[19] In a phase I study of the anti–PD-1 antibody nivolumab in 20 evaluable patients with EOC, the best overall response was 15%, which included 2 patients with durable complete responses; total disease control rate was 45%.[20] Similar activity was reported for the PD-L1–blocking antibodies avelumab and pembrolizumab, with response rates ranging from 11% to 17% and disease control rates of up to 65%.[21,22]. Larger studies using these agents are currently underway.
The combination of CTLA-4 and PD-1 blockade has been associated with additive and even synergistic activity in animal models. A recent phase III study evaluating combined CTLA-4 and PD-1 blockade (with ipilimumab and nivolumab, respectively) in patients with melanoma demonstrated enhanced response rate and progression-free survival compared with either agent alone, leading to recent US Food and Drug Administration approval of the combination for the treatment of melanoma,[23] although the regimen did result in high rates of grade 3 toxicity. An ongoing NRG Oncology Group randomized phase II study (ClinicalTrials.gov identifier: NCT02498600) is comparing the combination of nivolumab and ipilimumab vs nivolumab alone to determine whether the combination is also active and safe in patients with relapsed EOC.
Adoptive T-cell therapies
Adoptive cell therapies (ACTs) rely on the infusion of large numbers of autologous tumor-reactive T cells that have been isolated from tumors and expanded in vitro. Early studies reported significant efficacy for this approach in EOC,[24] although these studies were necessarily biased by the selection of patients from whom a sufficient quantity of TILs could be isolated. Additional studies using ACT in EOC are ongoing (ClinicalTrials.gov identifiers: NCT02482090, NCT01883297). As an alternative strategy, engineered T-cell technologies avoid the need for isolation of TILs. Using this strategy, peripheral-blood autologous lymphocytes are transduced either with a T-cell receptor that recognizes a specific tumor antigen MHC-peptide or with a chimeric antigen receptor (CAR) that recognizes a tumor-associated surface antigen. The efficacy of such approaches has been demonstrated in preclinical studies in which engineered T cells expressing a MUC16-specific CAR were associated with complete eradication of orthotopic ovarian xenografts.[25] A phase I study using this strategy is currently in development (ClinicalTrials.gov identifier: NCT02498912). Additional studies using T cells targeting other ovarian cancer–associated proteins—such as folate receptor alpha, mesothelin (ClinicalTrials.gov identifier: NCT01583686), and NY-ESO-1 (ClinicalTrials.gov identifiers: NCT01567891, NCT02457650)—are also upcoming.
Therapies to block other mechanisms of immune inhibition
Despite these findings, it is becoming increasingly apparent that the benefit of immune checkpoint blockade in EOC is not universal and that development of predictive biomarkers and combination therapies will be necessary. To this end, combination strategies using PD-1– and PD-L1–blocking antibodies together with antibodies targeting other mechanisms of T-cell activation (eg, glucocorticoid-induced tumor necrosis factor receptor–related protein [GITR], OX40), as well as antibodies targeting other immune checkpoints (eg, lymphocyte-activation gene [LAG]-3 and T-cell immunoglobulin and mucin domain–containing [TIM]-3), are already entering clinical trials in various tumor types. In addition, several immune inhibitory mechanisms have been demonstrated to be associated with poor prognosis in EOC, including tumor-infiltrating regulatory T cells (Tregs),[26] tumor-associated macrophages and myeloid-derived suppressor cells (MDSCs),[27] and expression of the enzyme indoleamine 2,3-dioxygenase (IDO) by the tumor or stromal cells.[28] There is thus a strong rationale for targeting these mechanisms in combination with PD-1/PD-L1 blockade, and studies are currently underway to evaluate these strategies in different tumor types.
Cervical Cancer
Cervical cancer is the fourth most common cancer in women; 528,000 new cases were diagnosed worldwide in 2012. For patients with advanced-stage, recurrent, or persistent cervical cancer, systemic cisplatin-based chemotherapy remains the cornerstone of treatment, although, unfortunately, most patients progress despite the use of different treatment combinations. After front-line therapy, single-agent chemotherapy regimens have only limited efficacy, highlighting an unmet clinical need for the development of new treatment strategies.
