

Abstract
Coxiella burnetii is a Gram-negative bacterium that causes Q fever in humans. Q fever is an atypical pneumonia transmitted through inhalation of contaminated aerosols. In mammalian lungs, C. burnetii infects and replicates in several cell types, including alveolar macrophages (AMs). The innate immunity and signaling pathways operating during infection are still poorly understood, in part because of the lack of relevant host cell models for infection in vitro. In the study described here, we investigated and characterized the infection of primary murine AMs by C. burnetii phase II in vitro. Our data reveal that AMs show a pronounced M2 polarization and are highly permissive to C. burnetii multiplication in vitro. Murine AMs present an increased susceptibility to infection in comparison to primary bone marrow-derived macrophages. AMs support more than 2 logs of bacterial replication during 12 days of infection in culture, similar to highly susceptible host cells, such as Vero and THP-1 cells. As a proof of principle that AMs are useful for investigation of C. burnetii replication, we performed experiments with AMs from Nos2−/− or Ifng−/− mice. In the absence of gamma interferon and nitric oxide synthase 2 (NOS2), AMs were significantly more permissive than wild-type cells. In contrast, AMs from Il4−/− mice were more restrictive to C. burnetii replication, supporting the importance of M2 polarization for the permissiveness of AMs to C. burnetii replication. Collectively, our data account for understanding the high susceptibility of alveolar macrophages to bacterial replication and support the use of AMs as a relevant model of C. burnetii growth in primary macrophages.
INTRODUCTION
The intracellular bacterial pathogen Coxiella burnetii is the causative agent of the zoonotic infection termed Q fever. Human infection with C. burnetii can lead to asymptomatic seroconversion or symptomatic Q fever, which often presents as an acute febrile illness. The nonspecific symptoms associated with acute infection indicate that it is likely significantly underdiagnosed. Acute infection is often self-limited, but in a minority of cases it can progress to a serious chronic infection that predominantly manifests as life-threatening endocarditis. Importantly, recent epidemiological data demonstrated the significant long-term health impact of Q fever, reporting that more than one in three patients continued to suffer from an impaired health status 24 months postdiagnosis (1).
Humans can become infected through the inhalation of contaminated aerosols, often from close contact with infected ruminants. Within the alveolar space, C. burnetii can infect a variety of cell types, including alveolar macrophages (AMs) (2). Essential to the capacity of C. burnetii to cause disease is the ability of the pathogen to replicate inside host cells within a unique lysosome-derived vacuole. The infectious, or small-cell variant, form of the bacterium enters the host cell and is passively trafficked through the endocytic pathway before reaching the hydrolytic and acidic confines of the lysosome. These conditions trigger the metabolic activation of C. burnetii and stimulate the conversion to a large-cell, replicative variant (3). This environment also triggers the active bacteria to assemble the Dot/Icm type IV secretion system that facilitates the translocation of over 130 effector proteins into the host cytoplasm (4). Collectively, the action of these effectors modulates the Coxiella-containing vacuole (CCV) to provide an environment conducive to replication (5). This intracellular life cycle holds true for both virulent (phase I) C. burnetii and avirulent (phase II) C. burnetii bacteria that have a lipopolysaccharide structure altered through genetic mutations that occur during serial passage in a nonimmunocompetent host (6).
It is clear that while development of the CCV is similar in many cell types, some cells, including primary peritoneal macrophages and bone marrow-derived macrophages (BMDMs), are intrinsically more restrictive to the intracellular replication of C. burnetii (7, 8). Furthermore, BMDMs from different inbred mouse strains vary in their ability to restrict C. burnetii infection, with A/J and BALB/c mice being more susceptible to C. burnetii phase II than many other mouse strains, including the C57BL/6 strain (9). The mechanisms through which different genetic backgrounds and cell types can control C. burnetii intracellular growth remain an area of interesting scientific pursuit.
AMs have long been considered the primary site of C. burnetii infection (2), and AMs from monkeys and humans have recently been used to explore the host-pathogen interactions that take place during C. burnetii infection. Cynomolgus monkey AMs were used to demonstrate the potent antiapoptotic activity associated with C. burnetii infection, although the replication dynamics of C. burnetii in this cellular model were not explored (10). More recently, human AMs, extracted from postmortem lung tissue samples, were shown to support the replication of different pathotypes of C. burnetii (11). In addition, this study demonstrated that both virulent (phase I) and avirulent (phase II) C. burnetii bacteria are able to infect human AMs. Interestingly, it was also observed that, within human AMs, C. burnetii more frequently forms multiple smaller CCVs rather than the large fusogenic vacuole observed in other cellular models of infection (11). Within a murine model of infection, it has been demonstrated that AMs are susceptible to C. burnetii infection (12), and an early study demonstrated that nitric oxide (NO) is produced by murine AMs in response to infection (13).
In the study described here, we investigated and characterized the infection of primary murine AMs by C. burnetii phase II in vitro. Our data reveal that AMs are permissive to C. burnetii multiplication. The AMs showed a pronounced M2 polarization and showed an increased susceptibility to infection in comparison to murine BMDMs. Finally, we investigated the contribution of signaling molecules to the susceptibility of AMs to C. burnetii, demonstrating the utility of AMs for investigation of the host restriction of C. burnetii infection in relevant host cells.
MATERIALS AND METHODS
Preparation of C. burnetii for infection.
Coxiella burnetii phase II Nine Mile strain (RSA439) bacteria were prepared from infected Vero cell monolayers as previously described (14). Confluent cultures of Vero cells were irradiated with 10 Gy 60Co to block cell division and maintained at 37°C in 5% CO2 in Dulbecco modified Eagle medium (DMEM; Gibco) with 10% fetal bovine serum (FBS; Gibco) and 1 mM l-glutamine (Sigma-Aldrich). Infection was carried out, and the cells were maintained under the above-described conditions for an additional 6 days. For the preparation of the Coxiella inoculum, infected Vero cells were lysed by homogenization in sterile distilled water. Cell lysates were clarified by centrifugation at 1,810 × g for 10 min at 4°C. The supernatant was extracted and centrifuged at 12,857 × g for 30 min at 4°C. The supernatant was discarded, and the pellet was resuspended in RPMI 1640 medium with 10% FBS. Bacterial aliquots were kept at −80°C. Prior to infection, aliquots were thawed at 37°C and subjected to sonication at 35 KHz for 3 min.
Mouse strains.
A/J, C57BL/6, Nos2−/−, Ifng−/−, and Il4−/− mice (all in the C57BL/6 mouse genetic background) were bred in the Animal Facilities of the Medical School of Ribeirão Preto (FMRP/USP). All animals were provided food and water ad libitum, and the temperature was controlled at 25°C. Mice were cared for according to the institutional guidelines on ethics in animal experiments (approved by the Comissão de Ética em Experimentação Animal da Faculdade de Medicina de Ribeirão Preto/CETEA, protocol number 097/2010).
Culture of mammalian cell lines.
