

Abstract
We used a murine model of acute, posttraumatic osteomyelitis to evaluate the virulence of two divergent Staphylococcus aureus clinical isolates (the USA300 strain LAC and the USA200 strain UAMS-1) and their isogenic sarA mutants. The results confirmed that both strains caused comparable degrees of osteolysis and reactive new bone formation in the acute phase of osteomyelitis. Conditioned medium (CM) from stationary-phase cultures of both strains was cytotoxic to cells of established cell lines (MC3TC-E1 and RAW 264.7 cells), primary murine calvarial osteoblasts, and bone marrow-derived osteoclasts. Both the cytotoxicity of CM and the reactive changes in bone were significantly reduced in the isogenic sarA mutants. These results confirm that sarA is required for the production and/or accumulation of extracellular virulence factors that limit osteoblast and osteoclast viability and that thereby promote bone destruction and reactive bone formation during the acute phase of S. aureus osteomyelitis. Proteomic analysis confirmed the reduced accumulation of multiple extracellular proteins in the LAC and UAMS-1 sarA mutants. Included among these were the alpha class of phenol-soluble modulins (PSMs), which were previously implicated as important determinants of osteoblast cytotoxicity and bone destruction and repair processes in osteomyelitis. Mutation of the corresponding operon reduced the cytotoxicity of CM from both UAMS-1 and LAC cultures for osteoblasts and osteoclasts. It also significantly reduced both reactive bone formation and cortical bone destruction by CM from LAC cultures. However, this was not true for CM from cultures of a UAMS-1 psmα mutant, thereby suggesting the involvement of additional virulence factors in such strains that remain to be identified.
INTRODUCTION
Staphylococcus aureus is a highly versatile pathogen capable of causing a remarkable array of human infections. One of the most devastating of these is osteomyelitis, which is extremely difficult to eradicate without extensive and often repetitive surgical debridement (1). Indeed, it has been suggested that, as with cancer, “remission” is a more appropriate term than “cure” in the context of osteomyelitis (2). Several factors contribute to this therapeutic recalcitrance, including the inability to diagnose the infection before it has progressed to a chronic stage in which the local vasculature is compromised, the formation of a bacterial biofilm that limits the efficacy of both conventional antibiotics and host defenses, the emergence of phenotypic variants within the biofilm (persister cells and small-colony variants) that exhibit metabolic traits that limit their antibiotic susceptibility, and the ability of the pathogens involved, including S. aureus, to invade and replicate within host cells, including osteoblasts (3–9). Collectively, these factors dictate that the clinical problem of osteomyelitis extends far beyond acquired resistance and the increasingly limited availability of effective antibiotics.
Our laboratories have placed a major emphasis on overcoming this problem by exploring alternative means for early diagnosis (3, 10), developing improved methods for localized antibiotic delivery for the prevention and treatment of infection (11–14), and identifying the bacterial factors that contribute to the prominence of S. aureus as an orthopedic pathogen. With respect to the last area of exploration, our studies have led us to place a primary emphasis on the staphylococcal accessory regulator (sarA), mutation of which limits biofilm formation to a greater degree than mutation of any other regulatory locus that we have examined (11, 15). The negative impact of mutated sarA on biofilm formation is also apparent in all S. aureus strains that we have examined, other than those with recognized regulatory defects (16, 17). Moreover, even in those cases in which a mutation enhanced biofilm formation, concomitant mutation of sarA reversed this effect (12, 15–17). We also confirmed that the limited ability of sarA mutants to form a biofilm can be correlated with increased susceptibility to diverse functional classes of antibiotics in vivo (18, 19). Additionally, mutation of sarA limits the ability of S. aureus to persist in the bloodstream and cause secondary infections, including hematogenous osteomyelitis (20, 21).
Taken together, these results suggest that sarA is a viable and perhaps preferred regulatory target in the context of biofilm-associated infections, including osteomyelitis. However, this conclusion must be interpreted with caution. For instance, under in vitro conditions, the relative impact of sarA versus that of the saePQRS (saeRS) regulatory locus on biofilm formation was recently shown to be dependent on the medium used to carry out the biofilm assay (22). Moreover, mutation of saeRS in the USA300 strain LAC was shown to limit virulence in a murine model of posttraumatic osteomyelitis owing to the increased production of the extracellular protease aureolysin, which results in the decreased accumulation of phenol-soluble modulins (PSMs) that would otherwise promote cytotoxicity for osteoblasts and bone destruction (23). A recent report also demonstrated that, under the hypoxic conditions encountered in bone, particularly as the infection progresses to a point that compromises the local blood supply, the srrAB regulatory locus plays a key role in S. aureus survival (24). Such results emphasize the complexity of the disease process in osteomyelitis and the fact that biofilm formation per se is not the only relevant consideration.
In this respect, it is important to note that the impact of mutating sarA has not been evaluated in the context of bone infection. It has been demonstrated that, at least under in vitro conditions, mutation of sarA results in a much greater increase in protease production than mutation of saeRS (12, 17) and that this can be correlated with the reduced accumulation of multiple virulence factors, including PSMs (20). Thus, it would be anticipated that mutation of sarA would also have a significant impact in this clinical context, but this has not been experimentally determined. Additionally, studies examining the role of different regulatory loci in a newly developed murine model of posttraumatic osteomyelitis have been limited to date to the USA300 strain LAC, which produces PSMs at high levels by comparison to many other strains of S. aureus (25–27). In this report, we address these issues by using the same murine model to assess the relative virulence of two genetically and phenotypically divergent strains of S. aureus and their isogenic sarA and psm mutants.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The S. aureus strains utilized in this study included a plasmid-cured, erythromycin-sensitive derivative of methicillin-resistant S. aureus (MRSA) USA300 strain LAC (28), USA200 methicillin-sensitive S. aureus osteomyelitis isolate UAMS-1 (29), and derivatives of each carrying mutations in sarA or the operon encoding the alpha class of PSMs (αPSMs). Mutants were generated by phage ϕ11-mediated transduction from mutants already on hand (19, 20, 23). Mutations in sarA and the psmα operon were genetically complemented using pSARA and pTXΔα as previously described (16, 27). Strains were maintained at −80°C in tryptic soy broth (TSB) containing 25% (vol/vol) glycerol. For analysis, strains were cultured from cold storage by plating on tryptic soy agar (TSA) with selection with the appropriate antibiotic. The following antibiotics were used at the indicated concentrations: chloramphenicol (Cm), 10 μg ml−1; erythromycin (Erm), 10 μg ml−1; kanamycin (Kan), 50 μg ml−1; neomycin (Neo), 50 μg/ml; and tetracycline (Tet), 5 μg ml−1.
