

Abstract
The Bacteroides fragilis enterotoxin (BFT), a virulence factor of enterotoxigenic B. fragilis (ETBF), interacts with intestinal epithelial cells and can provoke signals that induce mucosal inflammation. Although expression of heme oxygenase-1 (HO-1) is associated with regulation of inflammatory responses, little is known about HO-1 induction in ETBF infection. This study was conducted to investigate the effect of BFT on HO-1 expression in intestinal epithelial cells. Stimulation of intestinal epithelial cells with BFT resulted in upregulated expression of HO-1. BFT activated transcription factors such as NF-κB, AP-1, and Nrf2 in intestinal epithelial cells. Upregulation of HO-1 in intestinal epithelial cells was dependent on activated IκB kinase (IKK)–NF-κB signals. However, suppression of Nrf2 or AP-1 signals in intestinal epithelial cells did not result in significant attenuation of BFT-induced HO-1 expression. HO-1 induction via IKK–NF-κB in intestinal epithelial cells was regulated by p38 mitogen-activated protein kinases (MAPKs). Furthermore, suppression of HO-1 activity led to increased apoptosis in BFT-stimulated epithelial cells. These results suggest that a signaling pathway involving p38 MAPK–IKK–NF-κB in intestinal epithelial cells is required for HO-1 induction during exposure to BFT. Following this induction, increased HO-1 expression may regulate the apoptotic process in responses to BFT stimulation.
INTRODUCTION
Enterotoxigenic Bacteroides fragilis (ETBF) is associated with noninvasive diarrheal diseases (1, 2), inflammatory bowel diseases (1), and colorectal cancers (3–5). B. fragilis enterotoxin (BFT), a virulence factor of ETBF, is responsible for these diseases (1). BFT interacts with a single layer of intestinal epithelial cells and can provoke signals that induce mucosal inflammation (1, 6–9).
In mammalian cells, two genetically distinct isozymes of heme oxygenase (HO) have been clearly identified. HO-1 is inducible, whereas HO-2 is constitutively expressed. HO-1 catalyzes the degradation of free heme into carbon monoxide, biliverdin, and free iron (10, 11). Within mammalian cells, biliverdin reductase converts biliverdin to bilirubin. Pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), lipoteichoic acid, and peptidoglycan, as well as several proinflammatory cytokines, can induce HO-1 expression (12). Upregulated HO-1 expression can lead to adaptive immune responses that protect cells from immunopathogenesis or stress damage (12, 13). In addition, HO-1 expression is involved in clearance of pathogenic bacteria and downregulation of inflammatory responses. For example, HO-1 deficiency not only results in inadequate pathogen clearance (14) but also promotes the development of necrotizing enterocolitis-like intestinal injury in mice (15). HO-1 and HO-1-induced carbon monoxide can ameliorate intestinal inflammation through promotion of bacterial clearance (16). The HO-1/carbon monoxide pathway also suppresses Toll-like receptor 4 (TLR4) signaling, leading to downregulation of proinflammatory signaling induced by stimulation with LPS (17). Based on these findings, we hypothesized that the induction of HO-1 may regulate inflammatory responses induced by BFT. However, there are no reports regarding BFT-induced HO-1 expression.
Signals from transcription factors, including nuclear factor-κB (NF-κB), activator protein-1 (AP-1), and NF-E2-related factor 2 (Nrf2, or nuclear factor [erythroid-derived 2]-like 2 [NFE2L2]), regulate the expression of HO-1 (11). Stimulation of intestinal epithelial cells with BFT can activate NF-κB and AP-1 signaling (6–9, 18–20). We have previously demonstrated that exposure of intestinal epithelial cells to BFT results in delayed apoptosis, suggesting that protection of cells after BFT stimulation is related to the generation of signals that activate or suppress mucosal inflammation (21). These observations raise the possibility that signaling molecules that regulate HO-1 expression may be activated in BFT-exposed cells. However, there is no evidence that BFT-induced signaling results in HO-1 induction in intestinal epithelial cells. We therefore investigated HO-1 induction in response to stimulation of intestinal epithelial cells with BFT. We found that a signaling pathway involving p38 mitogen-activated protein kinases (MAPKs)–IκB kinase (IKK)–NF-κB in intestinal epithelial cells is required for HO-1 induction following exposure to BFT.
MATERIALS AND METHODS
Reagents.
LPS-free fetal bovine serum (FBS), antibiotics, l-glutamine, TRIzol, and Ca2+- and Mg2+-free Hanks' balanced salt solution (HBSS) were obtained from Gibco BRL (Gaithersburg, MD, USA). Collagenase XΙa, dispase, bovine serum albumin (BSA), soybean trypsin inhibitor, Dulbecco's modified Eagle's medium (DMEM), and cobalt protoporphyrin (CoPP) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Rabbit monoclonal antibodies (MAbs) against phospho-IκBα (clone 14D4) and phospho-IKKα/β (clone 16A6) and rabbit polyclonal antibodies (Abs) against phospho-p65, phospho-c-Jun, pan-extracellular signal-regulated kinase 1/2 (ERK1/2, p44/p42), phospho-ERK1/2, pan-p38, phospho-p38, pan-Jun N-terminal protein kinase (JNK; p54/p46), phospho-JNK, IKKα, and IKKβ were acquired from Cell Signaling Technology, Inc. (Beverly, MA, USA). Rabbit polyclonal Ab against phospho-Nrf2 was obtained from Bioss Antibodies, Inc. (Woburn, MA, USA). Rabbit polyclonal Abs against HO-1, Nrf2, p50, p52, p65, c-Rel, Rel B, c-Jun, c-Fos, Jun-B, Jun-D, and Fos-B were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse MAbs against actin (clone 2Q1055) and lamin B (clone B-10) and goat anti-mouse and anti-rabbit secondary Abs conjugated to horseradish peroxidase were also purchased from Santa Cruz Biotechnology. Alexa Fluor 488 and DyLight 549 secondary Abs were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and Abcam (Cambridge, MA, USA), respectively. Bay 11-7085, SB203580, PD98059, SP600125, and Hoechst 33258 were obtained from Calbiochem (La Jolla, CA, USA). SR11302 and tin protoporphyrin IX (SnPP) were acquired from Tocris Bioscience (Bristol, United Kingdom).
Purification of BFT and cell culture conditions.
BFT was purified from culture supernatants of a toxigenic strain of ETBF (ATCC 43858) as described previously (9, 19, 20, 22). The purity of BFT preparations was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The activity of LPS in BFT solutions (1 mg/ml) was less than 1 endotoxin unit/ml (Pyrosate test kit with a quantitative chromogenic Limulus amebocyte lysate test; Associates of Cape Cod, Inc., East Falmouth, MA, USA). Using an HEK-Blue LPS detection kit (InvivoGen, San Diego, CA, USA), with a detection limit of 3 ng/ml, the amount of LPS in BFT solutions (1 mg/ml) was found to be less than 3 ng/ml. BFT was frozen in aliquots at −80°C immediately after purification.
The murine intestinal epithelial cell line CMT-93 (ATCC CCL-223) was grown in DMEM with 10% FBS, antibiotics (100 units/ml of penicillin and 100 μg/ml of streptomycin), and glutamine (2 mM). CMT-93 cells were grown at 37°C with 5% CO2 as described previously (23). Cells were seeded at 0.5 × 106 to 2 × 106 cells per well onto six-well plates and allowed to attach overnight. After 12 h of serum starvation, cells were incubated with BFT.
Generation of primary murine intestinal epithelial cells.
Specific-pathogen-free C57BL/6 mice and breeding pairs of Nrf2−/− knockout mice were obtained from Orient Experimental Animals (Seoungnam, South Korea) and RIKEN BioResource Center (Tsukuba, Japan), respectively. Nrf2−/− knockout mice (RBRC 01390) were developed by Masayuki Yamamoto, Institute of Basic Medical Sciences and Center for Tsukuba Advanced Research Alliance, University of Tsukuba, Japan. The targeting vector containing a lacZ-neo cassette was transferred into E14 embryonic stem (ES) cells to replace a 1.2-kb segment containing the rest of the exon 5 coding sequence of the Nrf2 gene (24). All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Hanyang University. Experiments using Nrf2−/− knockout mice were approved by the Institutional Animal Care and Use Committee of Ewha Womans University. Primary murine colonic epithelial cells were isolated from specific-pathogen-free mice (8 to 12 week of age; body mass of 20 to 25 g), as described previously (23). Briefly, intestines were cut into 1-mm fragments and treated with HBSS containing an enzyme solution (60 units/ml collagenase XΙa, 0.02 mg/ml dispase, 2% BSA, and 0.2 mg/ml soybean trypsin inhibitor). FBS (10%) was added, followed by vigorous resuspension, and then supernatants were harvested in sedimentation medium (DMEM containing 10% sorbitol, 5% FBS, and antibiotics [100 units/ml of penicillin and 100 μg/ml of streptomycin]). Cells and small sheets of intestinal epithelium were separated from denser intestinal fragments, after which the epithelial fragments were centrifuged at 300 rpm for 3 min, and the pellet was resuspended in DMEM containing 2% sorbitol and 2.5% FBS. Cells were suspended in DMEM containing 10% FBS with antibiotics, plated on dishes coated with mouse fibronectin (3 μg/cm2; Innovative Research Inc., Novi, MI, USA), and incubated with 5% CO2 at 37°C. Cells were then cultivated in medium containing equal volumes of DMEM and Ham's 12 medium supplemented with FBS (10%) and antibiotics. The medium was replaced every other day. At least 90% of primary colonic epithelial cells were viable for 2 weeks in culture, as determined by trypan blue exclusion. This procedure was supported by Sang Hoon Lee of the University of California, Los Angeles, CA (25).