THP-1 cells were cultured in RPMI 1640 medium (Sigma-Aldrich) with 10% FBS, and Vero cells were maintained in DMEM (Gibco) with 10% FBS (Gibco) and 1 mM l-glutamine (Sigma-Aldrich) at 37°C in 5% CO2. For assays with THP-1 cells, differentiation into macrophage-like cells was mediated by the addition of 200 nM phorbol 12-myristate 13-acetate (PMA) to the culture overnight as described previously (10), and the medium was replaced with PMA-free medium prior to the infection. For infection of Vero cells, the cells were irradiated with 10 Gy 60Co and maintained overnight at 37°C in 5% CO2 prior to the infection.
Bone marrow-derived macrophages.
BMDMs were obtained and differentiated according to the protocol described previously (15). Briefly, bone marrow cells were harvested from femurs and differentiated with RPMI 1640 medium (Sigma-Aldrich) containing 20% FBS (Invitrogen) and 30% L-929 cell conditioned medium (LCCM) at 37°C in 5% CO2 (15). Macrophages were harvested, counted, and seeded in 24-well plates at a density of 105 cells/well 1 day prior to infection and kept in RPMI 1640 medium with 10% FBS and 5% LCCM.
Alveolar macrophages.
AMs were obtained from adult mice (age, 6 to 8 weeks) via bronchoalveolar lavage (BAL). The animals were euthanized, and the trachea was pierced with a sterile cannula (BD AngioCath). The lungs were washed four consecutive times with 1.0 ml of warm sterile phosphate-buffered saline (PBS) containing 5 mM EDTA, and the material was collected. Cells were transferred to a new sterile tube, washed twice in sterile PBS, and resuspended in RPMI 1640 medium with 10% FBS, 1 mM l-glutamine, and 100 U/ml penicillin-streptomycin. Cells were seeded in 24-well plates at a density of 105 cells/well. Macrophages were maintained in culture in RPMI 1640 medium with 10% FBS, 1 mM l-glutamine, and 100 U/ml penicillin-streptomycin at 37°C in 5% CO2. Culture medium was replaced two times before infection in antibiotic-free medium. Fresh media were added to the macrophage cultures at day 6 postinfection.
C. burnetii infection of cells.
Frozen stocks of C. burnetii were thawed at 37°C and treated in an ultrasound bath for 3 min to dissolve bacterial clumps. Tissue culture medium was aspirated from the cells and replaced with fresh medium before infection. Bacteria were homogenized with a pipette and applied directly to the cells at a multiplicity of infection (MOI) of 3 (except for the experiment whose results are presented in Fig. 1A, in which an MOI of 100 was used). The plates were centrifuged at 200 × g for 5 min at 25°C and placed at 37°C in 5% CO2. After 4 h, the culture medium was aspirated, the wells were washed with warm sterile PBS, and fresh culture medium was added. As an internal experimental control of C. burnetii replication, infected cells were treated with 20 μg/ml rifampin (RIF) to inhibit C. burnetii protein translation (16).
FIG 1.
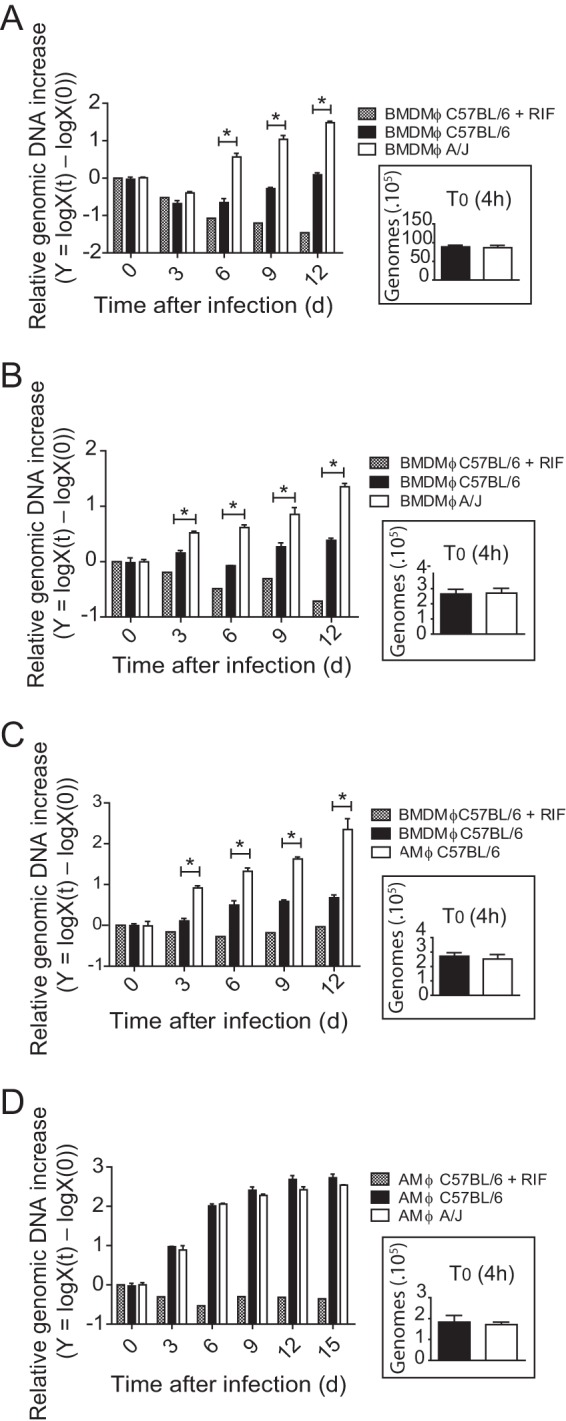
Alveolar macrophages are permissive to C. burnetii replication. BMDMs from C57BL/6 and A/J mice (A and B), BMDMs and AMs from C57BL/6 mice (C), or AMs from C57BL/6 and A/J mice (D) were infected with C. burnetii phase II (MOIs, 100 [A] and 3 [B to D]), and bacterial multiplication was evaluated by quantification of the genomic DNA by qPCR after 0 (4 h after infection), 3, 6, 9, 12 or 15 days (d) of infection. The y axis represents the difference in the base 10 logarithm between the genomic quantity X at time t [logX(t)] and the quantity X at time zero (T0) [logX(0)]. Cells treated with RIF were used as a negative control of bacterial replication. (Insets) Amount of C. burnetii genomic DNA present in cells after 4 h of infection. The data shown are the means ± standard errors of one representative experiment out of three performed. *, P < 0.05 (two-way ANOVA).
Fluorescence microscopy.