Preparation of conditioned medium.
Stationary-phase cultures were standardized to an optical density at 560 nm of 8.0. Cells were harvested by centrifugation, and the supernatants were filter sterilized. The culture medium was combined 1:1 with the appropriate cell culture medium containing 10% fetal bovine serum (FBS) and added to cell monolayers for cytotoxicity assays.
Cultivation of primary murine calvarial osteoblasts.
Murine primary calvarial osteoblasts were obtained from 3- to 5-day-old C57BL/6 mouse pups according to standard procedures (30), modified as follows: whole calvariae were dissected (the periosteum and endosteum were scraped off with a scalpel) and sequentially digested for 20 min at 37°C in alpha minimal essential medium (alpha-MEM) containing 0.1 mg/ml collagenase P (Roche), 0.04% trypsin-EDTA, and penicillin-streptomycin (166 U/ml and 166 μg/ml, respectively). The first 2 fractions of cells were discarded. Calvariae were further diced with sterile surgical scissors and digested in 1 ml of alpha-MEM with a double amount of collagenase and trypsin-EDTA for 1 h at 37°C with vigorous shaking every 15 to 20 min. Then, 3.75 ml of alpha-MEM containing 15% FBS and penicillin-streptomycin was added. After 24 h, the osteoblasts were washed with sterile phosphate-buffered saline (PBS) and expanded alpha-MEM containing 10% FBS, 2 mM glutamine, and penicillin and streptomycin (100 μg/ml and 100 μg/ml, respectively) for 2 to 4 days before passaging. Only early-passage osteoblasts grown in culture medium supplemented with 100 μg/ml of ascorbic acid were used for cytotoxicity assays.
Cytotoxicity assay.
Cytotoxicity assays with primary osteoblasts and established cell lines were done using the methods described above. MC3T3-E1 and RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC) and propagated according to ATCC recommendations. Cells were grown at 37°C in 5% CO2 with the replacement of medium every 2 or 3 days. For cytotoxicity assays, cells were seeded into black clear-bottom 96-well tissue culture-grade plates at a density of 10,000 cells per well for MC3T3-E1 cells, 50,000 cells per well for RAW 264.7 cells, or 10,000 cells per well for calvarial osteoblasts. After 24 h, the growth medium was removed and replaced with medium containing a 1:1 ratio of cell culture complete growth medium and S. aureus conditioned medium. The monolayers were incubated for an additional 24 h prior to removal of the medium and assessment of cell viability using calcein-AM to stain live cells (Thermo Fisher Scientific) according to the manufacturer's specifications. An Omega FLUOstar microplate reader (BMG Labtech) was used to determine the fluorescent intensity at 517 nm. The results of the microtiter plate assays were confirmed through fluorescence microscopy.
Cultivation and TRAP staining of primary osteoclasts.
Whole bone marrow was extracted from the tibia and femurs of one or two 8- to 10-week-old mice. Red blood cells were lysed in buffer (150 mM NH4Cl, 10 mM KNCO3, 0.1 mM EDTA, pH 7.4) for 5 min at room temperature. Bone marrow cells (5 × 106) were plated in a 100-mm petri dish and cultured in alpha-10 medium (alpha-MEM, 10% heat-inactivated FBS, and PSG [100 U/liter penicillin, 0.1 mg/liter streptomycin, and 2 mM l-glutamine]) containing 1/10 volume of conditioned medium (CM) supernatant from CMG 14-12 cells containing recombinant macrophage colony-stimulating factor (M-CSF) at 1 μg/ml for 4 to 5 days. Preosteoclasts and osteoclasts were generated by culturing bone marrow macrophages (BMMs) at a density of 160 BMMs/mm2 in 1/100 vol of CMG 14-12 culture supernatant and 100 ng/ml of recombinant RANKL. To determine cell viability, tartrate-resistant acid phosphatase (TRAP) staining was used to count the viable cells. BMMs were cultured on a 48-well tissue culture plate in alpha-10 medium with M-CSF and RANKL for 4 to 5 days. After medium replacement, the cells were treated with S. aureus culture supernatants diluted 1:1 in complete growth medium. The cells were then fixed with 4% paraformaldehyde–PBS and TRAP stained with NaK tartrate and naphthol AS-BI phosphoric acid (Sigma-Aldrich).
Murine model of acute posttraumatic osteomyelitis.
The murine model of acute posttraumatic osteomyelitis model was performed as previously described (23). Briefly, surgery was performed on the right hind limb of 8- to 10-week-old female C57BL/6 mice. Prior to surgery, the mice received 0.1 mg/kg of body weight buprenorphine via subcutaneous injection. Anesthesia was then maintained using isoflurane. The femur was exposed by blunt dissection, and a 1-mm unicortical bone defect was created at the lateral midshaft of the femur with a 21-gauge Precision Glide needle (Becton Dickinson). A bacterial inoculum of 1 × 105 CFU in 2 μl was delivered into the intramedullary canal. The muscle fasciae and skin were then closed with sutures, and the mice were allowed to recover from anesthesia. Infection was allowed to proceed for 14 days, at which time the mice were euthanized and the right femur was removed and subjected to micro-computed tomography (micro-CT) analysis. All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences and were performed according to NIH guidelines, the Animal Welfare Act, and U.S. federal law.
Micro-computed tomography.