Several immunotherapeutic strategies have been explored in cervical cancer, particularly in light of its association with human papillomavirus (HPV). The genotypes HPV-16 and HPV-18 account for the majority of cases of invasive cervical cancer. The HPV E6 and E7 oncoproteins, which are expressed intracellularly in HPV-associated cancers, represent a target for therapeutic vaccines. Several different types of vaccines have been explored for use in treating cervical cancer, including live vector–based vaccines, protein and peptide vaccines, nucleic acid–based vaccines, and DC vaccines.
Live vector–based vaccines are particularly effective, as they deliver antigen efficiently and can replicate within the cell. Live vector–based vaccines can be subdivided into those that use bacterial vectors and those that use viral vectors. The bacterial-based vector for HPV-associated cancers that is most advanced in clinical development is Listeria monocytogenes, a Gram-positive facultative intracellular bacterium. It preferentially infects APCs and can evade the phagosome through expression of listeriolysin O (LLO). For this reason, peptides from the vector are presented on MHC molecules and thus induce CD4+ and CD8+ T-cell responses.
A prospective phase II study using ADXS11-001 Listeria vector with and without cisplatin chemotherapy was conducted in India in 110 patients with recurrent cervical cancer previously treated with chemotherapy, radiotherapy, or both. The results, presented at the American Society of Clinical Oncology 2014 Annual Meeting, showed a 12-month survival rate of 36%; an 18-month survival rate of 28%; and an 11% response rate, which was independent of the use of cisplatin.[29] A Gynecologic Oncology Group (GOG) phase II trial (protocol 265) evaluating the use of ADXS11-001 in the treatment of persistent or recurrent cervical cancer (ClinicalTrials.gov identifier: NCT01266460) is ongoing.
Several viral vectors, the majority of which are vaccinia virus–based, have been tested in therapeutic HPV vaccines. Kaufmann et al conducted a study in women with International Federation of Gynecology and Obstetrics (FIGO) stage Ib or IIa cervical cancer to examine the safety and immunologic effects of vaccination with TA-HPV, a live recombinant vaccinia virus expressing modified forms of the HPV-16 and HPV-18 E6 and E7 proteins. Vaccination using this strategy has been associated with the development of HPV-specific T-cell responses.[30] Additional clinical studies evaluating the efficacy of viral vectors are certainly warranted.
The use of protein and peptide vaccines against HPV E6 and E7 proteins has also been explored. Early studies of peptide vaccines in cervical cancer showed mixed results. More recently, in a phase I study, 35 patients with late-stage cervical cancer were vaccinated with HPV-16 E6 and/or E7 in Montanide ISA 51 adjuvant in varying doses. The authors found that the vaccine was well tolerated and capable of inducing a broad IFNγ-associated T-cell response.[31] A phase I/II study is currently being conducted to determine the safety and immune effects of an HPV-16 E6/E7 long-peptide vaccine (ISA 101) at different doses, with or without IFNγ, in combination with carboplatin and paclitaxel in patients with advanced and recurrent cervical cancer (ClinicalTrials.gov identifier: NCT02128126). There has also been considerable interest in the SGN-00101 vaccine, which is based on a fusion protein consisting of heat-shock protein from Mycobacterium bovis and HPV-16 E7. This vaccine was associated with regression of lesions in small numbers of patients with grade II/III cervical intraepithelial neoplasia (CIN).[32]
Nucleic acid vaccines are an attractive therapeutic option, as they are relatively safe and stable, do not elicit neutralizing antibodies, and can sustain reasonable levels of antigen expression within cells. A phase I study of 15 women with grade II/III CIN was conducted to test the safety of and response to the ZYC101 vaccine, which contains a bacterial-expression plasmid encoding a 13–amino acid sequence that is highly homologous with HPV E7 within poly (lactide-co-glycolide) microparticles. There were no serious adverse events; 5 women (33%) had complete histologic responses, and 11 women (73%) had HPV-specific T-cell responses.[33] An updated version of this same vaccine was then studied in a phase II/III trial that recruited 161 subjects. Results indicated that the vaccine was highly tolerable and effective in women less than 25 years of age, with 70% CIN resolution.[34] A different DNA vaccine, VGX-3100, has been studied in women with grade II/III CIN. In a phase II randomized trial that recruited 167 women, the vaccine was administered with electroporation. In the modified intention-to-treat analysis, among the 114 women who received VGX-3100, 48.2% showed lesion regression, compared with 30% in the placebo group (P = .034).[35]
Lastly, DC vaccines represent a new area of clinical investigation, although their use has been limited, as implementation of these vaccines requires the use of autologous DCs. Two small clinical trials have been conducted to date evaluating DC-based HPV E6 and E7 vaccine therapy; both trials demonstrated evidence of T-cell response but no clinical benefit.[36,37]
ACT has been shown to be a promising salvage option for patients with advanced or recurrent metastatic cervical cancer. In a study by Stevanovic et al, HPV oncoprotein–reactive T-cell cultures generated from TILs were administered to nine women with metastatic cervical cancer who had received prior platinum-based chemotherapy; three of them attained an objective tumor response. Two patients with complete responses had ongoing remissions at 15 and 22 months, respectively.[38] Further studies assessing the efficacy of ACT are warranted, although such approaches are limited to centers specializing in this approach.