AMs from C57BL/6 mice were seeded at a density of 105 cells per well in a 24-well plate containing 13-mm glass coverslips. Cells were infected with wild-type C. burnetii bacteria or C. burnetii bacteria expressing mCherry at an MOI of 3 as previously described (17). After 4 h, 3 days, 6 days, 9 days, or 12 days of infection, cells were fixed with 3.5% paraformaldehyde containing 7.5% sucrose for 20 min and then washed twice with room temperature PBS. For the assays with wild-type C. burnetii bacteria, the cells were then permeabilized for 2 min with ice-cold 100% methanol, followed by two more washes with PBS. Cellular autofluorescence was removed by incubation with 0.1 M glycine in PBS for 10 min, and then the samples were blocked for 30 min in PBS containing 1% bovine serum albumin (BSA; Sigma-Aldrich) and 5% goat serum at room temperature. The cells were incubated with rabbit anti-C. burnetii (1:100) and rat anti-LAMP-1 (1:2) primary antibodies diluted in 1% BSA in PBS for 1 h, washed twice with room temperature PBS, and then incubated for 30 min in 1% BSA in PBS containing Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies (Invitrogen) at a concentration of 1:2,000. After three washes with room temperature PBS, cells were mounted with Prolong Gold antifade reagent containing DAPI (4′,6′-diamindino-2-phenylindole dilactate; Invitrogen). For the assays using mCherry-expressing C. burnetii, the cells were washed twice with room temperature PBS after the fixation and then directly mounted with Prolong Gold antifade reagent containing DAPI.
Multiphoton microscopy images were acquired using an LSM 780 Zeiss AxioObserver microscope equipped with a 63× oil immersion objective and analyzed using ImageJ software.
Transmission electron microscopy.
AMs were infected for 3 or 6 days, washed once with PBS, fixed for 1 h at room temperature with modified Karnovsky's fixative (2% formaldehyde, 2.5% glutaraldehyde in 0.1 M sodium cacodylate, pH 7.4), dislodged by scraping, and then processed for and analyzed by transmission electron microscopy as previously described (7).
Quantification of C. burnetii.
BMDMs, AMs, THP-1 cells, and irradiated Vero cells infected with C. burnetii were maintained in culture for up to 12 days. After 6 days of infection, cells were supplemented with an additional 500 μl of RPMI 1640 medium with 10% FBS and 5% LCCM for BMDMs, RPMI 1640 medium with 10% FBS for AMs and THP-1 cells, and DMEM (Gibco) with 10% FBS (Gibco) and 1 mM l-glutamine (Sigma-Aldrich) for irradiated Vero cells. At different times postinfection, cells were lysed with 1.0 ml of deionized water and cell lysates were combined with the culture supernatant from the respective wells. The material was centrifuged at 14,000 × g for 15 min at 4°C for deposition of the bacteria present in the samples. The DNA from C. burnetii was extracted using a Bacteria GenomicPrep Illustra kit (GE Healthcare), and the presence of genetic material was confirmed by spectrophotometry (determination of the absorbance at 260 nm). The primer pair dotA-for and dotA-rev (18) was used for quantitative PCR (qPCR), a reliable method for quantification of bacterial replication, as demonstrated by Brennan and Samuel (16) for analyses involving C. burnetii. The reaction was performed using a Platinum SYBR green qPCR Supermix-UDG with carboxy-X-rhodamine (ROX) kit (Invitrogen), and thermocycling reactions were performed according to the manufacturer's instructions. The samples were read on an ABI Prism 7000 thermocycler.
RT-PCR analysis.
mRNA from cultures of 106 AMs and BMDMs derived from C57BL/6 mice noninfected or infected for 12 h with C. burnetii at an MOI of 3 were extracted with the TRIzol reagent. RNA purification was done using an Illustra RNAspin minikit (GE Healthcare), according to the manufacturer's instructions, and genetic material was quantified using a NanoDrop spectrophotometer (Thermo Scientific). ImProm-II reverse transcriptase (Promega) was used to obtain cDNA. The real-time quantitative PCR assays were performed using the Platinum SYBR Green qPCR Supermix-UDG with carboxy-X-rhodamine kit (Invitrogen). Thermocycling reactions were performed according to the manufacturer's instructions, and the samples were read on a StepOnePlus real-time PCR system (Applied Biosystems). The expression of the target genes was calculated using the mean threshold cycle (CT) values of each sample, which were normalized to those for the housekeeping genes. The sequences of the primers used for reverse transcription-PCR (RT-PCR) are as follows: Gapdh-fwd, 5′-CATCACCATCTTCCAGGAGCG-3′; Gapdh-rev, 5′-ACGGACACATTGGGGGTAGG-3′; Nos2-fwd, 5′-CGAAACGCTTCSCTTCCAA-3′; Nos2-rev, 5′-TGAGCCTATATTGCTGTGGCT-3′; Ifn-fwd, 5′-GCATCTTGGCTTTGCAGCT-3′; Ifn-rev, 5′-CCTTTTTGCCCTTGCTGTTG-3′; Tnf-fwd, 5′-TGTGCTCAGAGCTTTCAACAA-3′; Tnf-rev, 5′-CTTGATGGTGCATGAGA-3′; Tlr2-fwd, 5′-AAGTCTCCGGAATTATCAGTCC-3′; Tlr2-rev, 5′-TGATGGATGTCGCGGAT-3′; Il4-fwd, 5′-AAGAGCATCATGCAAATGGA-3′; Il4-rev, 5′-TTAAAGCATGGTGGCTCAGTAC-3′; Arginase1-fwd, 5′-GTTCCCAGATGTACCAGGATTC-3′; Arginase1-rev, 5′-CGATGTCTTTGGCAGATATGC-3′; Fizz1-fwd, 5′-TGTGGCTTTGCCTGTGGAT-3′; Fizz1-rev, 5′-TCTTAGGACAGTTGGCAGCAG-3′; Ym1-fwd, 5′-GGGTTGGTTATGACAATGTCAG-3′; and Ym1-rev, 5′-TGAAGTCATCCATGTCCAGG-3′.
Flow cytometry for phenotypic analysis.
Single suspensions of AMs and BMDMs from C57BL/6 mice were stained with stains from the LIVE/DEAD fixable blue dead cell stain kit (Invitrogen) in PBS to exclude dead cells. Cells were stained with fluorochrome-conjugated antibodies against surface markers CD11b (clone M1/70), F4/80 (clone BM8), CD206 (clone C068C2), and CD86 (clone GL-1), purchased from BD Biosciences (San Diego, CA), and dectin-1/CLEC7A (CD369), obtained from BioLegend, in PBS containing 1% FBS for 30 min at 4°C and then washed. Staining was performed in the presence of 5 μg/ml of Fc Block (BD Biosciences). Cell acquisition was performed on a BD FACSCanto II flow cytometer using FACSDiva software (BD Biosciences). For each sample, at least 100,000 events were collected. Data were analyzed with FlowJo software (TreeStar).
Statistical analyses.
Statistical analyses were performed using GraphPad Prism (version 5) software. The results were analyzed by a statistical test for analysis of variance (ANOVA) followed by the Bonferroni posttest or Student's t test. Differences were considered statistically significant when the P value was <0.05.
RESULTS
Alveolar macrophages from C57BL/6 mice are permissive to replication of C. burnetii.