The analysis of cortical bone destruction and new bone formation was performed using micro-CT imaging with a Skyscan 1174 micro-CT (Bruker), and scans were analyzed using the manufacturer's analytical software. Briefly, axial images of each femur were acquired at a resolution of 6.7 μm at 50 kV and 800 μA through a 0.25-mm aluminum filter. Bones were visualized using a scout scan and then scanned in three sections as an oversize scan to image the entire femoral length. The volume of cortical bone was isolated in a semiautomated process per the manufacturer's instruction. Briefly, cortical bone was isolated from soft tissue and the background by global thresholding (low threshold, 89; high threshold, 255). The processes of opening, closing, dilation, erosion, and despeckling were configured using the bones from sham-treated controls to separate the new bone from the existing cortical bone, and a task list was created to apply the same process and values to all bones in the data set. After processing of the bones using the task list, the volume of interest (VOI) was corrected by drawing inclusive or exclusive contours on the periosteal surface. Cortical bone destruction analysis consisted of 600 slices centered on the initial surgical bone defect. Destruction was determined by subtraction of the volume of infected bones from the average bone volume from sham-treated controls. Reactive new bone formation was assessed by first isolating the region of interest (ROI) that contained only the original cortical bone (as described above). After cortical bone isolation the new bone volume was determined by subtraction of the original bone volume from the total bone volume. All calculations were performed on the basis of direct voxel counts.
Proteomic analysis.
Assessment of the secreted proteome of both S. aureus parent strains and their isogenic sarA mutants was performed in triplicate as previously described (20). Briefly, the lanes of SDS-polyacrylamide gels were divided into 20 slices and subjected to in-gel trypsin digestion. The gel slices were destained in 50% methanol, 100 mM ammonium bicarbonate, followed by reduction in 10 mM Tris(2-carboxyethyl)phosphine and alkylation in 50 mM iodoacetamide. The gel slices were then dehydrated in acetonitrile, followed by addition of 100 ng of sequencing-grade porcine trypsin (Promega, Madison, WI) in 100 mM ammonium bicarbonate and incubation at 37°C for 12 to 16 h. The peptide products were then acidified in 0.1% formic acid (Fluka, Milwaukee, WI). Tryptic peptides were analyzed by high-resolution tandem mass spectrometry (MS/MS) with a Thermo LTQ Orbitrap Velos mass spectrometer coupled to a Waters nanoAcquity liquid chromatography (LC) system. The proteins were identified from the MS/MS spectra by searching the UniprotKB USA300 (LAC) or MRSA252 (UAMS-1) database for the organism Staphylococcus aureus (2,607 entries) using the Mascot search engine (Matrix Science, Boston, MA).
Statistical analysis.
The results of both in vitro and in vivo experiments were tested for statistical significance using the Student t test. Comparisons were made between the two parent strains or between each parent strain and its appropriate isogenic mutant. P values of ≤0.05 were considered statistically significant.
RESULTS AND DISCUSSION
A primary focus of our laboratories has been on developing alternative strategies that can be used to overcome the therapeutic recalcitrance of orthopedic infections, including osteomyelitis. Despite the current prominence of hypervirulent isolates of the USA300 clonal lineage (25), it is imperative that the genetic and phenotypic diversity of different S. aureus strains be taken into account in this regard. Based on this, we chose to focus on the USA300 methicillin-resistant strain LAC and the USA200 methicillin-sensitive isolate UAMS-1, which have been shown to be distinct with respect to both gene content and overall transcriptional patterns by comparison to each other (29, 31). Of note is the fact that LAC and many other USA300 isolates express the accessory gene regulator (agr) at higher levels than strains like UAMS-1 and, consequently, produce extracellular toxins, including phenol-soluble modulins (PSMs), at higher levels (25, 27). At the same time, UAMS-1 (ATCC 49230) has a proven clinical provenance in the specific context of osteomyelitis, having been isolated directly from the bone of a patient during surgical debridement (32).
Thus, we used equivalent numbers (105 CFU) of LAC, UAMS-1, and their isogenic sarA mutants to infect mice via direct inoculation into the medullary canal via a unicortical defect (23). Femurs were harvested at 14 days postinfection and subjected to micro-CT analysis to assess cortical bone destruction and reactive new bone (callus) formation. Quantitative analysis was based on reconstructive evaluation of a series of images spanning from the prominence of the lessor trochanter to the distal femoral growth plate. This analysis confirmed that infection with either strain caused osteolysis at and around the site of inoculation and reactive new bone (callus) formation both proximally and distally to this site (Fig. 1). The prevalence of both of these phenotypes was elevated in mice infected with LAC by comparison to those infected with UAMS-1, although these differences were not statistically significant (Fig. 2).
FIG 1.

Bone destruction and reactive bone formation in osteomyelitis as a function of sarA. C57BL/6 mice (n = 5) were infected with LAC, UAMS-1 (U1), or their isogenic sarA mutants (ΔsarA). Femurs were harvested at 14 days after inoculation and subjected to micro-CT imaging analysis. Anteroposterior views of infected femurs are shown for comparison. Sham, results for mice subjected to the surgical procedure and injected with sterile PBS.
FIG 2.
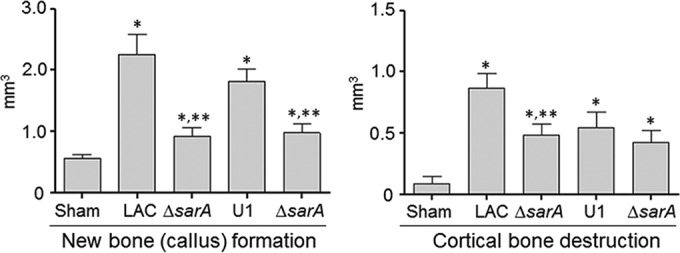
Quantitative analysis of micro-CT imaging. Images were analyzed for reactive new bone (callus) formation and cortical bone destruction in mice infected with LAC, UAMS-1 (U1), or their isogenic sarA mutants (ΔsarA). Sham, results of the same analysis with mice subjected to the surgical procedure and injected with sterile PBS. *, statistical significance compared to the results of sham treatment; **, statistical significance compared to the results for the isogenic parent strain.
In LAC, mutation of sarA limited both osteolysis and reactive new bone formation to a significant degree by comparison to those in the isogenic parent strain (Fig. 2). In UAMS-1, the impact of mutating sarA was statistically significant only in the context of reactive bone formation, with cortical bone destruction being reduced, but not to a significant degree. However, these results must be interpreted with caution, in that the surgical procedure itself involves the destruction of cortical bone to gain access to the intramedullary canal, thus complicating the analysis by comparison to that involving new bone formation.
