

Abstract
Two-partner secretion (TPS) systems export large TpsA proteins to the surface and extracellular milieu. In meningococci, three different TPS systems exist, and of these, TPS system 2 (TPS2) and TPS3 can be detected by the host's immune system. We evaluated the distribution of TPS systems among clinical isolates from two prospective cohort studies comprising 373 patients with meningococcal meningitis. TPS system 1 was present in 91% of isolates, and system 2 and/or 3 was present in 67%. The TPS system distribution was related to clonal complexes. Infection with strains with TPS2 and/or TPS3 resulted in less severe disease and better outcomes than infection with strains without these systems. Using whole-blood stimulation experiments, we found no differences in the host cytokine response between patients infected with TPS system 2 and 3 knockout strains and patients infected with a wild-type strain. In conclusion, meningococcal TPS system 2 and/or 3 is associated with disease severity and outcome in patients with meningitis.
INTRODUCTION
The two-partner secretion (TPS) pathway is a widespread protein secretion route in Gram-negative bacteria consisting of a large and secreted exoprotein (TpsA) typically of more than 100 kDa and an ∼60-kDa transporter protein (TpsB) (1). TpsA and TpsB translocate across the inner cytoplasmic membrane through the general secretion system (2). Subsequently, TpsB inserts into the outer membrane and facilitates the export of TpsA. TpsA targets TpsB using the so-called TPS domain, which is located at the N terminus of the periplasmic intermediate. Functions of TPS systems vary among different bacterial species and include hemolysis, cytotoxicity, and iron acquisition (3, 4). Neisseria meningitidis (the meningococcus) is a Gram-negative diplococcus and a major cause of bacterial meningitis and sepsis worldwide (5). Secreted products are considered to be important in the pathogenesis of meningococcal invasive disease (6). The TPS pathway is one of many protein export mechanisms in meningococci and comprises three distinct systems (1), of which one (TPS system 1 [TPS1]; see below) is generally present among meningococcal isolates (7, 8). This ubiquitous system had initially been indicated to contribute to adhesion to cultured host cells (9) or, alternatively, to prolonged intracellular survival (6, 7, 9). This TPS system was more recently also shown to play a role in maintaining meningococcal biofilms (10) and to act as a toxin-antitoxin system active in bacterial fratricide (11, 12). To date, it remains unclear how the different functions are related to each other.
Three TPS systems are known in the meningococcal genome: TPS1, encoded by tpsA1a, tpsA1b, tpsB1, and tpsB1tr; TPS2, encoded by tpsA2a, tpsA2b, and tpsB2; and TPS3, encoded by tpsA3 (the numbers in the gene and protein names refer to the systems) (8). However, the TPS domains of TpsA proteins of all three systems can be exported by TpsB2, whereas TpsB1 is capable of exporting only TpsA1 (1). During meningococcal infection, components of TPS2 and TPS3 can elicit an immune response, which suggests that antibodies against TPS systems may be potential vaccination components (8). Most TPS genes are unique, except for tpsA1b, a duplicate of tpsA1a, and tpsB1tr, a truncated and nonfunctional duplicate of tpsB1. TPS genes of the three systems are located within distinct genomic islands (8). Repetitive putative open reading frames (ORFs) encoding the C-terminal end of full-length TpsA proteins (tpsC cassettes) are present downstream of the tpsA1a, tpsA1b, and tpsA2a genes, possibly to enable variation of tpsA by recombination between these cassettes and full-length tpsA (12, 13).
We previously reported the frequency of tps genes in 88 clinical invasive N. meningitidis isolates from both sepsis and meningitis patients (8). With the current knowledge of the TpsA-TpsB specificity (1), 82% harbored a functional TPS1, 47% harbored a functional TPS2, and 20% harbored a functional TPS3. Another study reported the frequency of TPS genes in 822 meningococcal carrier isolates, determined using dot blot hybridization of tpsA genes: 77% harbored an intact TPS1, 40% harbored an intact TPS2, and 15% harbored an intact TPS3 (7). In both studies, isolates with TPS2 and/or TPS3 were linked to hyperinvasive clonal complexes (cc; defined as cc8, cc11, cc32, cc41/44, and cc269). Although these studies used different selections of isolates, the results suggest that meningococcal TPS systems are related to bacterial invasiveness or virulence. However, it does not indicate whether the TPS systems play a role in the severity of infection or the disease outcome. To establish such a link, we evaluated the distribution of TPS systems among clinical isolates from two prospective cohorts of bacterial meningitis patients and evaluated the association between these systems and meningococcal disease severity. Subsequently, we evaluated our findings with in vitro experiments using tps knockout meningococcal strains and complemented knockout strains.
(A part of the information presented here has previously been presented on a poster at the 47th Infectious Diseases Society of America [IDSA] Annual Meeting in Philadelphia, PA, USA, in 2009 [14] and on a poster at the 53rd Interscience Conference on Antimicrobial Agents and Chemotherapy [ICAAC] in Denver, CO, USA, in 2013 [15].)
MATERIALS AND METHODS
Meningitis cases.