In some experiments, cells were treated with an NF-κB essential modifier (NEMO) binding domain (NBD) peptide (200 μM; Peptron, Daejeon, South Korea) (23) for 1 h before the addition of BFT.
Quantitative reverse transcriptase PCR (RT-PCR).
Cells were treated with BFT, and then total cellular RNA was extracted using TRIzol. Reverse transcription and PCR amplification were performed as described previously (26). The primers and expected PCR product sizes were as follows: for mouse HO-1, 5′-AAG AGG CTA AGA CCG CCT TC-3′ (sense) and 5′-GTC GTG GTC AGT CAA CAT GG-3′ (antisense), 591 bp (GenBank accession number NM_010442.2; Mus musculus heme oxygenase 1 [Hmox1], mRNA) (27); for mouse β-actin, 5′-GTG GGC CGC TCT AGG CAC CAA-3′ (sense) and 5′-CTC TTT GAT GTC ACG CAC GAT TTC-3′ (antisense), 540 bp (GenBank accession number NM_007393.4; Mus musculus actin, beta [ActB], mRNA) (23). To quantify mRNA molecules, plasmids were constructed to encode standard RNAs as described previously (26, 28). Standard RNA molecules for mouse HO-1 and β-actin were generated by in vitro transcription using T7 RNA polymerase, as described previously (26, 28). The sizes of PCR products generated from standard RNAs for mouse HO-1 and β-actin are 478 bp and 746 bp, respectively.
To quantify the expressed mRNA molecules, serial dilutions of standard RNA molecules (between 104 and 109) were mixed with 1 μg of extracted sample (target) RNA. Reverse transcription was performed at 37°C for 60 min, followed by 95°C at 10 min. PCR amplification was performed in an Applied Biosystems thermal cycler (Applied Biosystems, Foster City, CA, USA). PCR amplification of HO-1 consisted of 23 cycles of 30 s of denaturation at 94°C, 30 s of annealing at 55°C, and 1 min of extension at 72°C. RNA isolated from CMT-93 cells stimulated with curcumin (30 μM) was used as a positive control for HO-1. Negative controls omitted the RNA from cDNA synthesis and PCR amplification. PCR products were separated on an agarose gel and visualized by ethidium bromide staining. The ratios of band intensities for each PCR product from the standard and target RNAs were plotted against the starting number of standard RNA molecules. When the ratio of band intensities equals 1, the number of target RNA molecules is equivalent to the number of standard RNA molecules (26, 28). Data are expressed as the number of target RNA molecules/microgram of total sample RNA.
EMSAs.
Cells were harvested, and nuclear extracts were prepared as described previously (9). The concentration of protein in extracts was determined using a Bradford assay (Bio-Rad, Hercules, CA, USA). Electrophoretic mobility shift assays (EMSAs) were performed according to the manufacturer's instructions (Promega, Madison, WI, USA). In brief, 5 μg of nuclear extract was incubated for 30 min at room temperature with γ-32P-labeled oligonucleotide probes (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ for the NF-κB binding site, 5′-CGC TTG ATG ACT CAG CCG GAA-3′ for the AP-1 binding site, and 5′-TGG GGA ACC TGT GCT GAG TCA CTG GAG-3′ for the Nrf2 binding site). After incubation, both bound DNA and free DNA were resolved on 5% polyacrylamide gels, as described previously (7, 9). Supershift assays were used to identify specific members of the NF-κB or AP-1 families activated by BFT stimulation. EMSAs were performed as described above, except that rabbit Abs (1 μg/reaction volume) against the NF-κB protein p50, p52, p65, c-Rel, or Rel B were added during the binding reaction period. For AP-1 supershift assays, rabbit Ab (1 μg/reaction volume) against c-Jun, c-Fos, Jun-B, Jun-D or Fos-B was used. For Nrf2 supershift assays, anti-Nrf2 Ab (1 μg/reaction) and an IgG isotype control Ab were used. A competition assay for Nrf2 signals was performed by adding a 100-fold excess of unlabeled probe (cold probe) prior to the addition of radiolabeled probe (hot probe) or a mutant probe to the reaction mixture. The sequence of the mutant oligonucleotide was 5′-TGG GGA ACC TGT GCT AGG TCA CTG GAG-3′ (the mutation is underlined). Oligonucleotide probes for the NF-κB or AP-1 binding assays were purchased from Promega. Oligonucleotides for the Nrf2 assay were obtained from Santa Cruz Biotechnology.
Transfection assay.
Lentiviral systems containing mammalian expression vectors were used to block NF-κB, AP-1, Nrf2, or MAPK activation, as described previously (23). Specifically, lentiviral systems containing mammalian expression vectors encoding a hemagglutinin (HA) epitope-tagged mutant IκBα (IκBα-AA) with substitutions of serine for alanine at positions 32 and 36, an HA epitope-tagged mutant c-Jun (TAM67) with deletions of amino acids at positions 3 to 122, and a hexahistidine (6×His) epitope-tagged mutant Erk2 with substitutions of lysine for arginine at position 52 were used to block NF-κB, AP-1, and ERK activation, respectively. Lentiviral systems containing mammalian expression vectors encoding a FLAG epitope-tagged mutant p38 with substitutions of threonine-X-tyrosine for alanine-X-phenylalanine and a FLAG epitope-tagged mutant JNK1 with substitutions of threonine for alanine at position 183 and tyrosine for phenylalanine at position 185 were used to block p38 and JNK activation, respectively. Viral systems were supported by BioCore in the Institute of Biomedical Science (Seoul, South Korea). Lentiviral vectors containing a plasmid expressing Nrf2 short hairpin RNA (shRNA) plasmid (mouse) or IKKβ shRNA plasmid (mouse) and control lentivirus were purchased from Santa Cruz Biotechnology. Transfection experiments were performed according to the manufacturer's instructions. A lentiviral packing kit and mouse HO-1 cDNA clone for lentiviral vectors were purchased from OriGene Technologies, Inc. (Rockville, MD, USA). Because the mouse HO-1 cDNA clone contains DDK (the peptide DYKDDDDK; FLAG tag), HO-1 expression can be verified using an anti-DDK MAb (IgG2a, TA50011-100; OriGene Technologies, Inc.). Transfection experiments were performed according to the manufacturer's instructions.
Small interfering RNAs (siRNAs) against the NF-κB p65 subunit gene and the c-Jun gene were designed as described previously (23). The siRNAs were synthesized by Qiagen (Valencia, CA, USA). A negative (nonsilencing) siRNA control (NS-RNA) was also purchased from Qiagen. Briefly, cells were cultured in six-well plates to 50% to 80% confluence and then transfected with an siRNA using Fugene 6 (Roche, Mannheim, Germany) as a transfection reagent, as described previously (23). Transfected cells were incubated for 48 h prior to the assay.
Immunoblotting and enzyme-linked immunosorbent assay (ELISA).
Cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed in 0.5 ml/well lysis buffer (150 mM NaCl, 20 mM Tris, pH 7.5, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 10 μg/ml aprotinin). Fifteen to 50 μg of protein per lane was size fractionated on a polyacrylamide minigel (Mini-Protein II; Bio-Rad) and electrophoretically transferred to a nitrocellulose membrane (0.1-μm pore size). Immunoreactive proteins to which primary Abs bound were visualized using goat anti-rabbit or anti-mouse secondary Abs conjugated to horseradish peroxidase, followed by enhanced chemiluminescence (ECL system; Amersham Life Science, Buckinghamshire, United Kingdom) and exposure to X-ray film (23).
The protein level of HO-1 following BFT stimulation was evaluated using a commercially available kit (R&D Systems, Inc., Minneapolis, MN, USA). An ELISA kit for the TransAM NF-κB family was obtained from Active Motif (Carlsbad, CA, USA). A PathScan phospho-IκBα kinase assay kit and p44/42 MAP kinase assay kit were purchased from Cell Signaling Technology (23). Each assay was performed according to the individual manufacturer's instructions.