We have previously shown that BMDMs from A/J and BALB/c are less restrictive to the replication of C. burnetii phase II than cells from mice with the C57BL/6 mouse genetic background. This was determined by assessing the formation of the large CCV and by the determination of the number of C. burnetii focus-forming units (9). To further confirm these data, we quantified the bacterial genomic DNA by quantitative PCR (qPCR) as a tool to assess the replication of C. burnetii in BMDMs as previously described (16). In agreement with previously published data (9), we found that BMDMs from C57BL/6 mice are highly restrictive to C. burnetii phase II, whereas BMDMs from A/J mice support some intracellular replication of C. burnetii phase II from days 3 to 12 postinfection. This was observed in experiments using high and low infection doses (MOIs, 100 and 3, respectively) (Fig. 1A and B, respectively). We continued the macrophage infection studies using an MOI of 3. Because most of the gene-deficient mice generated thus far were constructed in the C57BL/6 mouse background, we aimed to identify a primary cell that supported the replication of C. burnetii phase II in culture. It is known that C. burnetii infects macrophages and monocytes, with AMs considered the primary target cell of infection (10, 12, 19–22). Thus, we evaluated whether murine AMs from the C57BL/6 mouse strain support the replication of C. burnetii phase II in culture. Strikingly, when we assessed bacterial replication using qPCR, we found that, as opposed to BMDMs, the AMs were highly permissive to C. burnetii phase II replication (Fig. 1C). Of note, the internalization of bacteria by these different cells was similar, as measured by determination of the number of C. burnetii genomes associated with the cells after 4 h of infection (Fig. 1A to D, insets). As a negative control for bacterial replication, rifampin treatment was included to kill the C. burnetii bacteria in the macrophages. Further, when we compared the C. burnetii replication in AMs from C57BL/6 and A/J mice, we found that AMs from both strains were equally permissive for C. burnetii replication (Fig. 1D). These data suggest that the genetic mechanisms controlling the natural resistance of BMDMs to C. burnetii may be less effective or absent in AMs. Importantly, AMs emerge as a very interesting model of the primary macrophage that supports the intracellular replication of C. burnetii phase II in culture. As such, we decided to further characterize C. burnetii replication in AMs.
Alveolar macrophages accumulate large amounts of intracellular bacteria within the Coxiella-containing vacuole.
Given the robust replication of C. burnetii phase II in murine AMs (Fig. 1C and D), we aimed to assess bacterial replication in these macrophages by microscopy. AMs obtained from C57BL/6 mice were infected, and intracellular bacteria were visualized by immunofluorescence and transmission electron microscopy. Figure 2 illustrates the susceptibility of AMs over the first 6 days of C. burnetii phase II infection in culture. We detected an increased concentration of intracellular C. burnetii, stained in red, during the course of infection, suggesting that the bacteria can readily replicate in AMs. Further, the association of the bacteria with the lysosomal membrane marker LAMP-1 confirms that the bacteria replicate in the well-characterized lysosome-derived parasitophorous vacuole. During the procedures to stain the bacteria and LAMP-1, the AMs containing large spacious vacuoles were lost because these cells easily detach from the coverslips during the antibody washings. Thus, to assess the presence of typical large vacuoles in infected AMs, we used a strain of C. burnetii expressing mCherry (23). By examining the infected AMs using two-photon laser scanning fluorescence microscopy, we found a significant increase in bacterial replication in AMs from days 0 (4 h of infection) to 12 after infection (Fig. 2A and B). At day 6 of infection, we observed several cells containing many C. burnetii bacteria in large vacuoles (Fig. 2B). At day 9 of infection, the AMs were filled with C. burnetii bacteria, as illustrated by a merge image showing both differential interference contrast and fluorescence images (Fig. 2C). Intracellular bacteria were also observed by transmission electron microscopy (Fig. 3). At 3 days after infection, AMs showed large CCVs containing high numbers of large-cell variants of C. burnetii (Fig. 3A). At day 6 after infection, we detected vacuoles harboring a large amount of C. burnetii bacteria (Fig. 3B). Taken together, the results confirm that AMs support C. burnetii replication in culture and can be used as a useful model of the primary macrophage to study C. burnetii phase II replication in vitro.
FIG 2.
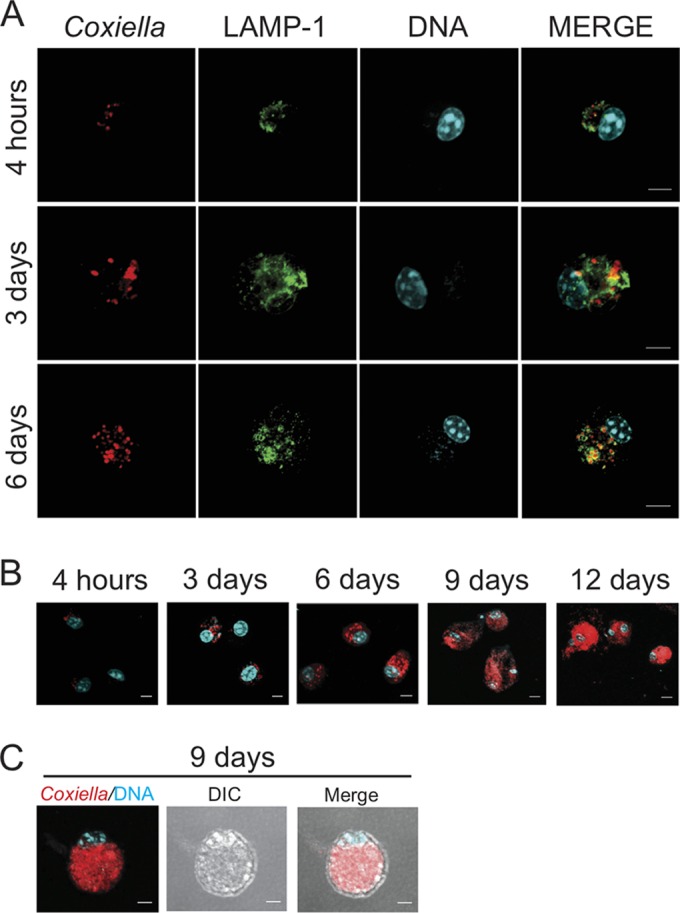
C. burnetii infection in alveolar macrophages assessed by fluorescence microscopy. AMs from C57BL/6 mice were infected with wild-type C. burnetii phase II (MOI, 3) for 4 h, 3 days, and 6 days (A) or infected with mCherry-expressing C. burnetii phase II for 4 h, 3 days, 6 days, 9 days, and 12 days (B). After the indicated times, cells were fixed, processed, and then examined using multiphoton fluorescence microscopy. (C) AM infected for 9 days with mCherry-expressing C. burnetii. DIC, differential interference contrast. Green, lysosomal membrane-associated protein LAMP-1; red, C. burnetii; cyan, DAPI-stained DNA. Bars, 5 μm.
FIG 3.