Nevertheless, these results suggest that the virulence factor(s) produced by S. aureus that contributes to bone remodeling in osteomyelitis is likely produced in larger amounts by LAC than UAMS-1 and that mutation of sarA limits the production and/or accumulation of these virulence factors in both strains. Thus, while mutation of sarA has been shown to limit biofilm formation both in vitro and in vivo to a degree that can be correlated with increased antibiotic susceptibility (15, 18, 19, 33) and to limit virulence in a murine model of bacteremia that can be correlated with a reduced capacity to cause hematogenous osteomyelitis (20, 21), this is the first demonstration that it also limits virulence in a relevant model of posttraumatic bone infection and, perhaps more importantly, that it does so in diverse clinical isolates.
Bone is a highly dynamic physiological environment in which constant remodeling occurs in response to biomechanical stresses and hormonal influences (34, 35). This remodeling process is mediated by osteoblasts and osteoclasts, with the first being responsible for new bone formation (ossification) and the second being responsible for bone resorption prior to osteoblast-mediated ossification. Osteocytes are terminally differentiated osteoblasts that become embedded within lacunae in the mineralized bone matrix; they extend long cytoplasmic processes through apertures of the lacunae that form a dense canalicular network inside the bone. They are the most abundant cell type in the adult skeleton and form an interconnected network that can coordinate the activity of osteoblasts and osteoclasts to facilitate bone repair and ultimately maintain its structural integrity (34, 35). Thus, disruption in the balance of osteoblast versus osteoclast function has the potential to compromise this integrity. For instance, bone destruction could result from increased osteoclast function or decreased osteoblast function. Conversely, new bone (callus) formation in the form of woven bone could result from increased osteoblast function or decreased osteoclast function.
To investigate whether osteoblasts and osteoclasts are directly affected by the secreted products of S. aureus, we evaluated the extent to which conditioned medium (CM) from cultures of LAC, UAMS-1, and their isogenic sarA mutants impact osteoblast and osteoclast viability. We chose to focus on CM based on a previous report demonstrating that the increased production of extracellular proteases in a LAC saeRS mutant limits the accumulation of important extracellular virulence factors that contribute to the bone destruction and repair process (23) and our studies demonstrating that mutation of sarA results in a greater increase in protease production than mutation of saeRS (12, 17). We initially focused on the preosteoblast cell line MC3T3-E1 because these cells have characteristics similar to those of primary calvarial osteoblasts and are derived from C57BL/6 mice, which is the same mouse strain used for our in vivo experiments. Similarly, we used the RAW 264.7 macrophage cell line as a surrogate for osteoclasts because they exhibit characteristics similar to those of bone marrow macrophages, the precursors of primary osteoclasts, but as an established cell line offered the advantage of ready accessibility and ease of manipulation.
CM from LAC (Fig. 3) and UAMS-1 (Fig. 4) cultures was cytotoxic for both MC3T3-E1 and RAW 264.7 cells, and in both strains, mutation of sarA limited this cytotoxicity. This was also true when the experiments were repeated using primary calvarial osteoblasts (Fig. 5) and when the results were assessed on the basis of the number of TRAP-positive multinucleated, primary bone marrow-derived osteoclasts (Fig. 6). When the results were assessed using primary osteoclasts, CM from LAC cultures appeared to be more cytotoxic for primary bone marrow-derived macrophages, although the difference did not reach statistical significance. The changes observed with each parent strain and its isogenic sarA mutants were consistent when both established cell lines and primary cells were used, and this finding is important, given that cell lines are much easier to maintain and more amenable to use in experiments. More importantly, these results are also consistent with the hypothesis that there is a cause-and-effect relationship between osteoblast and osteoclast cytotoxicity and bone destruction and repair processes in acute, posttraumatic osteomyelitis.
FIG 3.

Cytotoxicity of LAC assessed using established cell lines. MC3T3-E1 or RAW 264.7 cells were exposed to CM from cultures of LAC, its sarA mutant (ΔsarA), and its complemented sarA mutant (ΔsarAC). Viability was assessed after 24 h using Invitrogen Live calcein-AM staining (top) or fluorescence microscopy (bottom). The results of calcein-AM staining are reported as the average mean fluorescence intensity (MFI) ± standard deviation. *, statistical significance compared to the results observed with the isogenic parent strain; **, statistical significance compared to the results observed with the isogenic sarA mutant. DMEM, Dulbecco modified Eagle medium.
FIG 4.

Cytotoxicity of UAMS-1 assessed using established cell lines. MC3T3-E1 or RAW 264.7 cells were exposed to CM from cultures of UAMS-1 (U1), its sarA mutant (ΔsarA), and its complemented sarA mutant (ΔsarAC). Viability was assessed after 24 h using Invitrogen Live calcein-AM staining (top) or fluorescence microscopy (bottom). The results of calcein-AM staining are reported as the average mean fluorescence intensity (MFI) ± standard deviation. *, statistical significance compared to the results observed with the isogenic parent strain; **, statistical significance compared to the results observed with the isogenic sarA mutant.
FIG 5.

Cytotoxicity of conditioned medium for primary osteoblasts. Primary osteoblast cells were exposed to conditioned CM from cultures of the indicated strains, and viability was assessed after 24 h using Invitrogen Live calcein-AM staining (top) or fluorescence microscopy (bottom). The results of calcein-AM staining are reported as the average mean fluorescence intensity (MFI) ± standard deviation. *, statistical significance compared to the results observed with the isogenic parent strain.
FIG 6.
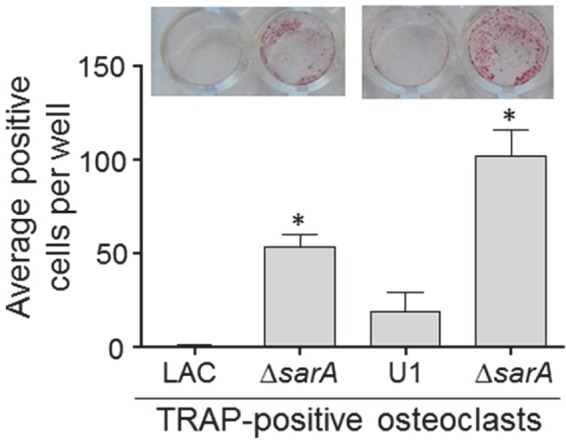
Cytotoxicity of conditioned medium for primary osteoclasts. Primary bone marrow-derived murine osteoclasts were exposed to CM from cultures of the indicated strains. After 12 h, viability was assessed by TRAP staining, with the graph representing the results of quantitative analysis of all replicates. (Inset) TRAP-positive multinucleated cells. *, statistical significance compared to the results observed with the isogenic parent strain.