The apparent immunogenicity of cervical cancer that results from the presence of foreign antigens is a good rationale for the evaluation of immune checkpoint–blocking antibodies in these patients; studies are currently underway (ClinicalTrials.gov identifiers: NCT01711515, NCT02257528) to address this question.
While it remains to be seen whether immune therapies will become standard treatment options for cervical cancer, early studies have shown promise. In particular, the ability of therapies such as DNA vaccines to induce regression of early lesions suggests that such strategies may be applicable in a more advanced setting, perhaps in combination with other therapeutics, such as chemotherapy and immune checkpoint–blocking agents. Many trials are ongoing and may provide further treatment options for patients with advanced-stage disease.
Endometrial Cancer
Endometrial cancer is the most common gynecologic malignancy. There are expected to be approximately 54,870 cases diagnosed in the United States in 2015, representing 3.3% of all new cancer cases.[39] These tumors generally present in the corpus uteri as localized disease (67%); the 5-year survival rate is 81.7%.[39] Patients with advanced or recurrent endometrial cancer have a poor prognosis, however, and there is an urgent need for new therapies.
Classification
The classification system for endometrial carcinomas was revised recently to reflect both histopathologic characteristics and genomic features. This allows subdivision into clinically relevant subsets, which may help clinicians tailor therapy, especially immunotherapy. The Cancer Genome Atlas (TCGA) classifies endometrial cancer into four distinct categories: POLE-ultramutated, microsatellite instability (MSI) hypermutated, copy number low, and copy number high.[40] POLE proofreading-mutant cancers in particular account for 7% to 12% of endometrial cancers and have an excellent prognosis. POLE proofreading-mutant and MSI-high endometrial cancers, such as those associated with Lynch syndrome, display a robust intratumoral T-cell response, with an enrichment of antigenic neopeptides.[41,42] Despite the increased number of immune infiltrates, MSI-high endometrial cancers do not appear to differ from microsatellite-stable cancers.[40] In fact, data suggest that some patients with MSI-high endometrial cancers may have a worsened prognosis, probably secondary to other immune inhibitory mechanisms in the tumor microenvironment, such as cyclo-oxygenase 2.[43] Nevertheless, the large number of potentially immunogenic neoantigenic peptides produced in these cancers provides a strong rationale for the development of immunotherapeutic strategies to treat endometrial cancer. By further sorting out the mutational background of endometrial cancer subtypes, future research may help guide treatment decisions and clinical trials of new immune-based agents.
Immunotherapeutic approaches
Due to a poor understanding of the interplay between the immune system and endometrial cancer, only a limited number of immunotherapeutic approaches have been tested in this cancer to date.
DC vaccines
Several studies have explored the use of DC vaccines in treating uterine cancer. In the most recent and largest study, six patients with uterine leiomyosarcoma or serous endometrial cancer were treated with autologous DCs electroporated with Wilms tumor 1 (WT1) microRNA. This approach was well tolerated and a transient oncologic or immunologic reaction was observed in three patients, all of whom were human leucocyte antigen (HLA)-A2–positive.[44] Other strategies for treating endometrial carcinoma and uterine sarcoma have used DCs loaded with whole tumor lysate. In vitro, these DCs are able to induce a strong CD8+ T-cell response against autologous tumor cells, although there was no evidence of clinical benefit in a phase I study.[45] These early studies indicate that the induction of immune responses with vaccination is possible in patients with endometrial cancer, highlighting a rationale for their therapeutic combination with other agents.