C. burnetii infection in alveolar macrophages assessed by transmission electron microscopy. AMs were infected with C. burnetii phase II (MOI, 3), fixed, and processed for transmission electron microscopy. Cells were fixed after 3 days (top) and 6 days (bottom) of infection. The panels on the right are enlarged images of the boxed regions in the panels on the left. Arrows, large-cell variant forms of C. burnetii. Bars, 1 μm.
Alveolar macrophages show a more pronounced polarization into M2 cells than bone marrow-derived macrophages.
To assess the mechanisms underlying the permissiveness of AMs to C. burnetii, we evaluated M1/M2 polarization, which has previously been shown to be important for C. burnetii replication and pathogenesis (24, 25). Initially, we evaluated the expression of genes known to be hallmarks of M1 and M2 polarization in AMs and BMDMs. By measuring gene expression in noninfected BMDMs and AMs, we found that the latter show a significantly reduced expression of Nos2, Tnfa, Ifng, and Tlr2 (genes associated with an M1 polarization phenotype) and an increased expression of Arginase1, Fizz1, and Ym1, which are associated with an M2 profile (Fig. 4). The AMs also showed a pronounced M2 polarization compared to that of BMDMs in response to C. burnetii infection. We detected reduced expression of Nos2, Tnfa, Ifng, and Tlr2 and increased expression of Il4, Arginase1, Fizz1, and Ym1 in AMs infected with C. burnetii compared to their expression in infected BMDMs (Fig. 4). These data are consistent with AMs being more susceptible because they polarize toward an M2 phenotype. Accordingly, M1 macrophages effectively trigger the production of tumor necrosis factor alpha (TNF-α), a cytokine known to facilitate the restriction of C. burnetii replication in macrophages (26). To further test this hypothesis, we evaluated the expression of surface markers known to be hallmarks of M2 macrophages, including mannose receptor (CD206) and dectin-1 (27). To evaluate protein expression, we performed fluorescence-activated cell sorting experiments in which we gated live macrophages and evaluated the expression of the cell surface markers CD206, dectin-1, and CD86 on cells double positive for CD11b and F4/80 (Fig. 5A and B). Using specific antibodies, we found that AMs showed increased expression of M2-associated molecules, such as CD206 (mannose receptor) and dectin-1 (CLEC7A), compared to BMDMs. This was evident under the uninfected condition (Fig. 5C) and in response to infection (Fig. 5D). Moreover, we measured the expression of the costimulatory molecule CD86, which was similarly expressed in AMs and BMDMs (Fig. 5C and D). These data are consistent with the hypothesis that AMs are more susceptible because of an M2 polarized phenotype, a characteristic that has previously been associated with permissiveness to C. burnetii replication (reviewed in reference 28).
FIG 4.

Alveolar macrophages exhibit higher levels of expression of genes related to the M2 phenotype. BMDMs and AMs from C57BL/6 mice were left uninfected or infected for 12 h with C. burnetii at an MOI of 3. Total RNA was extracted, and reverse transcription-PCR (RT-PCR) was performed for detection of the genes Nos2, Tnfa, Ifng, Tlr2, Il4, Arginase1, Fizz1, and Ym1. The data shown are the means ± standard errors from one representative experiment out of two performed. *, P < 0.05 comparing BMDMs and AMs; #, P < 0.05 comparing infected cells with the uninfected cells from the respective group (two-way ANOVA).
FIG 5.

Alveolar macrophages exhibit an M2 phenotype. (A and B) Gating strategy and frequency of noninfected BMDMs (A) and AMs (B). FSC, forward scatter; SSC, side scatter. (C and D) Surface phenotyping of BMDMs and AMs infected (for 24 h) (C) or not infected (D) with C. burnetii for CD86 and the M2 markers CD206 and dectin-1, as analyzed by flow cytometry. Cells were gated on live CD11b+ F4/80+ cells.
Alveolar macrophages deficient in NOS2 and IFN-γ are more susceptible to C. burnetii replication, whereas cells deficient in interleukin-4 (IL-4) are more restrictive to C. burnetii replication.
It is well established that nitric oxide (NO) is involved in the restriction of C. burnetii replication (8, 9, 29, 30). Thus, we assessed bacterial replication in AMs deficient in nitric oxide synthase 2 (NOS2; or inducible nitric oxide synthase [iNOS]) as a proof of principle that AMs can be used as a model to investigate the interactions of C. burnetii with primary mouse macrophages in the C57BL/6 mouse genetic background. In agreement with previous reports, we found that AMs from Nos2−/− mice are more permissive to C. burnetii phase II replication than AMs from wild-type C57BL/6 mice (Fig. 6A). We also evaluated bacterial replication in AMs from Ifng−/− mice because gamma interferon (IFN-γ) is known to be essential for NO production (31). Moreover, IFN-γ is well described to be involved in the immune response against C. burnetii (19, 32–40). We found that AMs from Ifng−/− mice are significantly more permissive to C. burnetii phase II replication than AMs from wild-type C57BL/6 mice (Fig. 6B). Collectively, these data support the use of AMs as a model of primary macrophages for investigations involving C. burnetii infections in primary macrophages from the C57BL/6 mouse, the strain mostly commonly used for the generation of transgenic mice. To further understand the effect of the M2 polarization of AMs that results in the increased susceptibility to C. burnetii, we performed experiments using AMs from mice deficient in IL-4, which is required for M2 differentiation (reviewed in reference 41). We found that AMs from Il4−/− mice are less permissive to C. burnetii replication than cells from C57BL/6 mice (Fig. 6C). The internalization of bacteria did not differ in AMs from C57BL/6, Nos2−/−, Ifng−/−, and Il4−/− mice, as measured by the number of C. burnetii bacteria in cultures infected for 4 h (Fig. 6A to C, insets). Collectively, these data support the hypothesis that the M1/M2 polarization underlies the increased susceptibility of AMs in comparison to BMDMs. Importantly, regardless of the mechanisms associated with the resistance of the BMDMs, these data support the use of AMs as an important and relevant host cell for evaluation of C. burnetii replication in primary macrophages.
FIG 6.