Immunofluorescence assay.
Cells were seeded (5 × 104 cells in 0.2 ml of RPMI 1640 medium/well) on eight-well poly-d-lysine-coated culture microslides (Santa Cruz). After samples were treated with BFT, the following method was used to evaluate HO-1 expression and phospho-p65 translocation. Cells were treated with 0.3% Triton X-100 in PBS for 30 min at room temperature, followed by incubation with goat anti-HO-1 and rabbit anti-phospho-p65 Abs as primary Abs for 2 h. In another experiment to evaluate HO-1 expression and phospho-Nrf2 translocation, cells were treated with goat anti-HO-1 and rabbit anti-phospho-Nrf2 Abs as primary Abs for 2 h. Cells were then treated with Alexa Fluor 488-conjugated secondary Ab (green) against goat IgG and DyLight 549-conjugated secondary Ab (red) against rabbit IgG for 1 h. Images were captured using a fluorescence microscope (DMI4000B; Leica Microsystems GmbH, Wetzlar, Germany).
Analysis of apoptosis.
For morphological assessment of cells undergoing apoptosis, cells were stained with the DNA dye Hoechst 33258 (5 μg/ml) and examined under a fluorescence microscope (DMI4000B). To determine DNA fragmentation, oligonucleosome release into the cytoplasm was assayed with a Cell Death Detection ELISA Plus kit (Roche Diagnostics) as described previously (21, 29). Caspase-3 activity was assayed with a colorimetric assay kit according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA) (21, 29).
Statistical analyses.
Data from quantitative RT-PCR assays are presented as means ± standard deviations (SD), and ELISA data are presented as means ± standard errors of the means (SEM). A Mann-Whitney t test was used for statistical analysis. P values of <0.05 were considered statistically significant.
RESULTS
BFT induces HO-1 upregulation in intestinal epithelial cells.
Stimulation of primary murine intestinal epithelial cells with BFT resulted in upregulated expression of HO-1 mRNA transcripts. A significant increase in HO-1 mRNA expression was first noted 1 h after treatment with BFT. Expression peaked approximately 6 h after stimulation and decreased to baseline levels at 24 h, as assessed by quantitative RT-PCR (Fig. 1A). Consistent with this, the expression of HO-1 proteins increased with BFT stimulation in primary intestinal epithelial cells (Fig. 1B, top panel) as assessed by immunoblotting. Similar results were also observed in the murine intestinal epithelial cell line CMT-93 (Fig. 1B, middle panel). Densitometric analysis showed that HO-1 protein expression in BFT-exposed primary intestinal epithelial cells was approximately 2-fold higher than that in the established CMT-93 cell line (Fig. 1B, bottom panel). The magnitude of HO-1 expression was dependent on the concentration of BFT used (Fig. 1C). The 50% effective concentration (EC50) of BFT was 99.8 ng/ml. Based on this result, 100 ng/ml BFT was used in subsequent experiments.
FIG 1.

HO-1 expression in intestinal epithelial cells stimulated with BFT. (A) Primary intestinal epithelial cells were treated with BFT (100 ng/ml) for the indicated periods of time. Levels of HO-1 and β-actin mRNAs were analyzed by quantitative RT-PCR. Values are expressed as means ± SD (n = 5). Asterisks indicate statistical significance in comparison to results with unstimulated controls (P < 0.05). (B) Primary intestinal epithelial cells (IECs) and CMT-93 cells were treated with BFT (100 ng/ml) for the indicated periods of time. Expression of HO-1 and actin proteins was analyzed by immunoblotting. Results are representative of more than three independent experiments. Densitometric analysis of expressed proteins is shown in the bottom panel. Values represent relative densities of each protein compared with the density of actin. (C) CMT-93 cells were treated with the indicated concentrations of BFT for 6 h. Expression of HO-1 (filled circles) and β-actin (open circles) mRNAs was analyzed by quantitative RT-PCR. Values are expressed as means ± SD (n = 5). *, P < 0.05, compared with results with untreated controls.
Activation of NF-κB is required to upregulate HO-1 expression in intestinal epithelial cells in response to BFT stimulation.
We previously demonstrated that transcription factors such as NF-κB and AP-1 are activated in BFT-stimulated intestinal epithelial cells (6–9, 18–20). Based on these results, we asked whether NF-κB activation by BFT stimulation is associated with HO-1 expression in intestinal epithelial cells. CMT-93 cells were transfected with lentivirus–IκBα-AA and then stimulated with BFT for 1 h, after which NF-κB DNA-binding activity was assessed by EMSA. In BFT-stimulated cells, transfection with lentivirus–IκBα-AA suppressed NF-κB activity to the control level. However, a control lentivirus containing a plasmid expressing green fluorescent protein (GFP) did not reduce NF-κB activation (Fig. 2A). In these experimental systems, cells transfected with lentivirus–IκBα-AA were stimulated with BFT, and the level of HO-1 mRNA was determined by quantitative RT-PCR. Transfection with lentivirus–IκBα-AA significantly suppressed HO-1 mRNA expression in CMT-93 cells under the BFT-stimulated condition (Fig. 2B). Consistent with these results, the expression of HO-1 proteins induced by BFT stimulation clearly decreased when NF-κB DNA-binding activity was blocked in CMT-93 cells (Fig. 2C). In addition, immunofluorescence microscopy showed that the protein expression of phospho-p65 and HO-1 increased in BFT-treated CMT-93 cells. Concurrently, phospho-p65 and HO-1 protein expression was significantly suppressed in cells transfected with lentivirus–IκBα-AA (Fig. 2D). Because activation of p65/p50 heterodimeric NF-κB in response to BFT stimulation was observed (Fig. 2E), another experiment was performed using p65 siRNA to suppress NF-κB activity. p65 siRNA almost completely suppressed nuclear phospho-p65 protein expression in BFT-stimulated CMT-93 cells (Fig. 2F, upper panel). In this experimental system, blocking NF-κB with p65 siRNA attenuated the BFT-induced increase in HO-1 expression in CMT-93 cells (Fig. 2F, lower panel).
FIG 2.

Effects of NF-κB suppression on HO-1 expression in CMT-93 cells stimulated with BFT. (A) CMT-93 cells were transfected with either lentivirus containing an IκBα superrepressor (IκBα-AA) or control virus (GFP). Transfected cells were stimulated with BFT (100 ng/ml) for 1 h. NF-κB binding activity was assayed by EMSA. Results are representative of three independent experiments. (B) Transfected CMT-93 cells were treated with BFT (100 ng/ml) for the indicated periods of time. Levels of HO-1 mRNA were analyzed by quantitative RT-PCR. Values are expressed as means ± SD (n = 5). β-Actin mRNA levels in each group remained relatively constant throughout the same periods (∼106 transcripts/μg total RNA). *, P < 0.05, compared with results with untransfected cells treated with BFT. (C) Transfected or untransfected cells were treated with BFT (100 ng/ml) for 6 h. Expression of HO-1 and actin proteins was analyzed by immunoblotting. Results are representative of more than three independent experiments. (D) Transfected CMT-93 cells were stimulated with BFT (100 ng/ml) for 6 h, and immunofluorescence microscopy was performed. Cells were stained with anti-HO-1 Ab (green), anti-phospho-p65 Ab (red), and 4′,6′-diamidino-2-phenylindole (DAPI) (blue; nucleus). Data are representative of at least five experiments. (E) Supershift assays using nuclear extracts from CMT-93 cells treated with BFT (100 ng/ml) for 1 h were performed. Results are representative of more than three independent experiments. (F) CMT-93 cells were transfected with NF-κB p65-specific siRNA or a nonsilencing siRNA (NS-RNA) as a control for 48 h, after which cells were combined with BFT (100 ng/ml) for 1 h. Nuclear extracts were analyzed by immunoblotting with the indicated Abs, as indicated. Transfected cells were stimulated with BFT (100 ng/ml) for 6 h, and expression of HO-1 and actin proteins was analyzed by immunoblotting. Results shown are representative of more than three independent experiments.
Stimulation of primary intestinal epithelial cells with BFT increased levels of phosphorylated IKKα/β (Fig. 3A). In this system, the addition of an IKK inhibitor, NBD peptide, into primary intestinal epithelial cells significantly attenuated the increased NF-κB activation and HO-1 expression induced by BFT stimulation (Fig. 3B). To confirm these results, CMT-93 cells were transfected with lentiviruses containing IKKβ shRNA to suppress IKK activity (Fig. 3C, upper panel). In this experimental system, the expression of HO-1 proteins induced by BFT stimulation was inhibited when lentiviruses containing IKKβ shRNA were transfected (Fig. 3C, lower panel). In addition, an IKKβ shRNA significantly reduced both NF-κB activation and HO-1 induction in BFT-stimulated CMT-93 cells as assessed by ELISA (Fig. 3D). These results suggest a connection between IKK–NF-κB-dependent signaling and HO-1 induction in BFT-stimulated intestinal epithelial cells.