Given the cytotoxicity of CM from both LAC and UAMS-1 cultures for osteoblasts and osteoclasts and the impact of both strains on bone destruction and repair processes, we examined the exoprotein profiles of each strain and their isogenic sarA mutants by in-gel tryptic digestion followed by GeLC-MS/MS. These studies revealed global differences between both LAC and UAMS-1 and their isogenic sarA mutants (see Table S1 in the supplemental material). A draft genome sequence of UAMS-1 has been published (36), but a fully annotated protein database is not yet available. Thus, on the basis of our studies demonstrating that they are closely related strains (31), identification of UAMS-1 proteins was based on comparisons to MRSA252 proteins. However, it should be noted that while these two strains are closely related, they are not identical. For instance, the MRSA252 genome, like that of LAC, does not encode the toxic shock syndrome toxin 1 (TSST-1) gene (tst), which is present in UAMS-1 (31).
Nevertheless, several particularly notable differences between LAC and UAMS-1 were identified (Table 1). For instance, the analyses confirmed that, unlike LAC, UAMS-1 does not produce LukD/LukE, the Panton-Valentine leucocidin (PVL), or alpha toxin, all of which are potentially important virulence factors in the phenotypes that we observed. However, LukD was present in increased amounts in a LAC sarA mutant relative to the amounts in its parent strain, while LukE was detected at very low levels in both strains (Table 1). Similarly, PVL was also present in increased amounts in a LAC sarA mutant relative to the amounts in its isogenic parent strain. This suggests that LukD/LukE or PVL is unlikely to contribute to the attenuation of a LAC sarA mutant. In contrast, alpha toxin was present in dramatically reduced amounts in a LAC sarA mutant (at ∼11% of the amount in the isogenic parent strain). This suggests that alpha toxin could contribute to both the enhanced virulence of LAC relative to that of UAMS-1 and the reduced virulence of a LAC sarA mutant (24). However, given its absence in UAMS-1, alpha toxin clearly does not contribute to the cytotoxicity or bone remodeling that we observed with this strain.
TABLE 1.
Relative production of select proteins in LAC, UAMS-1, and their isogenic sarA mutants
| Protein | Avg no. of spectral countsa |
|||
|---|---|---|---|---|
| LAC | LAC sarA mutant | UAMS-1 | UAMS-1 sarA mutant | |
| Alpha toxin | 1,019 | 117 | 0 | 0 |
| PVL (LukF) | 324 | 2,292 | 0 | 0 |
| PVL (LukS) | 229 | 1,458 | 0 | 0 |
| LukD | 104 | 576 | 0 | 0 |
| LukE | 23 | 3 | 0 | 0 |
| α1PSM | 102 | 15 | 56 | 13 |
| α2PSM | 32 | 0 | 0 | 0 |
| α3PSM | 12 | 0 | 0 | 0 |
| α4PSM | 112 | 5 | 64 | 14 |
| Delta toxin | 159 | 40 | 317 | 83 |
| Spa | 903 | 1 | 1,379 | 29 |
Results reflect the average number of spectral counts from triplicate samples as assessed by GeLC-MS/MS.
In general, these proteomics studies also confirmed the findings of our previous experiments (26) demonstrating that PSMs, specifically, the alpha class of PSMs (αPSMs), are present in increased levels in LAC relative to UAMS-1 and at reduced levels in both LAC and UAMS-1 sarA mutants relative to the levels in the isogenic parent strains (Fig. 7 and Table 1). In fact, in UAMS-1 the amounts of α2PSM and α3PSM were below the limit of detection of the assay. Nevertheless, the differences observed between UAMS-1 and its sarA mutant did reach statistical significance with respect to α1PSM and α4PSM, and statistically significant differences were observed between LAC and its sarA mutant with respect to all αPSMs (Fig. 7 and Table 1). These results are consistent with the results of our previous experiments, in which PSM levels were measured directly by high-pressure liquid chromatography (26). Moreover, previous studies employing a mutagenesis approach in LAC implicated αPSMs as key factors contributing to osteoblast cytotoxicity and bone remodeling in the same murine model that we employed in the experiments whose results are reported here (23).
FIG 7.
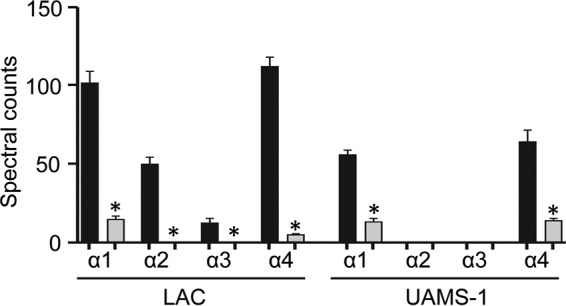
αPSM levels assessed by GeLC-MS/MS. Black bars, amounts of the indicated PSMs produced by LAC or UAMS-1; gray bars, amounts of the indicated PSMs produced by the isogenic sarA mutants. *, statistically significant difference for the indicated peptide compared to the amount of the same peptide observed in the isogenic parent strain.
Based on this, we examined the extent to which these peptides contribute to the phenotypes that we observed in each parent strain. In both LAC and UAMS-1, mutation of the operon encoding αPSMs resulted in a significant decrease in cytotoxicity for both MC3T3-E1 cells and RAW 264.7 cells (Fig. 8). This effect appeared to be greater for LAC than for UAMS-1, particularly when it was assessed using MC3T3-E1 cells. The cytotoxicity of both LAC and UAMS-1 αPSM mutants was also significantly reduced when it was assessed using primary osteoblasts and osteoclasts, and when it was assessed using calvarial osteoblasts, the impact of eliminating αPSM production was significantly greater for LAC than for UAMS-1 (Fig. 9). This is consistent with the observation that LAC produces PSMs at higher levels than UAMS-1 (26). Nevertheless, these results demonstrate that PSMs play an important role in mediating osteoblast and osteoclast cytotoxicity even in a strain like UAMS-1 that produces PSMs at relatively low levels, and they suggest that the reduced accumulation of αPSMs may be a primary factor contributing to the reduced virulence of both LAC and UAMS-1 sarA mutants in our model.