The Dutch Meningitis Cohort Study (October 1998 to April 2002) and the MeninGene Study (January 2006 to July 2014) were prospective nationwide observational cohort studies of adults with community-acquired bacterial meningitis in the Netherlands. Patients were included with the help of the Netherlands Reference Laboratory for Bacterial Meningitis (NRLBM). This laboratory received isolates from the cerebrospinal fluid (CSF) of 90% of all patients with bacterial meningitis in the Netherlands. Written informed consent was obtained from all patients or their legally authorized representatives. The study was approved by the Medical Ethics Committee of the Academic Medical Center, Amsterdam, the Netherlands.
Case record forms were used to collect data on the patients' history, symptoms and signs on admission, laboratory findings at admission, clinical course, outcome and neurologic findings at discharge, and treatment. The Glasgow Coma Scale (GCS) was used to determine the patients' conscious state on admission. Outcome was graded according to the Glasgow Outcome Scale (GOS) (16). A favorable outcome was defined as a score of 5, and an unfavorable outcome was defined as a score lower than 5.
Meningococcal typing.
N. meningitidis strains from patients with meningococcal meningitis were collected and stored at NRLBM. Serogrouping and multilocus sequence typing (MLST) were performed at NRLBM as previously described (17), and the results have been published previously (18, 19). The clonal complexes (cc) were assigned according to the MLST database, available at http://pubmlst.org/neisseria/.
PCR analysis of strains from the 1998 to 2002 cohort.
The distribution of genes encoding TpsA (tpsA1, tpsA2a, tpsA2b, tpsA3) and genes encoding TpsB (tpsB1, tpsB2) among the meningococcal isolates from the cohort studied from 1998 to 2002 (n = 254; referred to here as the 1998 to 2002 cohort) was determined by 6 different PCRs. The primer pairs used are displayed in Table S1 in the supplemental material. The primers targeting the tpsB genes were designed to produce full-length tpsB genes, meaning that the truncated form is not amplified in these PCRs. The tpsA1-specific PCR did not discriminate between a possible duplication of this ORF, as is present in the genome of N. meningitidis MC58 (tpsA1b).
Whole-genome sequencing of strains from the 2006 to 2014 cohort.
An in-house-adapted version of the Wizard Genomic DNA purification kit (Promega) was used to isolate chromosomal DNA. The genomes of the meningococcal isolates from the cohort studied from 2006 to 2014 (n = 115; referred to here as the 2006 to 2014 cohort) were sequenced at the Welcome Trust Sanger Institute using Illumina HiSeq 2000 analyzers, as was previously described (20).
TPS system definition.
For the 1998 to 2002 cohort, isolates with TPS system 1 were defined as isolates yielding an amplicon in the tpsA1 PCR and either the tpsB1 or the tpsB2 PCR. Isolates with TPS system 2 were defined as isolates yielding an amplicon in the tpsA2a and/or tpsA2b PCR in combination with an amplicon in the tpsB2 PCR. Isolates with TPS system 3 were defined as isolates yielding an amplicon in the tpsA3 and the tpsB2 PCRs. For the 2006 to 2014 cohort, TPS system genes were defined by BLAST analysis of the tps genes from meningococcal strain MC58, and the TPS systems were identified if the necessary tpsA and tpsB genes were present.
Meningococcal knockout and complemented knockout strains.
N. meningitidis knockout strain H44/76 (B:15:P1.7,16; cc32 [21]) tpsB2::ery lacks a functional TPS2 due to replacement of the tpsB2 gene by an erythromycin resistance (ery) cassette. Replacement of tpsB2 resulted from allelic exchange through homologous recombination using plasmid pKO-tpsB2::ery. This plasmid was constructed by replacing the kanamycin resistance (kan) cassette in pKO-tpsB2 (8) with the ery cassette of plasmid pFLOB4300 (22) using the PstI restriction sites present. The replacement resulted in strain H44/76 tpsB2::ery. The knockout strain H44/76 tpsA3::kan lacks a functional TPS3 due to replacement of the tpsA3 gene by a kan cassette (8). The knockout strain H44/76 tpsA3::kan tpsB2::ery (H44/76 ΔtpsA3 tpsB2) lacks both a functional TPS2 and a functional TPS3. In the H44/76 tpsB2::ery and the H44/76 tpsA3::kan tpsB2::ery strains, tpsB2 was complemented by transforming the knockout strain with plasmid pEN-TpsB2. The tpsB2 ORF was obtained by PCR using primers TCCGTGTATTGAATGCCCATTGATGA and GAGATCTGAATTCCGTTGATTTGACTATGCCGTTTTA (1). The resulting amplicon was digested by the restriction enzymes BamHI and EcoRI and ligated into pET11a (Invitrogen) that had been digested with the same enzymes. The ORF was subsequently inserted into the neisserial expression vector pEN300 (23) using restriction enzymes NdeI and AatII. The resulting plasmid (pEN-TpsB2) carried tpsB2 under the control of an IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible lac promoter and was introduced into strain H44/76 and its derivatives by transformation (1, 23).
Western blot analysis.
All Western blotting procedures were carried out as described earlier (1). Blots were incubated with antisera (anti-TPS1, anti-TPS2, and anti-TPSB2).
Blood stimulation experiments.