FIG 3.
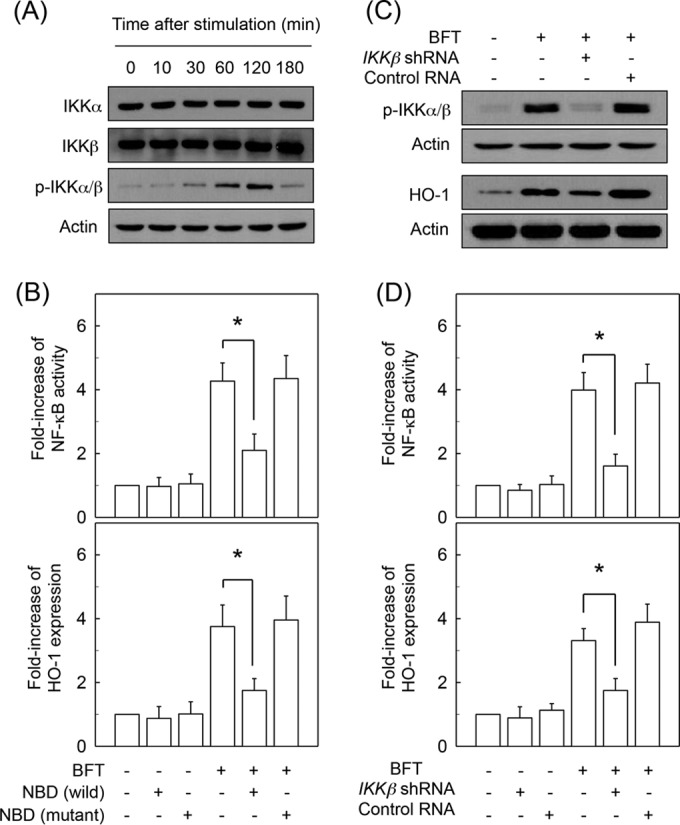
Effects of IKK activation on HO-1 expression in intestinal epithelial cells stimulated with BFT. (A) Primary intestinal epithelial cells were stimulated with BFT (100 ng/ml) for the indicated periods. Protein expression of IKKα, IKKβ, phospho-IKKα/β, and actin was assessed by immunoblot analysis. Results are representative of three independent experiments. (B) Primary intestinal epithelial cells were preincubated with NBD peptide (200 μM) for 1 h, and then BFT (100 ng/ml) was added for an additional 1 h (NF-κB) or 6 h (HO-1). Activity of NF-κB and expression of HO-1 protein were measured by ELISAs. Data are expressed as mean fold induction ± SEM relative to that of untreated controls (n = 5). (C) CMT-93 cells were transfected with lentiviral vectors containing IKKβ shRNA or a control shRNA plasmid. Transfected cells were stimulated with BFT (100 ng/ml) for 1 h (phospho-IKKα/β) or 6 h (HO-1). Expression of each protein was analyzed by immunoblotting. Results are representative of more than three independent experiments. (D) Culture conditions are identical to those described for panel C. Activity of NF-κB and expression of HO-1 protein were measured by ELISAs. Data are expressed as mean fold induction ± SEM relative to that of untreated controls (n = 5). *, P < 0.05.
AP-1 is not involved in induction of HO-1 in BFT-stimulated intestinal epithelial cells.
We next evaluated whether AP-1 activation by BFT stimulation is associated with HO-1 expression. Transfection with lentivirus containing a dominant negative plasmid expressing c-jun (lentivirus–dn-c-jun) appeared to suppress AP-1 DNA-binding activity to control levels in BFT-stimulated CMT-93 cells, while control lentivirus (GFP) did not reduce AP-1 activation (Fig. 4A). In addition, protein expression of nuclear phospho-c-Jun was suppressed in cells transfected with lentivirus-dn-c-jun. In these experimental systems, the levels of HO-1 mRNA were determined by quantitative RT-PCR. Transfection with lentivirus–dn-c-jun did not significantly change HO-1 mRNA expression in CMT-93 cells (Fig. 4B). Consistent with this, the expression of HO-1 proteins induced by BFT stimulation was not significantly affected in CMT-93 cells when AP-1 DNA-binding activity was suppressed (Fig. 4C). In another experiment, CMT-93 cells were transfected with siRNA against c-jun to suppress AP-1 activity because activation of c-Jun/c-Fos heterodimeric AP-1 was observed in response to BFT stimulation (Fig. 4D). The c-jun siRNA almost completely suppressed expression of nuclear phospho-c-Jun in CMT-93 cells (Fig. 4E), while c-jun siRNA did not change BFT-induced HO-1 expression (Fig. 4E).
FIG 4.

Effects of AP-1 suppression on HO-1 expression in intestinal epithelial cells stimulated with BFT. (A) CMT-93 cells were transfected with either lentivirus containing a dominant negative c-jun plasmid (dn-c-jun) or control virus (GFP). Transfected cells were stimulated with BFT (100 ng/ml) for 1 h. AP-1 binding activity was assayed by EMSA. Immunoblot results for concurrent phospho-c-Jun and lamin B in nuclear extracts under the same conditions are also provided. Results are representative of more than three independent experiments. (B) Transfected CMT-93 cells were treated with BFT (100 ng/ml) for the indicated periods of time. Levels of HO-1 mRNA were analyzed by quantitative RT-PCR. Values are expressed as means ± SD (n = 5). β-Actin mRNA levels in each group remained relatively constant for the same period (∼106 transcripts/μg total RNA). (C) Transfected or untransfected CMT-93 cells were treated with BFT (100 ng/ml) for 6 h. Expression of HO-1 and actin proteins was analyzed by immunoblotting. Results are representative of more than three independent experiments. (D) Supershift assays were performed using each Ab in CMT-93 cells stimulated with BFT (100 ng/ml) for 1 h. Results are representative of more than three independent experiments. (E) CMT-93 cells were transfected with siRNA against c-jun to suppress AP-1 activity or with a nonsilencing siRNA (NS-RNA) as a control for 48 h, after which cells were combined with BFT (100 ng/ml) for 1 h. Nuclear extracts were analyzed by immunoblotting with the indicated Abs. Transfected cells were stimulated with BFT (100 ng/ml) for 6 h. Expression of HO-1 and actin proteins was analyzed by immunoblotting, as indicated. Results shown are representative of more than three independent experiments. (F) Primary intestinal epithelial cells were preincubated with Bay 11-7082 (50 μM) or SR11302 (10 μM) for 30 min, followed by stimulation with BFT (100 ng/ml) for an additional 6 h. Expression level of HO-1 was measured by ELISA (means ± SEM; n = 5). *, P < 0.05, compared to results with BFT alone. NS, statistically nonsignificant. (G) Primary intestinal epithelial cells were treated with BFT (100 ng/ml) for 6 h. Expression of HO-1 and actin proteins was analyzed by immunoblotting. Results are representative of more than three independent experiments.
To confirm these results, primary intestinal epithelial cells were preincubated with the NF-κB inhibitor Bay 11-7082 or AP-1 inhibitor SR11302 for 30 min, followed by BFT treatment. An ELISA was used to determine the level of HO-1 expression. As shown in Fig. 4F, pretreatment of primary intestinal epithelial cells with Bay 11-7082 resulted in a significant decrease in HO-1 expression compared to the level with treatment with BFT alone. However, SR11302 did not significantly change BFT-induced HO-1 expression in intestinal epithelial cells. Combined treatment of primary intestinal epithelial cells with Bay 11-7082 and BFT clearly inhibited BFT-induced HO-1 protein expression compared to the level with BFT treatment alone, while combined treatment with SR11302 and BFT did not affect HO-1 protein expression (Fig. 4G).
Nrf2 activation is not associated with induction of HO-1 in BFT-stimulated intestinal epithelial cells.
Because the promoter region of HO-1 genes contains a binding site for Nrf2 (11), we evaluated whether BFT activates Nrf2 in intestinal epithelial cells. As shown in Fig. 5A, BFT increased Nrf2 DNA-binding activity in primary intestinal epithelial cells as assessed by EMSA. The activation of Nrf2 peaked at 1 to 6 h after stimulation and decreased thereafter. Similar results were observed in CMT-93 cells in response to BFT stimulation (Fig. 5B). The specificity of Nrf2 DNA-binding was confirmed using competition and supershift assays. As shown in Fig. 5C, the addition of excess Nrf2 oligomer (cold Nrf2) to nuclear extracts obtained from BFT-stimulated primary intestinal epithelial cells resulted in suppression of Nrf2 DNA binding. A supershift assay using nuclear extracts from primary intestinal epithelial cells also showed that the Nrf2 DNA binding disappeared upon treatment with anti-Nrf2 Ab (Fig. 5D).