Immune checkpoint blockade
The use of immune checkpoint blockade has not yet been widely explored in endometrial cancer. A recent study investigated whether cancers with mutations causing mismatch-repair deficiencies might be responsive to PD-1 blockade.[46] While this phase II study enrolled primarily patients with metastatic colorectal cancer, an extra cohort enrolled patients with other types of cancers, including two patients with mismatch-repair–deficient endometrial cancer. In all, there were seven patients with mismatch-repair–deficient noncolorectal cancer (ampullary or cholangiocarcinoma, endometrial [2 patients], small bowel, and gastric) and they showed an objective response rate (ORR) of 71% and a disease control rate of 71% after treatment with pembrolizumab. In comparison, the patients with mismatch-repair–deficient colorectal cancer had an ORR of 40% and a disease control rate of 90%, and the patients with mismatch-repair–proficient colorectal cancer had an ORR of 0% and a disease control rate of 11%.[46] In addition, tumors from patients with mismatch-repair deficiency contained a greater density of CD8+ lymphoid cells and had greater PD-L1 expression on TILs and tumor-associated macrophages. While these findings correlated with objective response and stable disease rates, they were not significantly associated with progression-free or overall survival.
Overall, this work suggests that hypermutated tumors, including endometrial cancers, show responsiveness to T-cell checkpoint immunotherapy. The question remains whether immune checkpoint blockade will be effective in treating endometrial cancers that are not hypermutated. However, given the features of the endometrial cancer microenvironment, which include multiple immunosuppressive mechanisms (eg, Tregs, overexpression of IDO),[47,48] and the positive prognostic value of tumor-infiltrating CD8+ cells,[49] it is likely that immunotherapies targeting these mechanisms will be of value in the treatment of endometrial cancer; clinical trials are certainly warranted.
Other Gynecologic Malignancies
There has been a paucity of immunotherapy trials in other gynecologic malignancies, such as ovarian germ cell/stromal cell tumors, uterine sarcomas, vulvar and vaginal cancers, and gestational trophoblastic disease. In most of these, however, analysis of the tumor microenvironment indicates that similar immune-activating and -inhibitory mechanisms are at play in controlling or facilitating tumor progression, providing a rationale for the evaluation of immunotherapeutic approaches in these cancers as well.
The Future of Immunotherapy in Gynecologic Malignancies
Recent years have seen many advances in immunotherapeutic approaches to various cancer types, and gynecologic malignancies are no exception. Promising early data reported with immune checkpoint inhibitors make it likely that these agents will eventually become part of the treatment arsenal for gynecologic cancers. These data also suggest, however, that checkpoint inhibitors are not universally effective as single agents, indicating a need for rationally designed treatment combinations. The optimal activation of antitumor immunity will probably involve targeting different components of the immune response, which are likely not to be universal, since mechanisms of immune evasion differ from patient to patient. Clinical trials incorporating appropriate biomarkers are likely to identify new immunotherapeutic approaches, will allow us to target these treatments to the appropriate patients, and will inform the development and use of combination therapies that may help overcome current limitations.
Acknowledgments
Financial Disclosure: Dr. Zamarin has received funding from the Foundation for Women’s Cancers (Judith Liebenthal Robinson Ovarian Cancer Foundation Award). Dr. Bourla is funded by the Mary Jane Milton Endowed Fellowship in Gynecologic Oncology and Immunotherapy.
References
- 1.American Cancer Society. Cancer Facts & Figures 2015. Atlanta: American Cancer Society; 2015. [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 4.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Ann Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nature Rev Imunol. 2013;13:227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 7.SEER Stat Fact Sheets: Ovary Cancer. http://seer.cancer.gov/statfacts/html/ovary.html. Accessed November 22, 2015.