N. meningitidis colonies were suspended in RPMI medium with 2% glucose and diluted to an optical density at 530 nm (OD530) of 0.05 in 40 ml RPMI medium. Bacteria were grown for 2.5 h to a mid-log growth phase, washed with 25 ml RPMI medium, and resuspended in 10 ml RPMI medium. Venous blood was drawn from seven healthy subjects, stored in sodium heparin tubes, and used directly. Meningococcal cultures with an OD530 of 0.01 (106 bacteria/ml) were added to blood in a 1:1 ratio in polypropylene tubes and grown for 4 h at 37°C in 5% CO2 with or without the presence of 1 mM IPTG. The number of CFU was determined in triplicate at time zero and 4 h. The interlukein-6 (IL-6) level was measured in duplicate by enzyme-linked immunosorbent assay (ELISA; Biosource-Invitrogen).
Statistics.
In the analysis of the association between the patient characteristics and the TPS system presence, we used the Mann-Whitney U test to identify differences between patient groups with respect to continuous variables. Dichotomous variables were compared by use of the χ2 test, and statistical tests were two-tailed, with a P value of <0.05 being regarded as significant.
RESULTS
Patient cohorts.
This study used meningococcal strains from two time periods. Strains from the first time period, October 1998 to April 2002, were from 258 episodes of community-acquired meningococcal meningitis in 258 patients. Detailed clinical and microbiological characteristics have been described previously (18, 24). Patient characteristics, their clinical course, and the causative bacterial strain isolated from the cerebrospinal fluid (CSF) were available for 254 (98.4%) of the 258 meningococcal meningitis episodes. The most prevalent clonal complexes were cc41/44 (41%; all strains but one were serogroup B), cc11 (24%; all strains were serogroup C), and cc32 (16%; all strains were serogroup B). All these clonal complexes are considered to be hyperinvasive because they are more frequently isolated from patients than from carriers (25). The case fatality rate was 7%, and 12% of patients had an unfavorable outcome.
In the second period analyzed, from 2006 to 2014, N. meningitidis was the causative organism in 150 of 1,412 episodes of community-acquired meningitis. The case fatality rate was 3%, and 13% of patients had an unfavorable outcome (19). In our analysis, we included 115 meningococcal isolates whose whole genomes were sequenced. The most prevalent clonal complexes were cc41/44 (32%), cc32 (23%), cc269 (9%), and cc213 (9%). In this cohort, the proportion of cc11 isolates dropped to 5%, due to a national serogroup C vaccination campaign in 2002.
tps genes in clinical isolates in the two patient cohorts.
Meningococcal serogroup data, sequence types, clonal complexes, and the tps gene/TPS system distribution in both cohorts are shown in Table S2 and Fig. S1 in the supplemental material. A summary of the TPS system distribution in all available cohorts is shown in Table S3 in the supplemental material. In the 1998 to 2002 cohort, 246 of 254 isolates (97%) harbored an intact TPS1 system. tpsA1 was absent in five isolates, and three isolates contained an insertion sequence (IS1301) in tpsB1 (all three isolates were sequence type 2704 [ST2704] from cc11). In 162 (64%) isolates, a complete TPS2 (tpsB2 and either tpsA2a or tpsA2b, or both) was present. In 52 isolates (20%), a functional TPS3 (tpsB2 and tpsA3) was present. Of the 87 strains that harbored TPS1 only, 71 (82%) were serogroup C strains, 59 (68%) belonged to hyperinvasive cc11, and 10 (11%) belonged to hyperinvasive cc8. Of note, none of the tested cc11 and cc8 isolates contained a TPS2 or TPS3 system. Of the 162 strains containing TPS system 2 and/or TPS system 3, 158 (98%) were serogroup B strains. In this subgroup, the most prevalent clonal complexes were hyperinvasive cc41/44 (n = 99), cc32 (n = 36), and cc269 (n = 16). The 52 isolates harboring all 3 systems belonged to hyperinvasive cc32 or cc269.
In the 2006 to 2014 cohort, 89 of 115 (77%) isolates harbored an intact TPS1 system (P < 0.0001 compared to the 1998 to 2002 cohort). A TPS2 system was present in 84 of 115 (73%) isolates, and a TPS3 system was present in 33 of 115 (29%) isolates. Only 8 of 115 (7%) isolates were serogroup C strains, and of these, 6 (all of which belonged to hyperinvasive cc11) did not contain any TPS system. Nonhyperinvasive clonal complexes that did not contain any TPS system were cc22 (n = 5), cc23 (n = 4), and cc174 (n = 3). These clonal complexes were absent in the first cohort. In 84/115 (73%) strains, either a TPS2 or a TPS3 system was present. Of the 31 isolates harboring all TPS systems, 30 belonged to hyperinvasive cc32 or cc269.
TPS systems and clinical outcome.
Patients infected with meningococci without a TPS system or harboring solely TPS1 presented with a lower GCS score (median, 12 [interquartile range {IQR}, 9 to 15] versus 14 [IQR, 11 to 15]; P = 0.006) and more often had focal neurological deficits upon presentation (31 of 122 [25%] versus 32 of 246 [13%] patients; P = 0.003) than patients infected with isolates that contained multiple TPS systems (Table 1). The proportion of patients with systemic complications (31 of 121 [26%] versus 34 of 245 [14%]; P = 0.006) and unfavorable outcomes (21 of 123 [17%] versus 23 of 246 [9.3%]; P = 0.031) was higher among the patients infected with meningococci without a TPS or harboring solely TPS1 than patients infected with isolates containing TPS system 2 and/or 3.