FIG 5.

Activation of Nrf2 in intestinal epithelial cells stimulated with BFT. (A and B) Primary intestinal epithelial cells (A) and CMT-93 cells (B) were treated with BFT (100 ng/ml) for the indicated periods of time. Nrf2 DNA-binding activity was assessed by EMSA. Results are representative of more than three independent experiments. (C and D) Nuclear extracts were obtained from primary intestinal epithelial cells treated with BFT (100 ng/ml) for 3 h. (C) Competition assays for Nrf2 signals. (D) Supershift assays for Nrf2 signals. Results are representative of more than three independent experiments. (E and F) Effects of Nrf2 suppression on HO-1 expression in CMT-93 cells stimulated with BFT. CMT-93 cells were transfected with Nrf2-specific shRNA or a control RNA (E). Transfected cells were combined with BFT (100 ng/ml) for 3 h. Nrf2 binding activity was assayed by EMSA, as indicated. Transfected cells were treated with BFT (100 ng/ml) for 6 h. Expression of HO-1 and actin proteins was analyzed by immunoblot. Results are representative of more than three independent experiments. Transfected CMT-93 cells were treated with BFT (100 ng/ml) for the indicated periods of time (F). The level of HO-1 mRNA was analyzed by quantitative RT-PCR. Values are expressed as means ± SD (n = 5). β-Actin mRNA levels in each group remained relatively constant throughout the same periods (∼106 transcripts/μg total RNA).
We next asked whether HO-1 induction was associated with Nrf2 activation in BFT-stimulated cells. Transfection with lentivirus containing Nrf2 shRNA was used to suppress Nrf2 activity in CMT-93 cells. As shown in Fig. 5E, Nrf2 shRNA almost completely suppressed Nrf2 activity in CMT-93 cells stimulated with BFT. Under these conditions, no difference in HO-1 protein expression levels was observed between CMT-93 cells transfected with Nrf2 shRNA and untransfected cells (Fig. 5E, bottom panel). To confirm this result, cells transfected with Nrf2 shRNA were stimulated with BFT. The level of HO-1 mRNA was then determined by quantitative RT-PCR. Results revealed that transfection with Nrf2 shRNA did not lead to statistically significant changes in HO-1 mRNA expression in CMT-93 cells under BFT-stimulated conditions (Fig. 5F). An additional experiment was then performed using immunofluorescence microscopy. This experiment showed that expression of phospho-Nrf2 and HO-1 proteins increased in BFT-treated CMT-93 cells, while Nrf2 shRNA transfection did not influence BFT-induced HO-1 protein expression (Fig. 6A).
FIG 6.
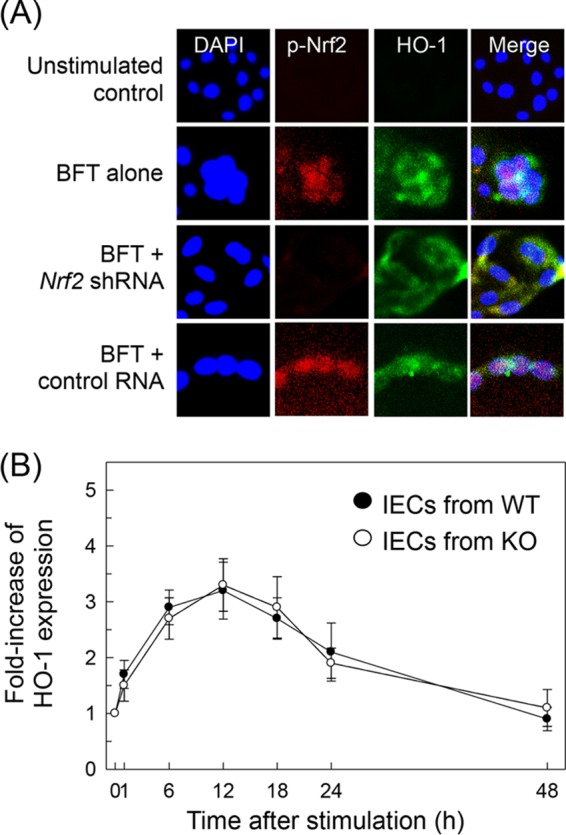
Effects of Nrf2 suppression on HO-1 expression in intestinal epithelial cells stimulated with BFT. (A) Nrf2 translocation and HO-1 expression in BFT-exposed CMT-93 cells. Cells were transfected with Nrf2-specific shRNA or a control RNA. Transfected CMT-93 cells were treated with BFT (100 ng/ml) for 6 h, and immunofluorescence microscopy was performed. Each group of cells was stained with anti-Nrf2 Ab (red), anti-HO-1 Ab (green), and 4′,6′-diamidino-2-phenylindole (DAPI) (blue; nucleus). Data are representative of at least five experiments. (B) HO-1 expression in cells derived from wild-type (WT) and Nrf2−/− knockout (KO) mice. Primary intestinal epithelial cells (IECs) derived from wild-type or Nrf2−/− knockout mice were stimulated with BFT (100 ng/ml) for the indicated periods of time. Expression of HO-1 protein in each panel was measured by ELISA (means ± SEM; n = 5).
To confirm these results, primary intestinal epithelial cells from Nrf2−/− knockout mice were used. As shown in Fig. 6B, BFT increased expression of HO-1 proteins in intestinal epithelial cells derived from wild-type mice. HO-1 expression in primary intestinal epithelial cells derived from Nrf2−/− knockout mice was not significantly altered compared to the expression level in intestinal epithelial cells derived from wild-type mice. These results indicate that Nrf2 signaling does not play a major role in the induction of HO-1 in BFT-exposed intestinal epithelial cells.
p38 MAPK is associated with HO-1 induction in BFT-stimulated intestinal epithelial cells.
BFT stimulation induced phosphorylation of MAPK proteins such as ERK1/2, p38, and JNK in intestinal epithelial cells. Thus, BFT activated phosphorylation of ERK1/2, p38, and JNK in primary intestinal epithelial cells (Fig. 7A). Similar results were obtained in CMT-93 cells (Fig. 7B). The effects of pretreatment with MAPK inhibitors on HO-1 expression in BFT-stimulated primary intestinal epithelial cells were assessed next. The following kinase inhibitors were used: PD98059, an inhibitor of MEK1/2, an MAPK that phosphorylates ERK1/2; pyridinyl imidazole inhibitor SB203580, which specifically inhibits p38; and SP600125, which inhibits JNK (22). Pretreatment of primary intestinal epithelial cells with PD98059 (≥50 μM), SB203580 (≥10 μM), or SP600125 (≥50 μM) for 30 min significantly inhibited BFT-induced activation of HO-1 (Fig. 7C).
FIG 7.

MAPK signals are associated with HO-1 expression in BFT-stimulated intestinal epithelial cells. (A and B) Primary intestinal epithelial cells (A) and CMT-93 cells (B) were stimulated with BFT (100 ng/ml) for the indicated periods of time. ERK1/2, p38, and JNK activities were measured by immunoblot analysis. Results are representative of three independent experiments. (C) Primary intestinal epithelial cells were preincubated with SB203580, PD98059, or SP600125 for 30 min and then stimulated with BFT (100 ng/ml) for another 6 h. Levels of HO-1 expression were determined by ELISA. Data are expressed as mean percent increase relative to the level in unstimulated controls ± SEM (n = 5). *, P < 0.05, compared to results with BFT alone.
In another experiment, lentiviral systems containing dominant negative plasmids were used. Phosphorylation of MAPK proteins was clearly suppressed in CMT-93 cells transfected with lentiviruses containing the various dominant negative plasmids (Fig. 8A). In these experimental systems, transfection with the lentivirus containing a dominant negative p38 plasmid (lentivirus–dn-p38) significantly decreased nuclear phospho-p65 protein expression following BFT stimulation (Fig. 8B, top panel). However, transfection with lentiviruses containing a dominant negative Erk2 plasmid (lentivirus–dn-Erk) or dominant negative JNK1 plasmid (lentivirus–dn-JNK) did not result in any change in nuclear phospho-p65 expression. In addition, transfection with lentivirus–dn-p38 resulted in significant suppression of HO-1 activity compared with activity in untransfected CMT-93 cells under BFT-treated conditions (Fig. 8B, bottom panel). To confirm p38 MAPK-induced NF-κB activation and HO-1 expression, IKK kinase activity was measured using a phospho-IκBα kinase assay kit. Transfection with lentivirus–dn-p38 significantly decreased phosphorylated IκBα activity in BFT-stimulated CMT-93 cells (Fig. 8C). Concurrently, lentivirus–dn-p38 significantly inhibited the expression of HO-1 following BFT stimulation (Fig. 8D). These results suggest that stimulation of intestinal epithelial cells with BFT activates a signaling cascade involving p38 MAPK, IKK, NF-κB, and HO-1.