FIG 8.
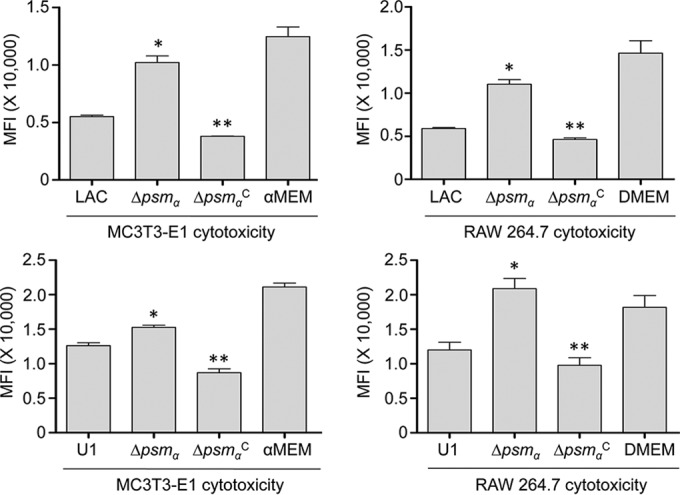
Cytotoxicity in established cell lines as a function of PSM production. MC3T3-E1 or RAW 264.7 cells were exposed to CM from cultures of LAC, UAMS-1 (U1), their isogenic αpsm mutants (Δpsmα), and complemented psm mutants (ΔpsmαC). Viability was assessed after 24 h using Invitrogen Live calcein-AM staining. Results of calcein-AM staining are reported as the average mean fluorescence intensity (MFI) ± standard deviation. *, statistical significance compared to the results observed with the isogenic parent strain; **, statistical significance compared to the results observed with the isogenic psm mutant.
FIG 9.
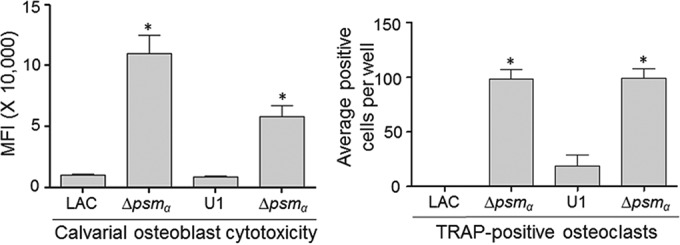
Impact of PSMs on cytotoxicity for primary osteoblasts and osteoclasts. Primary osteoblast cells were exposed to CM from cultures of the indicated strains. Viability was assessed after 24 h using Invitrogen Live calcein-AM staining. (Left) The results of calcein-AM staining are reported as the average mean fluorescence intensity (MFI) ± standard deviation. (Right) Primary bone marrow-derived murine osteoclasts were exposed to CM from cultures of the indicated strains. After 12 h, viability was assessed by TRAP staining, with the graph representing the results of quantitative analysis of all replicates. *, statistical significance compared to the results observed with the isogenic parent strain in both cell types.
To address this, we used our murine osteomyelitis model to compare each parent strain and its αPSM mutants. The results confirmed that eliminating the production of αPSMs in LAC significantly reduced both the reactive new bone formation and the cortical bone destruction observed in this model (Fig. 10). In contrast, neither of these parameters was significantly reduced in the UAMS-1 αPSM mutant in comparison to that in the isogenic parent strain. Thus, while these results suggest that PSMs play some role in the pathogenesis of acute, posttraumatic osteomyelitis even in strains like UAMS-1, they likely play a much more predominant role in defining USA300 strains like LAC. It is important to note in this regard that while the results observed with sarA mutants in vitro in the context of cytotoxicity (Fig. 3 to 6) were consistent with those observed in vivo in the overall context of bone remodeling (Fig. 2), this was not the case with a UAMS-1 αPSM mutant (Fig. 8 to 10). This may be due to the fact that PSMs can be inactivated when bound by host lipoproteins (37), an effect that would presumably be more evident in a strain like UAMS-1 that produces PSMs at relatively low levels.
FIG 10.

Impact of PSMs assessed by micro-CT. Images were analyzed for reactive new bone (callus) formation and cortical bone destruction in mice infected with LAC, UAMS-1 (U1), or their isogenic αpsm (Δpsmα) mutants. Sham refers to the results of the same analysis with mice subjected to the surgical procedure and injected with sterile PBS. *, statistical significance compared to the results observed with the sham treatment; **, statistical significance compared to the results observed with the isogenic parent strain.
The mechanistic basis for the role of PSMs in the pathogenesis of osteomyelitis also remains undetermined, but they are known to act as intracellular toxins that lyse osteoblasts, and this is particularly true for PSMs from hypervirulent strains of S. aureus, like LAC (5). PSMs have also been shown to induce the production of interleukin-8 (27), which in turn can promote osteoclast differentiation and activity (38). Taken together, these would presumably have the effect of increasing bone destruction by decreasing osteoblast activity while increasing osteoclast activity. It is difficult to envision how either would promote reactive bone formation, but it is noteworthy that this occurred at distinct sites distal to the inoculation site (Fig. 1). Together, these factors suggest the possibility that reactive bone formation is a downstream effect arising from the recruitment of osteoclasts to the site of infection and/or the systemic inflammatory response.
Finally, while our results demonstrate an important role for PSMs in the pathogenesis of osteomyelitis in LAC, they also suggest that other virulence factors play an important role in defining both the virulence of UAMS-1 and the attenuation of its isogenic sarA mutant. For instance, the fact that CM from a UAMS-1 αPSM mutant culture exhibited more cytotoxicity for primary osteoblasts than CM from a LAC αPSM mutant culture (Fig. 9) suggests that UAMS-1 produces a potentially relevant cytolytic factor that either is not produced by LAC or is produced in reduced amounts relative to the amounts in which it is produced by UAMS-1. Additionally, the fact that a UAMS-1 sarA mutant was less cytotoxic than a LAC sarA mutant (Fig. 3 to 6) suggests that the abundance of the relevant factor(s) is decreased in a UAMS-1 sarA mutant.