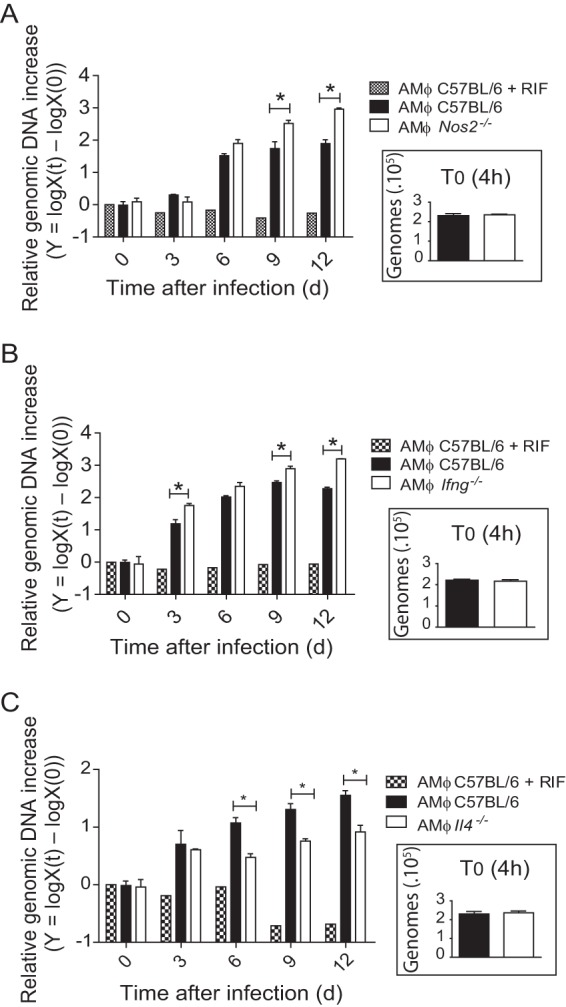
AMs support the assessment of innate immunity against C. burnetii phase II using knockout mice in the C57BL/6 mouse genetic background. Primary AMs from wild-type C57BL/6 mice and Nos2−/− (A), Ifng−/− (B), and Il4−/− (C) mice were infected with C. burnetii phase II (MOI, 3), and bacterial multiplication was evaluated by quantification of the genomic DNA by qPCR after 0 (4 h after infection), 3, 6, 9, and 12 days of infection. The y axis represents the difference in the base 10 logarithm between the genomic quantity X at time t [logX(t)] and the quantity X at time zero (T0) [logX(0)]. Cells treated with RIF were used as a negative control of bacterial replication. (Insets) Amount of C. burnetii genomic DNA present in cells after 4 h of infection. The data shown are the means ± standard errors from one representative experiment out of three independent experiments preformed. *, P < 0.05 (two-way ANOVA).
The permissiveness of mouse alveolar macrophages is comparable to that of highly susceptible immortalized cell lines.
Our results demonstrated that murine AMs are permissive for C. burnetii phase II replication in vitro. Thus, we decided to compare C. burnetii phase II replication in C57BL/6 mouse AMs with that in highly susceptible host cells, such as Vero cells (a green monkey kidney cell line) and cells of a monocytic cell line (THP-1) differentiated into macrophage-like cells. Both of these cell types are known to be very susceptible to C. burnetii replication (6, 14, 34). Figure 7 shows that at 6 days postinfection, murine AMs are as susceptible to C. burnetii phase II infection as Vero and THP-1 cells. At later time points after infection, the Vero and THP-1 cell lines supported bacterial replication more robust than that in primary AMs. Nonetheless, the comparable rate of replication during the first 6 days of infection reinforces the demonstration that murine AMs are indeed highly permissive for C. burnetii replication and can be used as a relevant host cell model for investigations involving the macrophage interaction with C. burnetii phase II in culture.
FIG 7.

Alveolar macrophages are permissive to C. burnetii phase II replication, similar to Vero and THP-1 cells, in vitro. C57BL/6 AMs, irradiated Vero cells, and THP-1 cells (previously differentiated overnight with 200 nM PMA) were infected with C. burnetii phase II (MOI, 3), and bacterial multiplication was evaluated by quantification of the genomic DNA by qPCR after 0 (4 h after infection), 3, 6, 9, and 12 days of infection. The y axis represents the difference in the base 10 logarithm between the genomic quantity X at time t [logX(t)] and the quantity X at time zero (T0) [logX(0)]. Cells treated with RIF were used as a negative control of bacterial replication. (Insets) Amount of C. burnetii genomic DNA present in cells after 4 h of infection. The data shown are the means ± standard deviations from one representative experiment out of three independent experiments preformed. *, P < 0.05 (two-way ANOVA).
DISCUSSION
C. burnetii is a unique intracellular bacterial pathogen capable of replicating to high numbers within a lysosome-derived vacuole. Humans can contract Q fever through the inhalation of contaminated aerosols and the subsequent intracellular replication of C. burnetii within AMs. As such, studying the interaction between this specific macrophage lineage and the pathogen provides an important, disease-relevant, in vitro model to understand the specific host-pathogen interaction that takes place during C. burnetii infection. Previous research has demonstrated that BMDMs from different mouse strains vary in their permissiveness to C. burnetii replication (42). BMDMs from A/J and BALB/c mice support the intracellular replication of C. burnetii, while BMDMs from C57BL/6 mice restrict bacterial replication. Here, we recapitulate these results and demonstrate that AMs are significantly more permissive to C. burnetii replication than BMDMs. This finding is most striking with C57BL/6 mice, in which BMDMs restrict the intracellular growth of C. burnetii, yet the bacteria readily replicate in AMs. Indeed, despite a remarkable difference in the replication of C. burnetii in BMDMs of C57BL/6 and A/J mice, there is no significant difference in the ability of C. burnetii to replicate in AMs from these two inbred mouse strains. This suggests that the genetic basis for C57BL/6 mouse BMDM restriction of C. burnetii infection reflects phenotypic features that may be specific for this cell type or that operate at a lower efficiency in AMs.
AMs are critical lung resident cells that, in healthy lungs, constitute the vast majority of the cellular content within alveoli (43). As such these cells are intricately essential for lung homeostasis and immune surveillance (44). Recent evidence indicates that AMs originate from fetal monocytes that are long living and are maintained by self-renewal rather than circulating bone marrow-derived monocytes (45). This tissue microenvironment has a significant epigenetic and transcriptomic impact on resident macrophages, rendering AMs phenotypically distinct from BMDMs (46–48). In light of our results, one can suggest that these phenotypic differences between BMDMs and AMs from C57BL/6 mice mediate a significant difference in the capacity to restrict the intracellular replication of C. burnetii. Interestingly, our data comparing M1/M2 polarization in BMDMs and AMs suggest that the M2 profile of AMs may underlie their increased susceptibility to infection. This hypothesis is supported by experiments using cells from Il4−/− mice. In the absence of IL-4, the AMs became significantly more resistant to infection, a feature that is consistent with the essential role of IL-4 in the induction of M2 polarization (41). It is likely that during coevolution with mammalian hosts, C. burnetii has evolved strategies to target and subvert specific responses in alveolar macrophages, a process that may be facilitated by the anti-inflammatory M2 characteristics of this cell type.