FIG 8.

Effects of MAPK suppression on HO-1 expression in CMT-93 cells stimulated with BFT. (A) Cells were infected with lentiviruses containing either a dominant negative or a control plasmid (GFP). Transfected CMT-93 cells were stimulated with BFT (100 ng/ml) for 30 min, after which immunoblotting was performed. Results are representative of three independent experiments. (B) Transfected CMT-93 cells were stimulated with BFT (100 ng/ml) for 1 h (phospho-p65) or 6 h (HO-1). Nuclear extracts were analyzed by immunoblotting with the indicated Abs. Expression of HO-1 and actin proteins was analyzed by immunoblotting. Results are representative of more than three independent experiments. (C) Transfected CMT-93 cells were stimulated with BFT (100 ng/ml) for the indicated periods of time. IKK kinase activity was measured using a phospho-IκBα kinase assay kit. Data are expressed as mean fold induction ± SEM of phosphorylated IκBα relative to the level in untreated controls (n = 5). (D) Transfected CMT-93 cells were stimulated with BFT (100 ng/ml) for 6 h. Expression of HO-1 protein was determined by ELISA. Data are expressed as the mean increase relative to the level in unstimulated controls ± SEM (n = 5). *, P < 0.05, compared to results with BFT alone.
BFT-induced HO-1 expression is associated with protection of intestinal epithelial cells.
Stimulation of intestinal epithelial cells with BFT for 48 h induced the nuclear fragmentation characteristics of apoptosis (Fig. 9A). Forty-eight hours after stimulation of primary intestinal epithelial cells with 1, 10, 100, and 500 ng/ml BFT, DNA fragmentation increased 1.0- ± 0.2-fold, 1.1- ± 0.3-fold, 1.7- ± 0.3-fold, and 3.7- ± 0.5-fold, respectively, relative to levels in unstimulated controls (means ± SEM; n = 3). Apoptosis of primary intestinal epithelial cells was not observed within 24 h poststimulation with BFT. Apoptosis was first apparent at 36 h after BFT stimulation, as assessed by quantitative analysis of DNA fragmentation (Fig. 9B). In contrast, HO-1 expression was upregulated in the early period of stimulation compared to the level of DNA fragmentation, indicating that apoptosis is likely a late response of epithelial cells to BFT stimulation compared to HO-1 expression.
FIG 9.

Effects of HO-1 suppression on apoptosis in intestinal epithelial cells stimulated with BFT. (A) Monolayers of CMT-93 cells were incubated with BFT (500 ng/ml) for 48 h and then stained with Hoechst dye 33258 (magnification, ×400). Apoptotic bodies and nuclear fragmentation are shown in BFT-treated cells (arrowheads). Results are representative of three independent experiments. (B) Time course of apoptosis and HO-1 expression in primary intestinal epithelial cells after treatment with BFT (500 ng/ml). Apoptosis was assessed using a cell death detection ELISA at the indicated times after BFT treatment. Numbers indicate DNA fragmentation expressed as the relative increase compared with the level in unstimulated controls (means ± SEM; n = 5). Levels of HO-1 protein were determined by ELISA. Data are expressed as the mean increase relative to the level in unstimulated controls ± SEM (n = 5). *, P < 0.05 (C) Primary intestinal epithelial cells were pretreated with SnPP (50 μM) for 18 h and then treated with BFT (500 ng/ml) for another 12 h. Oligonucleosome release into the cytoplasm was assayed by ELISA and is presented as the relative increase compared with the level in unstimulated controls (means ± SEM; n = 5). *, P < 0.05 (D) CMT-93 cells were transfected with HO-1-specific shRNA or a control RNA. Transfected cells were either left untreated or stimulated with BFT (500 ng/ml) for 12 h. Oligonucleosome release is presented as the relative increase compared to the level in untreated controls (means ± SEM; n = 5). Caspase-3 activity in cell extracts was assayed using a colorimetric assay kit and is expressed relative to the activity in untreated controls (means ± SEM; n = 5). *, P < 0.05. (E) Cell cultures were identical to those described for panel D. Transfected CMT-93 cells were stimulated with BFT (500 ng/ml) for 12 h, after which immunoblotting for HO-1 and actin proteins was performed. Results are representative of three independent experiments.
We next asked whether BFT-induced HO-1 expression is associated with early inhibition of apoptosis in intestinal epithelial cells. To evaluate this hypothesis, BFT-stimulated primary intestinal epithelial cells were pretreated with the pan-HO inhibitor SnPP (50 μM), and apoptosis was then measured. As shown in Fig. 9C, pretreatment with SnPP significantly increased DNA fragmentation and caspase-3 activity when apoptosis had not occurred 12 h after stimulation with BFT. SnPP is a pan-HO inhibitor and is also known to inhibit the HO-2 isoform (30). Therefore, an SnPP-induced increase in apoptosis was confirmed by another experiment using transfection of CMT-93 cells with HO-1 shRNA. Transfection with HO-1 shRNA significantly increased both DNA fragmentation and caspase-3 activity compared with activity in untransfected cells under BFT-stimulated conditions (Fig. 9D). In this system, expression of HO-1 proteins was almost completely suppressed in cells transfected with HO-1 shRNA (Fig. 9E). Treatment with the exogenous HO-1 inducer CoPP significantly decreased DNA fragmentation and caspase-3 activity 48 h after stimulation with BFT (Fig. 10A). To confirm these findings, CMT-93 cells were transfected with lentiviruses containing an HO-1-overexpressing plasmid (Fig. 10B). HO-1 overexpression significantly attenuated BFT-induced DNA fragmentation and caspase-3 expression in CMT-93 cells (Fig. 10C).
FIG 10.
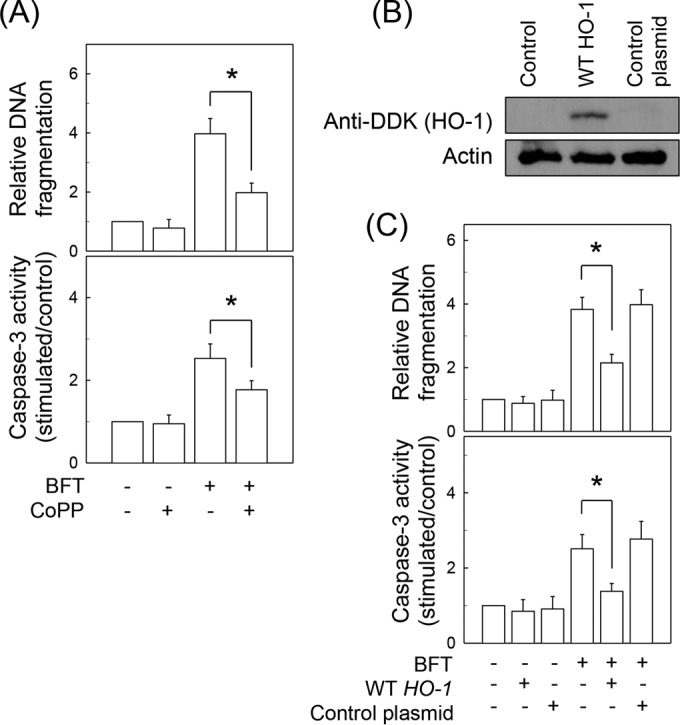
Effects of HO-1 overexpression on apoptosis in intestinal epithelial cells stimulated with BFT. (A) Primary intestinal epithelial cells were stimulated with BFT (500 ng/ml) for 12 h and then treated with CoPP (20 μM) for an additional 36 h. Oligonucleosome release into the cytoplasm was assayed by ELISA, and results are presented as the relative increase compared with the level in unstimulated controls (means ± SEM; n = 5). *, P < 0.05 (B) CMT-93 cells were transfected with lentivirus containing plasmids overexpressing wild-type (WT) HO-1. Transfected cells were stimulated with BFT (500 ng/ml) for 48 h, and HO-1 protein was then analyzed by immunoblotting using anti-DDK Ab. Results are representative of three independent experiments. (C) Cell cultures were identical to those described for panel B. Oligonucleosome release is presented as the relative increase compared to the level in untreated controls (means ± SEM; n = 5). Caspase-3 activity in cell extracts was assayed using a colorimetric assay kit and is expressed relative to the activity in untreated controls (means ± SEM; n = 5). *, P < 0.05.