One possibility is this regard are superantigens like TSST-1 and those encoded within the enterotoxin gene cluster (egc), which are produced by UAMS-1 but not LAC (39). However, while we did not detect TSST-1 in our proteomics analysis for the reasons discussed above, mutation of sarA has been shown to result in an increase in the production of TSST-1, albeit under in vitro conditions (40). One other possibility that does meet these criteria is protein A (Spa), which is present in both cell-associated and extracellular forms (41, 42) and was previously shown to bind to preosteoblastic cells via tumor necrosis factor alpha receptor 1, resulting in apoptosis and, ultimately, bone loss (43). Thus, protein A was present in increased amounts in UAMS-1 relative to the amounts in LAC (Spa in Table 1) and could have contributed to the virulence of UAMS-1, and elimination of PSM production in UAMS-1 had comparatively little impact in this model. The fact that the accumulation of Spa was reduced in a UAMS-1 sarA mutant could also account for why mutation of sarA had a comparable impact in both strains. At the same time, the generated sarA mutants of both strains still caused bone destruction and new bone formation to a degree that exceeded that observed with the sham-treated controls (Fig. 2). This is potentially important because it implicates virulence factors whose abundance is not impacted by mutation of sarA at the level of either their production or their accumulation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Otto for the kind gift of pTXΔα.
The content is solely the responsibility of the authors and does not represent the views of the NIH or the U.S. Department of Defense.
Funding Statement
Additional support was provided by core facilities supported by the Center for Microbial Pathogenesis and Host Inflammatory Responses (P20-GM103450) and the Translational Research Institute (UL1TR000039).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00152-16.
REFERENCES
- 1.Lew DP, Waldvogel FA. 2004. Osteomyelitis. Lancet 364:369–379. doi: 10.1016/S0140-6736(04)16727-5. [DOI] [PubMed] [Google Scholar]
- 2.Rao N, Ziran BH, Lipsky BA. 2011. Treating osteomyelitis: antibiotics and surgery. Plast Reconstr Surg 127(Suppl 1):177S–187S. doi: 10.1097/PRS.0b013e3182001f0f. [DOI] [PubMed] [Google Scholar]
- 3.Brown TL, Spencer HJ, Beenken KE, Alpe TL, Bartel TB, Bellamy W, Gruenwald JM, Skinner RA, McLaren SG, Smeltzer MS. 2012. Evaluation of dynamic [18F]-FDG-PET imaging for the detection of acute post-surgical bone infection. PLoS One 7:e41863. doi: 10.1371/journal.pone.0041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flannagan RS, Heit B, Heinrichs DE. 2015. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens 4:826–868. doi: 10.3390/pathogens4040826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasigade JP, Trouillet-Assant S, Ferry T, Diep BA, Sapin A, Lhoste Y, Ranfaing J, Badiou C, Benito Y, Bes M, Couzon F, Tigaud S, Lina G, Etienne J, Vandenesch F, Laurent F. 2013. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS One 8:e63176. doi: 10.1371/journal.pone.0063176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flannagan RS, Heit B, Heinrichs DE. 2016. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol 18:514–535. doi: 10.1111/cmi.12527. [DOI] [PubMed] [Google Scholar]
- 7.Scherr TD, Hanke ML, Huang O, James DB, Horswill AR, Bayles KW, Fey PD, Torres VJ, Kielian T. 2015. Staphylococcus aureus biofilms induce macrophage dysfunction through leukocidin AB and alpha-toxin. mBio 6:e01021-15. doi: 10.1128/mBio.01021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassat JE, Skaar EP. 2013. Recent advances in experimental models of osteomyelitis. Expert Rev Anti Infect Ther 11:1263–1265. doi: 10.1586/14787210.2013.858600. [DOI] [PubMed] [Google Scholar]
- 9.Hammer ND, Cassat JE, Noto MJ, Lojek LJ, Chadha AD, Schmitz JE, Creech CB, Skaar EP. 2014. Inter- and intraspecies metabolite exchange promotes virulence of antibiotic-resistant Staphylococcus aureus. Cell Host Microbe 16:531–537. doi: 10.1016/j.chom.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones-Jackson L, Walker R, Purnell G, McLaren SG, Skinner RA, Thomas JR, Suva LJ, Anaissie E, Miceli M, Nelson CL, Ferris EJ, Smeltzer MS. 2005. Early detection of bone infection and differentiation from post-surgical inflammation using 2-deoxy-2-[18F]-fluoro-d-glucose positron emission tomography (FDG-PET) in an animal model. J Orthop Res 23:1484–1489. [DOI] [PubMed] [Google Scholar]
- 11.Beenken KE, Spencer H, Griffin LM, Smeltzer MS. 2012. Impact of extracellular nuclease production on the biofilm phenotype of Staphylococcus aureus under in vitro and in vivo conditions. Infect Immun 80:1634–1638. doi: 10.1128/IAI.06134-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beenken KE, Mrak LN, Zielinska AK, Atwood DN, Loughran AJ, Griffin LM, Matthews KA, Anthony AM, Spencer HJ, Post GR, Lee CY, Smeltzer MS. 2014. Impact of the functional status of saeRS on in vivo phenotypes of Staphylococcus aureus sarA mutants. Mol Microbiol 92:1299–1312. doi: 10.1111/mmi.12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings JA, Carpenter DP, Troxel KS, Beenken KE, Smeltzer MS, Courtney HS, Haggard WO. 2015. Novel antibiotic-loaded point-of-care implant coating inhibits biofilm. Clin Orthop Relat Res 473:2270–2282. doi: 10.1007/s11999-014-4130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker AC, Beenken KE, Jennings JA, Hittle L, Shirtliff ME, Bumgardner JD, Smeltzer MS, Haggard WO. 2015. Characterization of local delivery with amphotericin B and vancomycin from modified chitosan sponges and functional biofilm prevention evaluation. J Orthop Res 33:439–447. doi: 10.1002/jor.22760. [DOI] [PubMed] [Google Scholar]