TABLE 1.
Associations between clinical characteristics of meningitis patients and meningococcal TPS system distribution
| Characteristic | Patients infected with isolates without an immunogenic TPS systema |
Patients infected with isolates with an immunogenic TPS systemb |
P value | ||
|---|---|---|---|---|---|
| Result | No. of patients | Result | No. of patients | ||
| Demographic characteristics | |||||
| Mean ± SD age (yr) | 38 ± 21 | 123 | 36 ± 18 | 246 | 0.63 |
| No. of patients with duration of symptoms of <24 h/total no. assessed (%) | 61/121 (50) | 115/236 (49) | 0.76 | ||
| Symptoms at presentation | |||||
| Median (25th–75th percentile) GCS score | 12 (9–15) | 121 | 14 (11–15) | 245 | 0.006 |
| No. of patients with rash/total no. assessed (%) | 71/123 (58) | 146/246 (59) | 0.77 | ||
| No. of patients with focal neurological deficitsc/total no. assessed (%) | 31/122 (25) | 32/246 (13) | 0.003 | ||
| Median (25th–75th percentile) diastolic blood pressure | 70 (60–84) | 120 | 70 (62–80) | 235 | 0.77 |
| Blood parameters | |||||
| No. of patients with positive blood culture/total no. assessed (%) | 75/116 (65) | 102/209 (49) | 0.006 | ||
| Median (25th–75th percentile) thrombocyte count (109/liter) | 166 (122–213) | 118 | 173 (139–225) | 232 | 0.14 |
| CSF parameters | |||||
| Median (25th–75th percentile) CSF leukocyte count (cells/mm3) | 4,998 (1,100–12,000) | 115 | 5,600 (1,742–13,325) | 233 | |
| Median (25th–75th percentile) CSF/blood glucose ratio | 0.081 (0.01–0.35) | 112 | 0.80 (0.01–0.31) | 219 | 0.84 |
| No. of patients with the following outcome parameters/total no. assessed (%): | |||||
| Any systemic complication | 31/121 (26) | 34/245 (14) | 0.006 | ||
| Unfavorable outcomed | 21/123 (17) | 23/246 (9.3) | 0.031 | ||
Isolates without TPS2 and/or TPS3. Data are for 123 patients, unless indicated otherwise.
Isolates with TPS2 and/or TPS3. Data are for 246 patients, unless indicated otherwise.
Defined as aphasia, monoparesis, hemiparesis. or cranial nerve palsies.
An unfavorable outcome was a Glasgow Outcome Scale score of 1 to 4.
In the 1998 to 2002 cohort, we previously described a polymorphism at position 184 in the meningococcal factor H binding protein (fHbp) to be associated with cc11 and an unfavorable outcome in this cohort (26). There is considerable overlap in the distribution between cc11, the factor fHbpD184 polymorphism, and TPS systems, raising the possibility that this polymorphism rather than the TPS system is important for the clinical outcome (Fig. 1). However, in analyses that excluded the patients who were infected with meningococci with fHbpD184, the difference in the proportion of meningococci with no TPS or solely TPS1 and meningococci with multiple TPS systems remained detectable between patients with a GCS score of <14 or a GCS score of ≥14: 38 of 60 (63%) versus 68 of 151 (45%) (P = 0.016). The difference in the proportion of patients with any systemic complication showed a similar trend: 14 of 60 (23.3%) versus 19 of 151 (12.6%) (P = 0.052).
FIG 1.
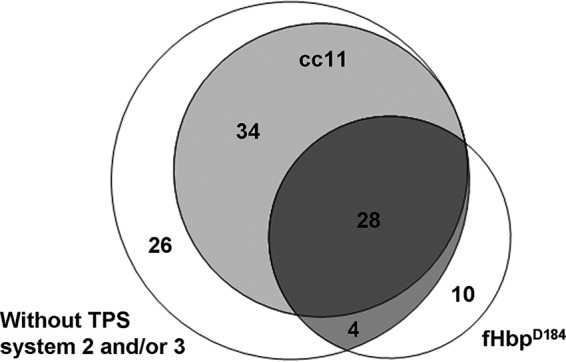
Venn diagram of meningococcal genetic factors associated with clinical outcome. The overlap of isolates from clonal complex 11, isolates with the fHbpD184 polymorphism, and isolates without immunogenic TPS systems is shown for our cohort of 254 patients with bacterial meningitis studied from 1998 to 2002 (total n = 102).
tps knockout strain and cytokine induction.
Next, we performed whole-blood stimulation experiments in which we compared the growth of meningococcal strain H44/76, which has all three TPS systems intact, and isogenic TPS system 2 and 3 knockout strains and the induction of IL-6 (one of the acute-phase mediators of the immune response to infection) by these strains (Fig. 2). We also included strains in which the knocked out gene was complemented by the original gene on a plasmid. The TpsA protein expression profile of all created strains was tested by Western blotting and was consistent with the genetic background (see Fig. S2 in the supplemental material). The growth of all tested bacterial strains in RPMI medium was similar (see Fig. S2 in the supplemental material). After 4 h, we observed no difference in the growth of the tested knockout mutants and the wild type or the complemented strains in whole blood from each of the seven donors. However, bacterial growth results were different between donors: in samples from three persons, all strains grew exponentially. In samples from three other persons, no growth was observed and the number of viable bacteria in the inoculum and after 4 h of incubation remained equal. In the sample from one person, all strains were killed and the IL-6 concentration in the blood of this person was therefore not measured. In the blood samples from the six remaining test subjects, the IL-6 concentrations induced after 4 h of growth showed no differences among the tested strains. There was no correlation between the number of CFU and IL-6.