DISCUSSION
ETBF is a noninvasive bacteria that produces BFT, which is known to provoke the pathogenic effects associated with ETBF infection (1). It has been postulated that ETBF-derived BFT must first act on and stimulate intestinal epithelial cells to regulate intestinal inflammation. Our experiments revealed that treatment of intestinal epithelial cells with BFT induced an increase in HO-1 expression at the protein and mRNA levels. In our previous studies, interleukin-8 (IL-8) and human β-defensin-2 (hBD-2) mRNA expression were first observed within 1 to 3 h after BFT stimulation (9, 20). In the present study, HO-1 mRNA expression was noted within 1 h. These findings suggest that upregulation of HO-1 in intestinal epithelial cells may be one of the early responses to BFT stimulation.
Transcription factors such as NF-κB, AP-1, and Nrf2 regulate a variety of inflammatory responses (11, 31–33). The promoter region of HO-1 contains binding sites for these transcription factors. We previously demonstrated that stimulation of intestinal epithelial cells with BFT activated NF-κB and AP-1 signaling (6–9, 18–20). The present study demonstrated that Nrf2 signaling was also activated in intestinal epithelial cells stimulated with BFT. It remains controversial as to whether HO-1 expression in stimulated intestinal epithelial cells is associated with NF-κB or Nrf2. For example, the expression of HO-1 was found to be dependent on Nrf2 signaling in intestinal epithelial cells (34–36). However, tumor necrosis factor alpha (TNF-α) was shown to increase HO-1 protein levels in the Caco-2 cell line, and Nrf2 suppression did not affect TNF-α-induced HO-1 protein expression in these cells (37). In the present study, suppression of NF-κB activity by either transfection of intestinal epithelial cells with lentivirus–IκBα-AA or with p65 siRNA significantly inhibited BFT-induced HO-1 expression. However, suppression of AP-1 or Nrf2 signals did not result in a significant change in HO-1 expression in BFT-treated intestinal epithelial cells. Therefore, this NF-κB-dependent and both AP-1- and Nrf2-independent expression of HO-1 may be a unique characteristic of intestinal epithelial cells stimulated with ETBF-derived BFT. Further studies are required to clarify if HO-1 is induced in intestinal epithelial cells during the course of in vivo infection with ETBF.
Although MAPK signaling is important for HO-1 expression (11, 38, 39), there is no evidence of BFT-induced MAPK and NF-κB activation leading to HO-1 expression. In the present study, pretreatment of primary intestinal epithelial cells with the p38 inhibitor SB203580 was superior to treatment with the ERK inhibitor PD98059 or the JNK inhibitor SP600125 at inhibiting HO-1 expression. In addition, suppression of p38 MAPK signals in BFT-treated cells using the lentiviral transfection systems resulted in significant inhibition of IKK–NF-κB activation and HO-1 expression. These results suggest that BFT-exposed intestinal epithelial cells activate a signaling cascade involving p38 MAPKs, leading to NF-κB activation and finally HO-1 induction.
Apoptosis is inhibited by HO-1 metabolites. For example, when HO-1 activity was blocked by SnPP or when the action of carbon monoxide was inhibited by hemoglobin, HO-1 no longer prevented apoptosis in endothelial cells although these reagents did not affect antiapoptotic action (40). Treatment with biliverdin or bilirubin also inhibited apoptosis in serum-deprived Caco-2 cells (41). Therefore, metabolites generated by HO-1 may reduce the rate of apoptosis in BFT-stimulated intestinal epithelial cells. Induction of HO-1 plays an important protective role in acute and chronic inflammation of the gastrointestinal tract (42–44). With respect to BFT-exposed intestinal epithelial cells, delayed loss of cell viability and apoptosis of detached intestinal epithelial cells were observed following stimulation of T84 cells or human colon biopsy specimens with BFT (1). We previously demonstrated that expression of cellular inhibitor of apoptosis protein-2 (c-IAP2) increases during the early period of BFT stimulation, resulting in inhibition of apoptotic cell death (21). In the present study, suppression of HO-1 in intestinal epithelial cells augmented apoptotic cell death under BFT stimulation. Therefore, upregulation of both c-IAP2 and HO-1 in intestinal epithelial cells may contribute to inhibition of apoptotic cell death during the early period of BFT stimulation.
In the present study, intestinal epithelial cell apoptosis was observed 36 to 48 h after stimulation, when BFT-induced HO-1 expression reached the baseline level. In addition, exogenously induced HO-1 significantly attenuated BFT-induced apoptosis when BFT-induced HO-1 expression returned to the baseline level. Based on these results, HO-1 induction in BFT-exposed intestinal epithelial cells appears to be transient. This temporary delay of apoptosis may be purposeful. The conferred survival advantage may allow BFT-exposed cells to induce inflammatory responses in the mucosal layer. Furthermore, delayed loss of cell viability and apoptosis after ETBF infection may be important to the host by providing sufficient time for intestinal epithelial cells to generate signals to prevent bacterial colonization. Consistent with this, expression of antimicrobial peptide/proteins, such as hBD-2 (20) and lipocalin-2 (19), was enhanced in intestinal epithelial cells during BFT stimulation. Based on these results, it is plausible that an ETBF-infected host may adapt to BFT as a counter measure.
ETBF clinical illnesses are typically self-limited with watery diarrhea. Persistent diarrhea (>14 days) has been reported in a minority (0 to 22%) of human patients (1). In an animal study, wild-type ETBF induced acute persistent colitis in specific-pathogen-free mice and rapidly resulted in lethal colitis in germfree mice (45). In addition, pharmacologic doses of BFT have been reported to induce fluid accumulation in ligated murine intestinal loops (8). Moreover, there are possibly differences between mice and humans with regard to the function of colonic epithelial cells. Therefore, species-specific differences in HO-1 induction between murine and human intestinal epithelial cells may exist. Because this study was performed exclusively in murine colonic epithelial cells, further studies are required to clarify the species-specific differences that contribute to HO-1 induction in BFT-stimulated cells.
In summary, we demonstrated that exposure of intestinal epithelial cells to BFT resulted in rapid activation of MAPK signaling. Activated MAPK signals led to the induction of HO-1 molecules via the IKK–NF-κB signaling pathway in intestinal epithelial cells. The resulting increase in HO-1 expression may regulate the apoptotic process in response to BFT stimulation.
ACKNOWLEDGMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2015R1D1A1A01058565) and by a grant from the Medical Research Center (2008-0062287), funded by the NRF of the Ministry of Science, ICT and Future Planning, Republic of Korea.
None of the authors of this study has any financial or commercial conflicts of interest.