- 8.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 9.Liao JB, Disis ML. Therapeutic vaccines for ovarian cancer. Gynecol Oncol. 2013;130:667–73. doi: 10.1016/j.ygyno.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Monk BJ, Brady WE, Lankes HA, et al. VTX-2337, a TLR8 agonist, plus chemotherapy in recurrent ovarian cancer: preclinical and phase I data by the Gynecologic Oncology Group. J Clin Oncol. 2013;31(suppl) abstr 3077. [Google Scholar]
- 12.Zitvogel L, Galluzzi L, Kepp O, et al. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–14. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 13.Alberts DS, Hannigan EV, Liu PY, et al. Randomized trial of adjuvant intraperitoneal alpha-interferon in stage III ovarian cancer patients who have no evidence of disease after primary surgery and chemotherapy: an intergroup study. Gynecol Oncol. 2006;100:133–8. doi: 10.1016/j.ygyno.2005.07.117. [DOI] [PubMed] [Google Scholar]
- 14.Kim KH, Dmitriev IP, Saddekni S, et al. A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol Oncol. 2013;130:518–24. doi: 10.1016/j.ygyno.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards RP, Gooding W, Lembersky BC, et al. Comparison of toxicity and survival following intraperitoneal recombinant interleukin-2 for persistent ovarian cancer after platinum: twenty-four-hour versus 7-day infusion. J Clin Oncol. 1997;15:3399–407. doi: 10.1200/JCO.1997.15.11.3399. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez RD, Sill MW, Davidson SA, et al. A phase II trial of intraperitoneal EGEN-001, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: a gynecologic oncology group study. Gynecol Oncol. 2014;133:433–8. doi: 10.1016/j.ygyno.2014.03.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zamarin D, Postow MA. Immune checkpoint modulation: rational design of combination strategies. Pharmacol Ther. 2015;150:23–32. doi: 10.1016/j.pharmthera.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008;105:3005–10. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. New Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamanishi J, Mandai M, Ikeda T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015 Sep 8; doi: 10.1200/JCO.2015.62.3397. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Disis ML, Patel MR, Pant S, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with previously treated, recurrent or refractory ovarian cancer: a phase Ib, open-label expansion trial. J Clin Oncol. 2015;33(suppl) abstr 5509. [Google Scholar]
- 22.Varga A, Piha-Paul SA, Ott PA, et al. Antitumor activity and safety of pembrolizumab in patients (pts) with PD-L1 positive advanced ovarian cancer: Interim results from a phase Ib study. J Clin Oncol. 2015;33(suppl) abstr 5510. [Google Scholar]
- 23.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:1270–1. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujita K, Ikarashi H, Takakuwa K, et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1:501–7. [PubMed] [Google Scholar]
- 25.Chekmasova AA, Rao TD, Nikhamin Y, et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res. 2010;16:3594–606. doi: 10.1158/1078-0432.CCR-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 27.Reinartz S, Schumann T, Finkernagel F, et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: correlation of CD163 expression, cytokine levels and early relapse. Int J Cancer. 2014;134:32–42. doi: 10.1002/ijc.28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inaba T, Ino K, Kajiyama H, et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115:185–92. doi: 10.1016/j.ygyno.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 29.Basu P, Mehta AO, Jain MM, et al. ADXS11-001 immunotherapy targeting HPV-E7: Final results from a phase 2 study in Indian women with recurrent cervical cancer. J Clin Oncol. 2014;32(suppl 5) abstr 5610. [Google Scholar]
- 30.Kaufmann AM, Stern PL, Rankin EM, et al. Safety and immunogenicity of TA-HPV, a recombinant vaccinia virus expressing modified human papillomavirus (HPV)-16 and HPV-18 E6 and E7 genes, in women with progressive cervical cancer. Clin Cancer Res. 2002;8:3676–85. [PubMed] [Google Scholar]