FIG 2.
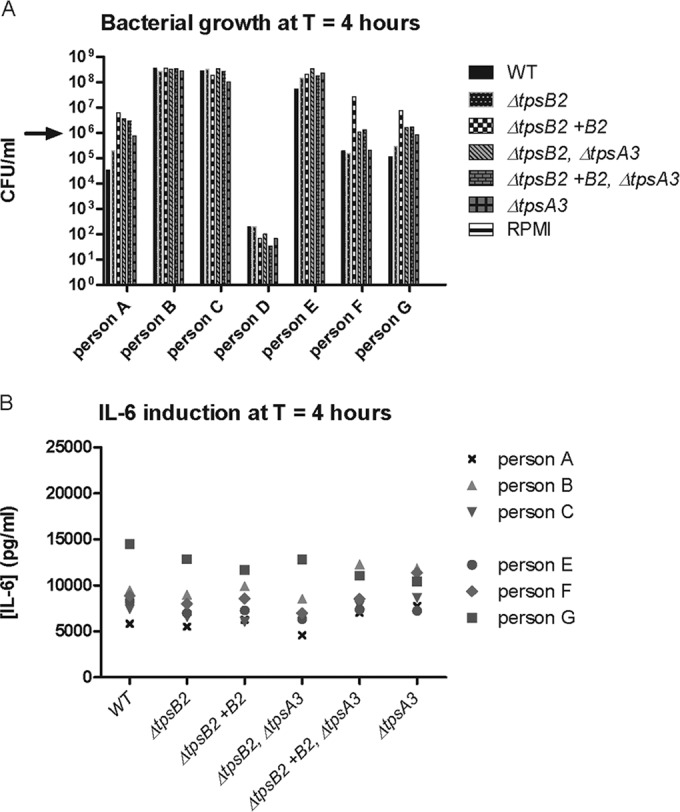
Meningococcal blood stimulation experiments. (A) Growth of the N. meningitidis H44/76 wild type (WT) and several TPS system 2 and 3 knockout and complemented knockout strains in human blood samples. Arrow, starting inoculum. Three different patterns of growth in blood from the different people were observed among the isolates. No differences in growth were observed among the meningococcal strains. Negative control samples without bacteria (RPMI medium only) showed no bacterial growth. (B) IL-6 induction in blood samples from 6 persons after 4 h of growth. IL-6 induction by wild-type, several TPS system 2 and 3 knockout, and complemented knockout strains was compared. We observed no difference in IL-6 induction. T, time.
tps genome signature.
TPS systems 1 and 2 are encoded within genomic islands that carry several traits that suggest the influence of recombination events that may influence strain-to-strain variation, e.g., the presence of tpsC cassettes and a high abundance of sequence repeats (8). Horizontally acquired genes can be detected by an aberrant dinucleotide composition (genome signature) compared to the rest of the genome (27). We characterized the genome signature of all tps genes by determining the dinucleotide relative abundance (δ*) in the genome of N. meningitidis strain MC58 (cc32) (28). The genome signatures of tpsA3 and all tpsB genes were similar to those of the rest of the genome, whereas the genome signatures of all tpsA genes from strains with TPS systems 1 and 2 (tpsA1a, tpsA1b, tpsA2a, tpsA2b) were highly dissimilar from those of the rest of the genome: over 96% of the genome fragments yielded a lower δ* score than tpsA1a, tpsA1b, tpsA2a, or tpsA2b (Table 2). This supports a role for recombination with heterologous DNA fragments to vary the tpsA sequences.
TABLE 2.
Genome signature of tps genes
| Gene | % genomic fragments with a lower δ*a |
|---|---|
| tpsB1 | 49.4 |
| tpsB1tr | 41.9 |
| tpsA1 | 96.0 |
| tpsA1b | 98.4 |
| tpsB2 | 64.5 |
| tpsA2a | 99.3 |
| tpsA2b | 97.9 |
| tpsA3 | 44.1 |
DISCUSSION
Our results show that the presence of functional meningococcal TPS2 and TPS3 is associated with a better clinical outcome in meningococcal meningitis. Patients infected with meningococci harboring no TPS or solely TPS1 were admitted to the hospital with more severe disease and had a higher risk of an unfavorable outcome than patients infected with meningococci harboring TPS2 and/or TPS3. This contrasts with findings that these systems are immunogenic in humans and thought to contribute to virulence, since they are more often found in hyperinvasive strains, as described earlier (8) and corroborated by the results presented here. Unfortunately, our first attempts to address the underlying biological mechanism in our blood stimulation experiments failed, since the growth characteristics and IL-6 responses were similar between isogenic strains that either carried or lacked the three TPS systems. It therefore remains to be shown why TPS systems 2 and 3 are more often found in hypervirulent clonal complexes but lead to a more favorable outcome.