REFERENCES
- 1.Sears CL. 2009. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 22:349–369. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wick EC, Sears CL. 2010. Bacteroides spp. and diarrhea. Curr Opin Infect Dis 23:470–474. doi: 10.1097/QCO.0b013e32833da1eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC, Platz EA, Pardoll DM, Sears CL. 2015. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis 60:208–215. doi: 10.1093/cid/ciu787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sears CL, Geis AL, Housseau F. 2014. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest 124:4166–4172. doi: 10.1172/JCI72334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL. 2009. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JM, Lee DH, Kim JS, Lee JY, Park HG, Kim YJ, Oh YK, Jung HC, Kim SI. 2009. 5,7-Dihydroxy-3,4,6-trimethoxyflavone inhibits the inflammatory effects induced by Bacteroides fragilis enterotoxin via dissociating the complex of Hsp90 and IκBα and IκB kinase-gamma in intestinal epithelial cell culture. Clin Exp Immunol 155:541–551. doi: 10.1111/j.1365-2249.2008.03849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JM, Jung HY, Lee JY, Youn J, Lee CH, Kim KH. 2005. Mitogen-activated protein kinase and activator protein-1 dependent signals are essential for Bacteroides fragilis enterotoxin-induced enteritis. Eur J Immunol 35:2648–2657. doi: 10.1002/eji.200526321. [DOI] [PubMed] [Google Scholar]
- 8.Kim JM, Lee JY, Yoon YM, Oh YK, Kang JS, Kim YJ, Kim KH. 2006. Bacteroides fragilis enterotoxin induces cyclooxygenase-2 and fluid secretion in intestinal epithelial cells through NF-κB activation. Eur J Immunol 36:2446–2456. doi: 10.1002/eji.200535808. [DOI] [PubMed] [Google Scholar]
- 9.Kim JM, Oh YK, Kim YJ, Oh HB, Cho YJ. 2001. Polarized secretion of CXC chemokines by human intestinal epithelial cells in response to Bacteroides fragilis enterotoxin: NF-κB plays a major role in the regulation of IL-8 expression. Clin Exp Immunol 123:421–427. doi: 10.1046/j.1365-2249.2001.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu XM, Peyton KJ, Durante W. 2013. Physiological cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am J Physiol Heart Circ Physiol 304:H1634–H1643. doi: 10.1152/ajpheart.00872.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. 2010. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol 80:1895–1903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Ma X, You X, Zeng Y, He J, Liu L, Deng Z, Jiang C, Wu H, Zhu C, Yu M, Wu Y. 2013. Mycoplasma fermentans MALP-2 induces heme oxygenase-1 expression via mitogen-activated protein kinases and Nrf2 pathways to modulate cyclooxygenase 2 expression in human monocytes. Clin Vaccine Immunol 20:827–834. doi: 10.1128/CVI.00716-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raval CM, Lee PJ. 2010. Heme oxygenase-1 in lung disease. Curr Drug Targets 11:1532–1540. doi: 10.2174/1389450111009011532. [DOI] [PubMed] [Google Scholar]
- 14.Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, Soares MP, Otterbein LE. 2014. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest 124:4926–4940. doi: 10.1172/JCI72853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulz S, Wong RJ, Jang KY, Kalish F, Chisholm KM, Zhao H, Vreman HJ, Sylvester KG, Stevenson DK. 2013. Heme oxygenase-1 deficiency promotes the development of necrotizing enterocolitis-like intestinal injury in a newborn mouse model. Am J Physiol Gastrointest Liver Physiol 304:G991–G1001. doi: 10.1152/ajpgi.00363.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onyiah JC, Sheikh SZ, Maharshak N, Otterbein LE, Plevy SE. 2014. Heme oxygenase-1 and carbon monoxide regulate intestinal homeostasis and mucosal immune responses to the enteric microbiota. Gut Microbes 5:220–224. doi: 10.4161/gmic.27290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. 2009. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol 182:3809–3818. doi: 10.4049/jimmunol.0712437. [DOI] [PubMed] [Google Scholar]
- 18.Kim JM, Cho SJ, Oh YK, Jung HY, Kim YJ, Kim N. 2002. Nuclear factor-κB activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin Exp Immunol 130:59–66. doi: 10.1046/j.1365-2249.2002.01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo do Y, Ko SH, Jung J, Kim YJ, Kim JS, Kim JM. 2013. Bacteroides fragilis enterotoxin upregulates lipocalin-2 expression in intestinal epithelial cells. Lab Invest 93:384–396. doi: 10.1038/labinvest.2013.1. [DOI] [PubMed] [Google Scholar]
- 20.Yoon YM, Lee JY, Yoo D, Sim YS, Kim YJ, Oh YK, Kang JS, Kim S, Kim JS, Kim JM. 2010. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/IκB kinase/NF-κB-dependent pathway. Infect Immun 78:2024–2033. doi: 10.1128/IAI.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JM, Lee JY, Kim YJ. 2008. Inhibition of apoptosis in Bacteroides fragilis enterotoxin-stimulated intestinal epithelial cells through the induction of c-IAP-2. Eur J Immunol 38:2190–2199. doi: 10.1002/eji.200838191. [DOI] [PubMed] [Google Scholar]
- 22.Roh HC, Yoo do Y, Ko SH, Kim YJ, Kim JM. 2011. Bacteroides fragilis enterotoxin upregulates intercellular adhesion molecule-1 in endothelial cells via an aldose reductase-, MAPK-, and NF-κB-dependent pathway, leading to monocyte adhesion to endothelial cells. J Immunol 187:1931–1941. doi: 10.4049/jimmunol.1101226. [DOI] [PubMed] [Google Scholar]
- 23.Ko SH, Jeon JI, Kim H, Kim YJ, Youn J, Kim JM. 2014. Mitogen-activated protein kinase/IκB kinase/NF-κB-dependent and AP-1-independent CX3CL1 expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. J Mol Med 92:411–427. doi: 10.1007/s00109-013-1117-y. [DOI] [PubMed] [Google Scholar]
- 24.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/Small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 25.Choi YJ, Im E, Chung HK, Pothoulakis C, Rhee SH. 2010. TRIF mediates Toll-like receptor 5-induced signaling in intestinal epithelial cells. J Biol Chem 285:37570–37578. doi: 10.1074/jbc.M110.158394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park H, Kim NI, Kim JM, Kim JS, Oh YK, Kim YJ, Kim N, Jung HC, Song IS. 2006. Expression of eotaxin in gastric epithelial cells stimulated with Helicobacter pylori vacuolating cytotoxin. J Bacteriol Virol 36:11–20. doi: 10.4167/jbv.2006.36.1.11. [DOI] [Google Scholar]
- 27.Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. 2006. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res 66:11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 28.Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF. 1995. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest 95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JM, Kim JS, Kim N, Ko SH, Jeon JI, Kim YJ. 2015. Helicobacter pylori vacuolating cytotoxin induces apoptosis via activation of endoplasmic reticulum stress in dendritic cells. J Gastroenterol Hepatol 30:99–108. doi: 10.1111/jgh.12663. [DOI] [PubMed] [Google Scholar]
- 30.Zakhary R, Gaine SP, Dinerman JL, Ruat M, Flavahan NA, Snyder SH. 1996. Heme oxygenase 2: endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc Natl Acad Sci U S A 93:795–798. doi: 10.1073/pnas.93.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin HS, Park JK, Jo EK. 2014. Toll-like receptors and NOD-like receptors in innate immune defense during pathogenic infection. J Bacteriol Virol 44:215–225. doi: 10.4167/jbv.2014.44.3.215. [DOI] [Google Scholar]
- 32.Pedruzzi LM, Stockler-Pinto MB, Leite M Jr, Mafra D. 2012. Nrf2-keap1 system versus NF-κB: the good and the evil in chronic kidney disease? Biochimie 94:2461–2466. doi: 10.1016/j.biochi.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 33.Yuk JM, Jo EK. 2011. Toll-like receptors and innate immunity. J Bacteriol Virol 41:225–235. doi: 10.4167/jbv.2011.41.4.225. [DOI] [Google Scholar]
- 34.Lee SH, Sohn DH, Jin XY, Kim SW, Choi SC, Seo GS. 2007. 2′,4′,6′-Tris(methoxymethoxy) chalcone protects against trinitrobenzene sulfonic acid-induced colitis and blocks tumor necrosis factor-alpha-induced intestinal epithelial inflammation via heme oxygenase 1-dependent and independent pathways. Biochem Pharmacol 74:870–880. doi: 10.1016/j.bcp.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 35.Liu C, Zhu C, Wang G, Xu R, Zhu Y. 2015. Higenamine regulates Nrf2-HO-1-Hmgb1 axis and attenuates intestinal ischemia-reperfusion injury in mice. Inflamm Res 64:395–403. doi: 10.1007/s00011-015-0817-x. [DOI] [PubMed] [Google Scholar]
- 36.Takagi T, Naito Y, Yoshikawa T. 2009. The expression of heme oxygenase-1 induced by lansoprazole. J Clin Biochem Nutr 45:9–13. doi: 10.3164/jcbn.SR09-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kathiria AS, Butcher MA, Hansen JM, Theiss AL. 2013. Nrf2 is not required for epithelial prohibitin-dependent attenuation of experimental colitis. Am J Physiol Gastrointest Liver Physiol 304:G885–G896. doi: 10.1152/ajpgi.00327.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al-Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W, Sathish JG. 2014. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem 289:16442–16451. doi: 10.1074/jbc.M113.532069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy Chowdhury S, Sengupta S, Biswas S, Sinha TK, Sen R, Basak RK, Adhikari B, Bhattacharyya A. 2014. Bacterial fucose-rich polysaccharide stabilizes MAPK-mediated Nrf2/Keap1 signaling by directly scavenging reactive oxygen species during hydrogen peroxide-induced apoptosis of human lung fibroblast cells. PLoS One 9:e113663. doi: 10.1371/journal.pone.0113663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. 2000. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 192:1015–1026. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Busserolles J, Megías J, Terencio MC, Alcaraz MJ. 2006. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int J Biochem Cell Biol 38:1510–1517. doi: 10.1016/j.biocel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Drechsler Y, Dolganiuc A, Norkina O, Romics L, Li W, Kodys K, Bach FH, Mandrekar P, Szabo G. 2006. Heme oxygenase-1 mediates the anti-inflammatory effects of acute alcohol on IL-10 induction involving p38 MAPK activation in monocytes. J Immunol 177:2592–2600. doi: 10.4049/jimmunol.177.4.2592. [DOI] [PubMed] [Google Scholar]
- 43.Lee TS, Chau LY. 2002. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med 8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 44.Naito Y, Takagi T, Higashimura Y. 2014. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys 564:83–88. doi: 10.1016/j.abb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, Rabizadeh S, Golub JE, Mathews LE, Shin J, Sartor RB, Golenbock D, Hamad AR, Gan CM, Housseau F, Sears CL. 2009. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun 77:1708–1718. doi: 10.1128/IAI.00814-08. [DOI] [PMC free article] [PubMed] [Google Scholar]