In support of our data showing the increased replication of C. burnetii in AMs, it was previously reported that tissue-resident AMs are susceptible to C. burnetii replication in vivo (12). Moreover, human AMs were shown to support C. burnetii replication in culture (11). In addition, a system that allows infection of ex vivo human lung tissue was recently reported. It was found that within human lung tissue, C. burnetii preferentially replicated in human AMs (49). In this context, our data provide a definitive demonstration that mouse AMs can be used as a relevant host cell for investigations of the interactions of C. burnetii with host cells in vitro. C57BL/6 mouse AMs support the intracellular replication of C. burnetii, and the bacteria replicate in a manner that is comparable to that in widely used immortalized cell lines that are highly susceptible to C. burnetii replication. Over the first 6 days after infection, we found that C. burnetii phase II replicates similarly in AMs, THP-1 cells, and Vero cells. At days 9 and 12, we detected higher bacterial loads in Vero and THP-1 cells, which are possibly caused by the saturation of the host cells available for bacterial replication. This hypothesis is supported by the experiments performed with AMs infected with mCherry-expressing C. burnetii. After 9 days of infection, we visualized AMs filled with bacteria, which were contained in large vacuoles that occupy the entire macrophage cytoplasm (Fig. 2B and C). We did not detect a reduction of bacterial loads from days 9 to 12, a feature that does not support the restriction of bacterial replication in the AMs at later stages of infection.
C57BL/6 mouse AMs can serve as an important host cell for the investigation of host factors that aid or restrict the intracellular replication of C. burnetii. Here we have confirmed the importance of both nitric oxide and IFN-γ in restricting the ability of C. burnetii to replicate. Significantly higher levels of C. burnetii were recovered from both Nos2−/− and Ifng−/− mouse AMs than from wild-type C57BL/6 mouse AMs. Production of nitric oxide and its reactive nitrogen intermediates is an important cellular response that acts against respiratory pathogens (50). At high levels, nitric oxide has a cytotoxic effect and can participate in the control of intracellular pathogen replication. Indeed, several studies have demonstrated that nitric oxide has the capacity to restrict the intracellular replication of C. burnetii (8, 9, 29, 30, 51). Our data support these findings, demonstrating that AMs from C57BL/6 mice lacking Nos2 support higher levels of C. burnetii replication than wild-type cells. Similarly, the absence of IFN-γ, which is critical for nitric oxide production, is known to be important to host restriction of C. burnetii replication. Importantly, by using AMs from Nos2−/− and Ifng−/− mice, we validated the use of AMs as a relevant host cell for the in vitro infection setting. Our characterization of this model of infection demonstrates that studying the interaction between C. burnetii and AMs will allow intricate investigation of both host and bacterial factors that are important for pathogenesis and host resistance during Q fever.
ACKNOWLEDGMENTS
We are grateful to Maira C. Nakamura and Victoria Maria dos Santos for technical assistance. We also thank Maria D. S. Ferreira, José A. Maulin, and Roberta R. Rosales for technical assistance in the institutional facilities for electron microscopy (M. D. S. Ferreira and J. A. Maulin) and multiphoton microscopy (R. R. Rosales).
We declare no conflict of interest in relation to this work.
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grants 2013/08216-2, 2014/50268-2, and 2014/04684-4), Conselho Nacional do Desenvolvimento Cientifico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo ao Ensino, Pesquisa e Assistência do Hospital das Clínicas da FMRP/USP (FAEPA), and the Australian National Health and Medical Research Council (NHMRC; grants 1062383 and 1063646 to H.J.N.). T.D.F. and L.M.M. are supported by a fellowship from CAPES. L.D.C., J.M.R., and D.S.L. are supported by fellowships from FAPESP. H.J.N. is a visiting professor from the Science without Borders Program (CNPq; grant 401577/2014-7). D.S.Z. is a research fellow from CNPq, Brazil.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.van Loenhout JA, Wielders CC, Morroy G, Cox MJ, van der Hoek W, Hautvast JL, Paget WJ, van der Velden J. 2015. Severely impaired health status of non-notified Q fever patients leads to an underestimation of the true burden of disease. Epidemiol Infect 143:2580–2587. doi: 10.1017/S0950268814003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khavkin T, Tabibzadeh SS. 1988. Histologic, immunofluorescence, and electron microscopic study of infectious process in mouse lung after intranasal challenge with Coxiella burnetii. Infect Immun 56:1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seshadri R, Hendrix LR, Samuel JE. 1999. Differential expression of translational elements by life cycle variants of Coxiella burnetii. Infect Immun 67:6026–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton HJ, McDonough JA, Roy CR. 2013. Effector protein translocation by the Coxiella burnetii Dot/Icm type IV secretion system requires endocytic maturation of the pathogen-occupied vacuole. PLoS One 8:e54566. doi: 10.1371/journal.pone.0054566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffatt JH, Newton P, Newton HJ. 2015. Coxiella burnetii: turning hostility into a home. Cell Microbiol 17:621–631. doi: 10.1111/cmi.12432. [DOI] [PubMed] [Google Scholar]
- 6.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zamboni DS, Mortara RA, Freymuller E, Rabinovitch M. 2002. Mouse resident peritoneal macrophages partially control in vitro infection with Coxiella burnetii phase II. Microbes Infect 4:591–598. doi: 10.1016/S1286-4579(02)01577-0. [DOI] [PubMed] [Google Scholar]
- 8.Zamboni DS, Rabinovitch M. 2003. Nitric oxide partially controls Coxiella burnetii phase II infection in mouse primary macrophages. Infect Immun 71:1225–1233. doi: 10.1128/IAI.71.3.1225-1233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamboni DS, Rabinovitch M. 2004. Phagocytosis of apoptotic cells increases the susceptibility of macrophages to infection with Coxiella burnetii phase II through down-modulation of nitric oxide production. Infect Immun 72:2075–2080. doi: 10.1128/IAI.72.4.2075-2080.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voth DE, Howe D, Heinzen RA. 2007. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun 75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. 2013. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol 15:1012–1025. doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calverley M, Erickson S, Read AJ, Harmsen AG. 2012. Resident alveolar macrophages are susceptible to and permissive of Coxiella burnetii infection. PLoS One 7:e51941. doi: 10.1371/journal.pone.0051941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshiie K, Matayoshi S, Fujimura T, Maeno N, Oda H. 1999. Induced production of nitric oxide and sensitivity of alveolar macrophages derived from mice with different sensitivity to Coxiella burnetii. Acta Virol 43:273–278. [PubMed] [Google Scholar]