- 31.Kenter GG, Welters MJ, Valentijn AR, et al. Phase I immunotherapeutic trial with long peptides spanning the E6 and E7 sequences of high-risk human papillomavirus 16 in end-stage cervical cancer patients shows low toxicity and robust immunogenicity. Clin Cancer Res. 2008;14:169–77. doi: 10.1158/1078-0432.CCR-07-1881. [DOI] [PubMed] [Google Scholar]
- 32.Roman LD, Wilczynski S, Muderspach LI, et al. A phase II study of Hsp-7 (SGN-00101) in women with high-grade cervical intraepithelial neoplasia. Gynecol Oncol. 2007;106:558–66. doi: 10.1016/j.ygyno.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 33.Sheets EE, Urban RG, Crum CP, et al. Immunotherapy of human cervical high-grade cervical intraepithelial neoplasia with microparticle-delivered human papillomavirus 16 E7 plasmid DNA. Am J Obstet Gynecol. 2003;188:916–26. doi: 10.1067/mob.2003.256. [DOI] [PubMed] [Google Scholar]
- 34.Garcia F, Petry KU, Muderspach L, et al. ZYC101a for treatment of high-grade cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2004;103:317–26. doi: 10.1097/01.AOG.0000110246.93627.17. [DOI] [PubMed] [Google Scholar]
- 35.Trimble CL, Morrow MP, Kraynyak KA, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015 Sep 16; doi: 10.1016/S0140-6736(15)00239-1. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrara A, Nonn M, Sehr P, et al. Dendritic cell-based tumor vaccine for cervical cancer II: results of a clinical pilot study in 15 individual patients. J Cancer Res Clin Oncol. 2003;129:521–30. doi: 10.1007/s00432-003-0463-5. [DOI] [PubMed] [Google Scholar]
- 37.Santin AD, Bellone S, Palmieri M, et al. Human papillomavirus type 16 and 18 E7-pulsed dendritic cell vaccination of stage IB or IIA cervical cancer patients: a phase I escalating-dose trial. J Virol. 2008;82:1968–79. doi: 10.1128/JVI.02343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevanovic S, Draper LM, Langhan MM, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33:1543–50. doi: 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.SEER Stat Fact Sheets: Endometrial Cancer. http://seer.cancer.gov/statfacts/html/corp.html. Accessed November 22, 2015.
- 40.Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Gool IC, Eggink FA, Freeman-Mills L, et al. POLE Proofreading Mutations Elicit an Antitumor Immune Response in Endometrial Cancer. Clin Cancer Res. 2015;21:3347–55. doi: 10.1158/1078-0432.CCR-15-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howitt BE, Shukla SA, Sholl LM, et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015 doi: 10.1001/jamaoncol.2015.2151. Accessed November 22, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Suemori T, Susumu N, Iwata T, et al. Intratumoral CD8+ Lymphocyte Infiltration as a Prognostic Factor and Its Relationship With Cyclooxygenase 2 Expression and Microsatellite Instability in Endometrial Cancer. Int J Gynecol Cancer. 2015;25:1165–72. doi: 10.1097/IGC.0000000000000482. [DOI] [PubMed] [Google Scholar]
- 44.Coosemans A, Vanderstraeten A, Tuyaerts S, et al. Wilms’ Tumor Gene 1 (WT1)–loaded dendritic cell immunotherapy in patients with uterine tumors: a phase I/II clinical trial. Anticancer Res. 2013;33:5495–500. [PubMed] [Google Scholar]
- 45.Santin AD, Bellone S, Ravaggi A, et al. Induction of tumour-specific CD8(+) cytotoxic T lymphocytes by tumour lysate-pulsed autologous dendritic cells in patients with uterine serous papillary cancer. Br J Cancer. 2002;86:151–7. doi: 10.1038/sj.bjc.6600026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ino K, Yamamoto E, Shibata K, et al. Inverse correlation between tumoral indoleamine 2,3-dioxygenase expression and tumor-infiltrating lymphocytes in endometrial cancer: its association with disease progression and survival. Clin Cancer Res. 2008;14:2310–7. doi: 10.1158/1078-0432.CCR-07-4144. [DOI] [PubMed] [Google Scholar]
- 48.Giatromanolaki A, Bates GJ, Koukourakis MI, et al. The presence of tumor-infiltrating FOXP3+ lymphocytes correlates with intratumoral angiogenesis in endometrial cancer. Gynecol Oncol. 2008;110:216–21. doi: 10.1016/j.ygyno.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 49.Kondratiev S, Sabo E, Yakirevich E, et al. Intratumoral CD8+ T lymphocytes as a prognostic factor of survival in endometrial carcinoma. Clin Cancer Res. 2004;10:4450–6. doi: 10.1158/1078-0432.CCR-0732-3. [DOI] [PubMed] [Google Scholar]