Thus far, functional analyses of meningococcal TPS systems have focused on the TpsA proteins (also known as HrpA proteins) of strains with TPS system 1 (7, 9, 10, 12), while those of strains with TPS systems 2 and 3 have not been addressed. The overall homology between secreted TpsAs of different systems is primarily based upon the TPS domain, important for secretion, and not the functional parts downstream of that N-terminal domain (6, 8, 29). Nevertheless, the additional systems could have a comparable function. A function as an adhesin seems less likely, since adherence to cultured epithelial cell lines mediated by TpsA1s required strains that lacked both a capsule and lipopolysaccharide molecules of normal length (9). Furthermore, biofilm formation appeared to be independent of adhesive functions in TpsA1s (10). Both functions would also improve the virulence of a bacterial strain. Prolonged intracellular survival and the observation that meningococci that lack a TpsA1 are more often found to be associated with the endocytic pathway (9), as well as its fratricide functions (12), could result from TpsA toxin activity, which resides in the C-terminal region of the protein (11). Strikingly, one of the two system 2 TpsAs includes a VENN motif also found in TpsA1 and is instrumental in toxic activity (11, 30). Of note, toxins with a VENN motif are often endonucleases targeting DNA or RNA. Furthermore, both genetic islands that encode systems 2 and 3 include a gene encoding an acyltransferase of a type that is often needed to activate other bacterial toxins (6) (results not shown).
In our collection of clinical CSF isolates, TPS1 was almost uniformly present in the first cohort (97%), whereas the rate at which it was present in the second cohort decreased to 77%, due to the higher prevalence (17% versus 4%) of strains without TPS systems (see Fig. S1C in the supplemental material). The high rate of TPS1 in our first cohort is in contrast to the frequency of TPS1 (82%) that was reported in a previous study of meningococcal disease isolates covering strains recovered from 2001 to 2005 (8). This might be explained by the higher proportion of serogroup Y and W strains in that collection (7 of 13 strains were from one of these serogroups, whereas 2 of 8 strains from the 1998 to 2002 cohort were from one of these serogroups).
In the same study, the prevalence of TPS systems capable of inducing an immune response (TPS system 2 or 3) was 47%, which is markedly lower than the 64% found in the 1998 to 2002 cohort and the 73% found in the 2006 to 2014 cohort. In our two cohorts, 92% and 69% of the CSF isolates were from hypervirulent clonal complexes, respectively. Remarkably, we could not detect TPS system 2 and/or 3 in hypervirulent cc11 and cc8 strains, whereas the prevalence of these systems in the other hypervirulent clonal complex strains was 81 to 100%. The combined data from the available studies show that cc8 isolates (n = 17) never contained TPS2 and/or 3, whereas these systems rarely appeared in cc11 isolates (2 isolates out of 81 total isolates tested were positive) (8, 9). Overall, the TPS system distribution is highly related to specific clonal complexes. In the first cohort, of 235 isolates belonging to the 5 most prevalent clonal complexes, only 13 (6%) had an aberrant distribution of TPS systems. In the second cohort, this proportion was 16%.
The genes encoding TPS systems are located on genomic islands. This is in line with the observed genomic dissimilarity of the tpsA genes of TPS systems 1 and 2, suggesting horizontal gene transfer as the mechanism of TPS system acquisition. In contrast, the genome signature of the two tpsB genes is not aberrant from that of the genome. System 3 appears to be encoded on a genetic island that is always found to be inserted in the same chromosomal location and flanked by a repeat of 52 nucleotides that is present in a single copy in strains that do not include the systems. However, the genome signature of this genomic island does not differ from that of the genome, suggesting an early acquisition and a sufficiently long period of amelioration. Alternatively, system 3 is part of the genuine meningococcal genome and may be lost to increase virulence. The tpsA genes vary in sequence at their 3′ end, and the genomic islands of both TPS system 1 and 2 strains contain tpsC cassettes that appear to be silent variants of the 3′ end of the functional tpsA genes, suggesting the existence of recombination events that appear to occur at a low frequency (6, 12). The position of the islands for system 1 and 2 strains, however, appears, again, to be conserved between strains, suggesting that the observed association with clinical outcome is not caused by flanking genes (8, 31). Furthermore, these genetic islands appear to be dedicated to the TPS systems and do not encode additional virulence factors.
All tested meningococcal wild-type and knockout strains induced a similar IL-6 response in our blood stimulation experiments. Remarkably, the size of the response did not correlate with bacterial growth, even though at 4 h there was a 1,000-fold difference in the number of CFU between samples from different donors. Therefore, it is likely that in our experiments the IL-6 immune response is unrelated to the presence of TpsA but is already maximally stimulated by other meningococcal factors. In future research, the levels of a larger panel of cytokines/chemokines in blood samples from more persons should be tested to fully investigate whether TpsA has a role in stimulating proinflammatory cytokines. However, the functions of TPS systems 2 and 3 appeared to influence the host defenses and their signaling routes in such a way that the effects of the induction of proinflammatory cytokines were reduced, as suggested by the less severe disease outcome. Previously, TPS system components elicited an antibody response in 4 out of 17 serum samples from patients who were recovering from a meningococcal infection (8). Only a small portion of the population is probably capable of eliciting an antibody response against TPS system components or the responses are masked by immunodominant antigens that are more strongly expressed.