- 14.Zamboni DS, Mortara RA, Rabinovitch M. 2001. Infection of Vero cells with Coxiella burnetii phase II: relative intracellular bacterial load and distribution estimated by confocal laser scanning microscopy and morphometry. J Microbiol Methods 43:223–232. doi: 10.1016/S0167-7012(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 15.Marim FM, Silveira TN, Lima DS Jr, Zamboni DS. 2010. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One 5:e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennan RE, Samuel JE. 2003. Evaluation of Coxiella burnetii antibiotic susceptibilities by real-time PCR assay. J Clin Microbiol 41:1869–1874. doi: 10.1128/JCM.41.5.1869-1874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cunha LD, Ribeiro JM, Fernandes TD, Massis LM, Khoo CA, Moffatt JH, Newton HJ, Roy CR, Zamboni DS. 2015. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat Commun 6:10205. doi: 10.1038/ncomms10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carey KL, Newton HJ, Luhrmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amara AB, Bechah Y, Mege JL. 2012. Immune response and Coxiella burnetii invasion. Adv Exp Med Biol 984:287–298. doi: 10.1007/978-94-007-4315-1_15. [DOI] [PubMed] [Google Scholar]
- 20.Hackstadt T, Williams JC. 1981. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A 78:3240–3244. doi: 10.1073/pnas.78.5.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shannon JG, Heinzen RA. 2008. Infection of human monocyte-derived macrophages with Coxiella burnetii. Methods Mol Biol 431:189–200. [DOI] [PubMed] [Google Scholar]
- 23.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-11. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benoit M, Barbarat B, Bernard A, Olive D, Mege JL. 2008. Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol 38:1065–1070. doi: 10.1002/eji.200738067. [DOI] [PubMed] [Google Scholar]
- 25.Mehraj V, Textoris J, Ben Amara A, Ghigo E, Raoult D, Capo C, Mege JL. 2013. Monocyte responses in the context of Q fever: from a static polarized model to a kinetic model of activation. J Infect Dis 208:942–951. doi: 10.1093/infdis/jit266. [DOI] [PubMed] [Google Scholar]
- 26.Bradley WP, Boyer MA, Nguyen HT, Birdwell LD, Yu J, Ribeiro JM, Weiss SR, Zamboni DS, Roy CR, Shin S. 2016. Primary role for Toll-like receptor-driven tumor necrosis factor rather than cytosolic immune detection in restricting Coxiella burnetii phase II replication within mouse macrophages. Infect Immun 84:998–1015. doi: 10.1128/IAI.01536-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez FO, Gordon S, Locati M, Mantovani A. 2006. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 28.Benoit M, Desnues B, Mege JL. 2008. Macrophage polarization in bacterial infections. J Immunol 181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 29.Brennan RE, Russell K, Zhang G, Samuel JE. 2004. Both inducible nitric oxide synthase and NADPH oxidase contribute to the control of virulent phase I Coxiella burnetii infections. Infect Immun 72:6666–6675. doi: 10.1128/IAI.72.11.6666-6675.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howe D, Barrows LF, Lindstrom NM, Heinzen RA. 2002. Nitric oxide inhibits Coxiella burnetii replication and parasitophorous vacuole maturation. Infect Immun 70:5140–5147. doi: 10.1128/IAI.70.9.5140-5147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinakar C, Malur A, Raychaudhuri B, Buhrow LT, Melton AL, Kavuru MS, Thomassen MJ. 1999. Differential regulation of human blood monocyte and alveolar macrophage inflammatory cytokine production by nitric oxide. Ann Allergy Asthma Immunol 82:217–222. doi: 10.1016/S1081-1206(10)62600-2. [DOI] [PubMed] [Google Scholar]
- 32.Turco J, Thompson HA, Winkler HH. 1984. Interferon-gamma inhibits growth of Coxiella burnetii in mouse fibroblasts. Infect Immun 45:781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dellacasagrande J, Capo C, Raoult D, Mege JL. 1999. IFN-gamma-mediated control of Coxiella burnetii survival in monocytes: the role of cell apoptosis and TNF. J Immunol 162:2259–2265. [PubMed] [Google Scholar]
- 34.Ghigo E, Capo C, Tung CH, Raoult D, Gorvel JP, Mege JL. 2002. Coxiella burnetii survival in THP-1 monocytes involves the impairment of phagosome maturation: IFN-gamma mediates its restoration and bacterial killing. J Immunol 169:4488–4495. doi: 10.4049/jimmunol.169.8.4488. [DOI] [PubMed] [Google Scholar]
- 35.Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun 75:3245–3255. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochoa-Reparaz J, Sentissi J, Trunkle T, Riccardi C, Pascual DW. 2007. Attenuated Coxiella burnetii phase II causes a febrile response in gamma interferon knockout and Toll-like receptor 2 knockout mice and protects against reinfection. Infect Immun 75:5845–5858. doi: 10.1128/IAI.00901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong X, Meng Y, Wang X, Qi Y, Li J, Duan C, Wen B. 2012. Mice immunized with bone marrow-derived dendritic cells stimulated with recombinant Coxiella burnetii Com1 and Mip demonstrate enhanced bacterial clearance in association with a Th1 immune response. Vaccine 30:6809–6815. doi: 10.1016/j.vaccine.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Schoffelen T, Sprong T, Bleeker-Rovers CP, Wegdam-Blans MC, Ammerdorffer A, Pronk MJ, Soethoudt YE, van Kasteren ME, Herremans T, Bijlmer HA, Netea MG, van der Meer JW, Joosten LA, van Deuren M. 2014. A combination of interferon-gamma and interleukin-2 production by Coxiella burnetii-stimulated circulating cells discriminates between chronic Q fever and past Q fever. Clin Microbiol Infect 20:642–650. doi: 10.1111/1469-0691.12423. [DOI] [PubMed] [Google Scholar]
- 39.Schoffelen T, Self JS, Fitzpatrick KA, Netea MG, van Deuren M, Joosten LA, Kersh GJ. 2015. Early cytokine and antibody responses against Coxiella burnetii in aerosol infection of BALB/c mice. Diagn Microbiol Infect Dis 81:234–239. doi: 10.1016/j.diagmicrobio.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoffelen T, Wegdam-Blans MC, Ammerdorffer A, Pronk MJ, Soethoudt YE, Netea MG, van der Meer JW, Bleeker-Rovers CP, van Deuren M. 2015. Specific in vitro interferon-gamma and IL-2 production as biomarkers during treatment of chronic Q fever. Front Microbiol 6:93. doi: 10.3389/fmicb.2015.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon S. 2003. Alternative activation of macrophages. Nat Rev Immunol 3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 42.Zamboni DS. 2004. Genetic control of natural resistance of mouse macrophages to Coxiella burnetii infection in vitro: macrophages from restrictive strains control parasitophorous vacuole maturation. Infect Immun 72:2395–2399. doi: 10.1128/IAI.72.4.2395-2399.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kopf M, Schneider C, Nobs SP. 2015. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol 16:36–44. doi: 10.1038/ni.3052. [DOI] [PubMed] [Google Scholar]
- 44.Morales-Nebreda L, Misharin AV, Perlman H, Budinger GR. 2015. The heterogeneity of lung macrophages in the susceptibility to disease. Eur Respir Rev 24:505–509. doi: 10.1183/16000617.0031-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. 2013. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. 2014. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. 2014. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gordon S, Pluddemann A, Martinez Estrada F. 2014. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev 262:36–55. doi: 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graham JG, Winchell CG, Kurten RC, Voth DE. 2016. Development of an ex vivo tissue platform to study the human lung response to Coxiella burnetii. Infect Immun 84:1438–1445. doi: 10.1128/IAI.00012-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nathan C, Shiloh MU. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A 97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill J, Samuel JE. 2011. Coxiella burnetii acid phosphatase inhibits the release of reactive oxygen intermediates in polymorphonuclear leukocytes. Infect Immun 79:414–420. doi: 10.1128/IAI.01011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]