- 15.Atwood DN, Loughran AJ, Courtney AP, Anthony AC, Meeker DG, Spencer HJ, Gupta RK, Lee CY, Beenken KE, Smeltzer MS. 2015. Comparative impact of diverse regulatory loci on Staphylococcus aureus biofilm formation. Microbiologyopen 4:436–451. doi: 10.1002/mbo3.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beenken KE, Mrak LN, Griffin LM, Zielinska AK, Shaw LN, Rice KC, Horswill AR, Bayles KW, Smeltzer MS. 2010. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS One 5:e10790. doi: 10.1371/journal.pone.0010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mrak LN, Zielinska AK, Beenken KE, Mrak IN, Atwood DN, Griffin LM, Lee CY, Smeltzer MS. 2012. saeRS and sarA act synergistically to repress protease production and promote biofilm formation in Staphylococcus aureus. PLoS One 7:e38453. doi: 10.1371/journal.pone.0038453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss EC, Spencer HJ, Daily SJ, Weiss BD, Smeltzer MS. 2009. Impact of sarA on antibiotic susceptibility of Staphylococcus aureus in a catheter-associated in vitro model of biofilm formation. Antimicrob Agents Chemother 53:2475–2482. doi: 10.1128/AAC.01432-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss EC, Zielinska A, Beenken KE, Spencer HJ, Daily SJ, Smeltzer MS. 2009. Impact of sarA on daptomycin susceptibility of Staphylococcus aureus biofilms in vivo. Antimicrob Agents Chemother 53:4096–4102. doi: 10.1128/AAC.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zielinska AK, Beenken KE, Mrak LN, Spencer HJ, Post GR, Skinner RA, Tackett AJ, Horswill AR, Smeltzer MS. 2012. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol Microbiol 86:1183–1196. doi: 10.1111/mmi.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loughran AJ, Atwood DN, Anthony AC, Harik NS, Spencer HJ, Beenken KE, Smeltzer MS. 2014. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen 3:897–909. doi: 10.1002/mbo3.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zapotoczna M, McCarthy H, Rudkin JK, O'Gara JP, O'Neill E. 2015. An essential role for coagulase in Staphylococcus aureus biofilm development reveals new therapeutic possibilities for device-related infections. J Infect Dis 212:1883–1893. doi: 10.1093/infdis/jiv319. [DOI] [PubMed] [Google Scholar]
- 23.Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP. 2013. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe 13:759–772. doi: 10.1016/j.chom.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, Hinger SA, Aysanoa EE, Blanchard C, Dunman PM, Wasserman GA, Chen J, Shopsin B, Gilmore MS, Skaar EP, Cassat JE. 2015. Bacterial hypoxic responses revealed as critical determinants of the host-pathogen outcome by TnSeq analysis of Staphylococcus aureus invasive infection. PLoS Pathog 11:e1005341. doi: 10.1371/journal.ppat.1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, Chambers HF, Lu Y, Otto M. 2009. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci U S A 106:5883–5888. doi: 10.1073/pnas.0900743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zielinska AK, Beenken KE, Joo HS, Mrak LN, Griffin LM, Luong TT, Lee CY, Otto M, Shaw LN, Smeltzer MS. 2011. Defining the strain-dependent impact of the staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J Bacteriol 193:2948–2958. doi: 10.1128/JB.01517-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 28.Wormann ME, Reichmann NT, Malone CL, Horswill AR, Grundling A. 2011. Proteolytic cleavage inactivates the Staphylococcus aureus lipoteichoic acid synthase. J Bacteriol 193:5279–5291. doi: 10.1128/JB.00369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cassat J, Dunman PM, Murphy E, Projan SJ, Beenken KE, Palm KJ, Yang SJ, Rice KC, Bayles KW, Smeltzer MS. 2006. Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiology 152:3075–3090. doi: 10.1099/mic.0.29033-0. [DOI] [PubMed] [Google Scholar]
- 30.Robey PG, Termine JD. 1985. Human bone cells in vitro. Calcif Tissue Int 37:453–460. doi: 10.1007/BF02557826. [DOI] [PubMed] [Google Scholar]
- 31.Cassat JE, Dunman PM, McAleese F, Murphy E, Projan SJ, Smeltzer MS. 2005. Comparative genomics of Staphylococcus aureus musculoskeletal isolates. J Bacteriol 187:576–592. doi: 10.1128/JB.187.2.576-592.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beenken KE, Blevins JS, Smeltzer MS. 2003. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun 71:4206–4211. doi: 10.1128/IAI.71.7.4206-4211.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldring SR. 2015. The osteocyte: key player in regulating bone turnover. RMD Open 1:e000049. doi: 10.1136/rmdopen-2015-000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldring SR. 2015. Inflammatory signaling induced bone loss. Bone 80:143–149. doi: 10.1016/j.bone.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 36.Sassi M, Sharma D, Brinsmade SR, Felden B, Augagneur Y. 2015. Genome sequence of the clinical isolate Staphylococcus aureus subsp. aureus strain UAMS-1. Genome Announc 3(1):e01584-14. doi: 10.1128/genomeA.01584-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Surewaard BG, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA. 2012. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog 8:e1002606. doi: 10.1371/journal.ppat.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ. 2003. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone 33:28–37. doi: 10.1016/S8756-3282(03)00086-3. [DOI] [PubMed] [Google Scholar]
- 39.King JM, Kulhankova K, Stach CS, Vu BG, Salgado-Pabün W. 2016. Phenotypes and virulence among Staphylococcus aureus USA100, USA200, USA300, USA400, and USA600 clonal lineages. mSphere 1(3):e00071–16. doi: 10.1128/mSphere.00071-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andrey DO, Jousselin A, Villanueva M, Renzoni A, Monod A, Barras C, Rodriguez N, Kelley WL. 2015. Impact of the regulators sigB, rot, sarA and sarS on toxic shock tst promoter and TSST-1 expression in Staphylococcus aureus. PLoS One 10:e135579. doi: 10.1371/journal.pone.0135579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards AM, Bowden MG, Brown EL, Laabei M, Massey RC. 2012. Staphylococcus aureus extracellular adherence protein triggers TNF alpha release, promoting attachment to endothelial cells via protein A. PLoS One 7:e43046. doi: 10.1371/journal.pone.0043046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Halloran DP, Wynne K, Geoghegan JA. 2015. Protein A is released into the Staphylococcus aureus culture supernatant with an unprocessed sorting signal. Infect Immun 83:1598–1609. doi: 10.1128/IAI.03122-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Widaa A, Claro T, Foster TJ, O'Brien FJ, Kerrigan SW. 2012. Staphylococcus aureus protein A plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS One 7:e40586. doi: 10.1371/journal.pone.0040586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.