An association between cc11 and outcome was observed previously, although at that time the reason remained unknown (18). Due to serogroup C vaccination in the Netherlands in 2002, the incidence of cc11 meningitis has decreased from 24 to 5%. More recently, the meningococcal factor H binding protein D184 polymorphism was associated with an unfavorable outcome (26). The considerable overlap of these traits with the TPS system distribution might suggest that the observed association with outcome is a genetic linkage effect. However, when we reanalyzed our data by excluding the data for patients who were infected with meningococci with fHbpD184, the difference in the proportion of patients with a GCS score of <14 remained between patients infected with meningococci without TPS systems or harboring solely TPS1 and those infected with meningococci with multiple TPS systems.
Our study provides insights into the presence of the different types of TPS systems and their effects on disease outcomes and reveals a high frequency of certain TPS system types in strains belonging to hyperinvasive clonal complexes, but also a less severe disease outcome when these TPS system types are present. This is relevant to the discussion of the hyperinvasiveness of some clonal complexes. Because of the natural variation in the distribution of these systems among strains, further epidemiological surveys as well as analysis of the biological role of the systems remain warranted.
Supplementary Material
ACKNOWLEDGMENTS
D. van de Beek and A. van der Ende participate in the Seventh Framework Programme of the European Commission, proposal/contract no. 279185 (EUCLIDS—the genetic basis of meningococcal and other life-threatening bacterial infections of childhood), and A. van der Ende participated in the Sixth Framework Programme of the European Commission, proposal/contract no. 512061 (Network of Excellence European Virtual Institute for Functional Genomics of Bacterial Pathogens). This publication made use of the Neisseria Multilocus Sequence Typing website (http://pubmlst.org/neisseria/) developed by Keith Jolley and sited at the University of Oxford. Development of this site has been funded by the Wellcome Trust and the European Union.
This study has been funded by grants from the Netherlands Organization for Health Research and Development (ZonMw; NWO-Veni grant 2006 [916.76.023] and NWO-Vidi grant 2010 [016.116.358] to D. van de Beek), Academic Medical Center (AMC) Fellowship 2008 to D. van de Beek, and AMC Ph.D. Scholarship 2008 to J. R. Piet. The Netherlands Reference Laboratory for Bacterial Meningitis is supported by the National Institute of Public Health and Environmental Protection, Bilthoven, the Netherlands.
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00160-16.
REFERENCES
- 1.ur Rahman S, van Ulsen P. 2013. System specificity of the TpsB transporters of coexpressed two-partner secretion systems of Neisseria meningitidis. J Bacteriol 195:788–797. doi: 10.1128/JB.01355-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevalier N, Moser M, Koch HG, Schimz KL, Willery E, Locht C, Jacob-Dubuisson F, Muller M. 2004. Membrane targeting of a bacterial virulence factor harbouring an extended signal peptide. J Mol Microbiol Biotechnol 8:7–18. [DOI] [PubMed] [Google Scholar]
- 3.Jacob-Dubuisson F, Guerin J, Baelen S, Clantin B. 2013. Two-partner secretion: as simple as it sounds? Res Microbiol 164:583–595. doi: 10.1016/j.resmic.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 4.van Ulsen P, Rahman S, Jong WS, Daleke-Schermerhorn MH, Luirink J. 2014. Type V secretion: from biogenesis to biotechnology. Biochim Biophys Acta 1843:1592–1611. doi: 10.1016/j.bbamcr.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Stephens DS, Greenwood B, Brandtzaeg P. 2007. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 369:2196–2210. doi: 10.1016/S0140-6736(07)61016-2. [DOI] [PubMed] [Google Scholar]
- 6.van Ulsen P, Tommassen J. 2006. Protein secretion and secreted proteins in pathogenic Neisseriaceae. FEMS Microbiol Rev 30:292–319. doi: 10.1111/j.1574-6976.2006.00013.x. [DOI] [PubMed] [Google Scholar]
- 7.Schmitt C, Turner D, Boesl M, Abele M, Frosch M, Kurzai O. 2007. A functional two-partner secretion system contributes to adhesion of Neisseria meningitidis to epithelial cells. J Bacteriol 189:7968–7976. doi: 10.1128/JB.00851-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Ulsen P, Rutten L, Feller M, Tommassen J, van der Ende A. 2008. Two-partner secretion systems of Neisseria meningitidis associated with invasive clonal complexes. Infect Immun 76:4649–4658. doi: 10.1128/IAI.00393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tala A, Progida C, De Stefano M, Cogli L, Spinosa MR, Bucci C, Alifano P. 2008. The HrpB-HrpA two-partner secretion system is essential for intracellular survival of Neisseria meningitidis. Cell Microbiol 10:2461–2482. doi: 10.1111/j.1462-5822.2008.01222.x. [DOI] [PubMed] [Google Scholar]
- 10.Neil RB, Apicella MA. 2009. Role of HrpA in biofilm formation of Neisseria meningitidis and regulation of the hrpBAS transcripts. Infect Immun 77:2285–2293. doi: 10.1128/IAI.01502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poole SJ, Diner EJ, Aoki SK, Braaten BA, t'Kint de Roodenbeke C, Low DA, Hayes CS. 2011. Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet 7:e1002217. doi: 10.1371/journal.pgen.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arenas J, Schipper K, van Ulsen P, van der Ende A, Tommassen J. 2013. Domain exchange at the 3′ end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC Genomics 14:622. doi: 10.1186/1471-2164-14-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bentley SD, Vernikos GS, Snyder LA, Churcher C, Arrowsmith C, Chillingworth T, Cronin A, Davis PH, Holroyd NE, Jagels K, Maddison M, Moule S, Rabbinowitsch E, Sharp S, Unwin L, Whitehead S, Quail MA, Achtman M, Barrell B, Saunders NJ, Parkhill J. 2007. Meningococcal genetic variation mechanisms viewed through comparative analysis of serogroup C strain FAM18. PLoS Genet 3:e23. doi: 10.1371/journal.pgen.0030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piet JR, van der Ende A, van de Beek D. 2009. Abstr 47th Annu Meet Infect Dis Soc Am, abstr 1269. [Google Scholar]
- 15.Piet JR, van Ulsen P, van der Ende A, van de Beek D. 2013. Abstr 53rd Intersci Conf Antimicrob Agents Chemother, abstr 1141. [Google Scholar]
- 16.Jennett B, Teasdale G. 1981. Management of head injuries, 2nd ed F. A. Davis, Philadelphia, PA. [Google Scholar]
- 17.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heckenberg SG, de Gans J, Brouwer MC, Weisfelt M, Piet JR, Spanjaard L, van der Ende A, van de Beek D. 2008. Clinical features, outcome, and meningococcal genotype in 258 adults with meningococcal meningitis: a prospective cohort study. Medicine (Baltimore) 87:185–192. doi: 10.1097/MD.0b013e318180a6b4. [DOI] [PubMed] [Google Scholar]
- 19.Bijlsma MW, Brouwer MC, Kasanmoentalib ES, Kloek AT, Lucas MJ, Tanck MW, van der Ende A, van de Beek D. 2015. Community-acquired bacterial meningitis in adults in the Netherlands, 2006-14: a prospective cohort study. Lancet Infect Dis 16:339–347. doi: 10.1016/S1473-3099(15)00430-2. [DOI] [PubMed] [Google Scholar]
- 20.Chewapreecha C, Harris SR, Croucher NJ, Turner C, Marttinen P, Cheng L, Pessia A, Aanensen DM, Mather AE, Page AJ, Salter SJ, Harris D, Nosten F, Goldblatt D, Corander J, Parkhill J, Turner P, Bentley SD. 2014. Dense genomic sampling identifies highways of pneumococcal recombination. Nat Genet 46:305–309. doi: 10.1038/ng.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holten E. 1979. Serotypes of Neisseria meningitidis isolated from patients in Norway during the first six months of 1978. J Clin Microbiol 9:186–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston DM, Cannon JG. 1999. Construction of mutant strains of Neisseria gonorrhoeae lacking new antibiotic resistance markers using a two gene cassette with positive and negative selection. Gene 236:179–184. doi: 10.1016/S0378-1119(99)00238-3. [DOI] [PubMed] [Google Scholar]
- 23.van Ulsen P, van Alphen L, ten Hove J, Fransen F, van der Ley P, Tommassen J. 2003. A neisserial autotransporter NalP modulating the processing of other autotransporters. Mol Microbiol 50:1017–1030. doi: 10.1046/j.1365-2958.2003.03773.x. [DOI] [PubMed] [Google Scholar]
- 24.van de Beek D, de Gans J, Spanjaard L, Weisfelt M, Reitsma JB, Vermeulen M. 2004. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med 351:1849–1859. doi: 10.1056/NEJMoa040845. [DOI] [PubMed] [Google Scholar]
- 25.Caugant DA. 1998. Population genetics and molecular epidemiology of Neisseria meningitidis. APMIS 106:505–525. doi: 10.1111/j.1699-0463.1998.tb01379.x. [DOI] [PubMed] [Google Scholar]
- 26.Piet JR, Brouwer MC, Exley R, van der Veen S, van de Beek D, van der Ende A. 2012. Meningococcal factor H binding protein fHbpd184 polymorphism influences clinical course of meningococcal meningitis. PLoS One 7:e47973. doi: 10.1371/journal.pone.0047973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Passel MW, Bart A, Thygesen HH, Luyf AC, van Kampen AH, van der Ende A. 2005. An acquisition account of genomic islands based on genome signature comparisons. BMC Genomics 6:163. doi: 10.1186/1471-2164-6-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Passel MW, Luyf AC, van Kampen AH, Bart A, van der Ende A. 2005. Deltarho-web, an online tool to assess composition similarity of individual nucleic acid sequences. Bioinformatics 21:3053–3055. doi: 10.1093/bioinformatics/bti460. [DOI] [PubMed] [Google Scholar]
- 29.ur Rahman S, Arenas J, Ozturk H, Dekker N, van Ulsen P. 2014. The polypeptide transport-associated (POTRA) domains of TpsB transporters determine the system specificity of two-partner secretion systems. J Biol Chem 289:19799–19809. doi: 10.1074/jbc.M113.544627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, Low DA. 2010. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joseph B, Schwarz RF, Linke B, Blom J, Becker A, Claus H, Goesmann A, Frosch M, Muller T, Vogel U, Schoen C. 2011. Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome. PLoS One 6:e18441. doi: 10.1371/journal.pone.0018441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.