

Abstract
Human purinergic G protein‐coupled receptor P2Y1 (P2Y1R) is activated by adenosine 5′‐diphosphate (ADP) to induce platelet activation and thereby serves as an important antithrombotic drug target. Crystal structures of P2Y1R revealed that one ligand (MRS2500) binds to the extracellular vestibule of this GPCR, whereas another (BPTU) occupies the surface between transmembrane (TM) helices TM2 and TM3. We introduced a total of 20 μs all‐atom long‐timescale molecular dynamic (MD) simulations to inquire why two molecules in completely different locations both serve as antagonists while ADP activates the receptor. Our results indicate that BPTU acts as an antagonist by stabilizing extracellular helix bundles leading to an increase of the lipid order, whereas MRS2500 blocks signaling by occupying the ligand binding site. Both antagonists stabilize an ionic lock within the receptor. However, binding of ADP breaks this ionic lock, forming a continuous water channel that leads to P2Y1R activation.
Keywords: activation mechanism, allosteric ligand, GPCR, molecular switches, P2Y1R
G‐protein‐coupled receptors (GPCRs) participate in a wide spectrum of physiological functions by transmitting signals from an extracellular binding site to the cytoplasm. Thus, GPCRs are among the most important targets for modern therapeutics, constituting at least one third of all marketable drugs.1 While there is considerable interest in understanding how drugs that bind to different regions of the same GPCR can produce identical responses, the molecular basis of this phenomenon remains obscure. Herein, we used P2Y1R, a family A GPCR, to investigate this question. Human purinergic GPCRs are divided into two subfamilies, P2Y1R‐like receptors coupled to Gq proteins, and P2Y12R‐like receptors coupled to Gi proteins.2 Both are activated by ADP to trigger glutamate release, which plays a crucial role in thrombus formation.2 Moreover, blockade of either receptor significantly decreases ADP‐induced platelet aggregation. However, inhibitors of P2Y1Rs offer a safety advantage over P2Y12R inhibitors by reducing the liability of bleeding.2, 3
The P2Y1R complex crystal structures show that there are two allosteric antagonists that bind at two different regions of the receptor: 1) MRS2500 (Scheme 1; Supporting Information, Figure S1), completely blocks ADP‐induced platelet aggregation, effectively decreases arterial thrombosis,4 and binds on the surface of the ECL2 loop. 2) BPTU substantially reduces platelet aggregation5 and binds between two helix bundles.
Scheme 1.
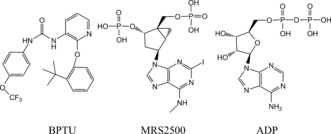
Molecules used in the MD simulations.
To address structural and mechanistic questions about P2Y1R, we performed a total of 20 μs atomic‐level MD simulations (Table S1) on the human P2Y1 receptor, starting from its crystal structures (PDB: 4XNW, 4XNV):2 P2Y1R 1) bound to BPTU (P2Y1R*‐BPTU); 2) bound to MRS2500 (P2Y1R‐MRS2500); and 3) bound to agonist (P2Y1R‐ADP, P2Y1R*‐ADP; Figure 1). From these simulations, we conclude that the two different allosteric antagonists exert their effects by either stabilizing part of the extracellular helix bundles, which lead to an increase in the lipid order (BPTU), or occupying the ligand‐binding site (MRS2500). Both antagonists stabilize an ionic lock within the receptor. In contrast, the agonist molecule ADP induces breakage of the ionic lock and then formation of a continuous water channel that results in the activation of P2Y1R.6
Figure 1.
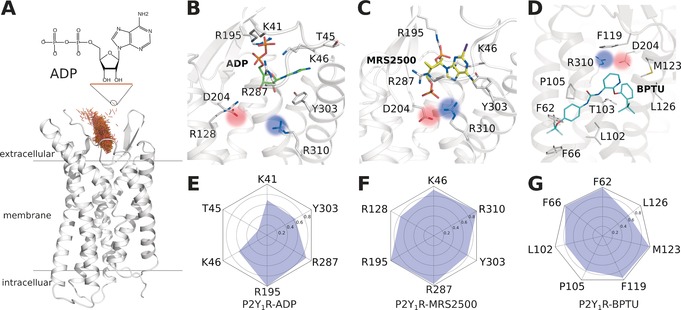
The ligand binding modes of P2Y1R at the end of MD simulations. A) The entrance pathway of ADP into the receptor represented as superposition of the mass centers of ADP over a time period of 0.2 μs (brown points). B) ADP binding mode. C) MRS2500 binding mode. D) BPTU binding mode. E–G) Interaction fingerprint of P2Y1R with bound ADP (E), MRS2500 (F), and BPTU (G).
To sample the binding mode of the agonist molecule, we placed an ADP at the P2Y1R extracellular vestibule entrance, approximately 15 Å from the orthosteric site. Then we performed 6×2 μs all‐atom long‐timescale MD simulations for this system (Figure 1 A,B,E). The final poses of the ligands converged well in each simulation (Figure S2). The aromatic purine ring of ADP engaged in π‐π stacking with Y3037.32, whereas its ribose sugar ring formed an H‐bond network with Y3037.32 through the bulk water molecules. Strong interactions occurred between the negatively charged pyrophosphate and several positively charged residues, including K411.41, K461.46, R195ECL2, and R2876.38. This observed binding mode is consistent with extensive mutagenesis data indicating that mutations of these residues decrease the binding affinity of ADP.2, 7 We then executed 2×2 μs simulations for two additional antagonist‐bound systems, namely P2Y1R‐MRS2500 and P2Y1R*‐BPTU. In antagonist‐bound P2Y1R‐MRS2500 (Figure 1 C,F), the MRS2500 ligand located in a space similar to that of P2Y1R‐ADP. The substituted purine ring of MRS2500 engaged in σ‐π stacking with Y3037.32. Additionally, the 3′‐phosphate formed an ionic lock with K461.46 and R195ECL2, whereas the 5′‐phosphate formed an ionic lock with R2876.38 and R3107.39. In the other antagonist‐bound system, P2Y1R*‐BPTU (Figure 1 D,G), the ligand was located at the transmembrane (TM) helix surface far away from the classic ADP‐ligand‐binding site. The antagonist molecule BPTU was mainly stabilized by hydrophobic interactions with several residues, including F621.43, F661.47, L1022.55, P1052.58, F119ECL1, M1233.24, and L1263.27. Previously, mutagenesis studies of these helix bundles revealed a reduced P2Y1R‐binding affinity for both antagonist ligands.2, 8
Interestingly, we observed that an ionic lock between D204ECL2 and R3107.39 in agonist‐bound P2Y1R, was broken during the MD simulations (Figure 1 B and Figure S3 B,D). Both residues were confirmed by mutagenesis studies5, 6, 7 as playing essential roles in P2Y1R activation. To validate whether this observation is a unique feature of agonist‐bound P2Y1R, we analyzed two antagonist‐bound systems, namely P2Y1R‐MRS2500 and P2Y1R*‐BPTU (Figure S3 A,C). We found that this ionic lock was stabilized throughout the entire MD simulation in both P2Y1R‐MRS2500 and P2Y1R*‐BPTU, implying that the ionic lock is crucial for P2Y1R activation.
We noted that the size of the binding pocket in the agonist‐bound system differed from those in the antagonist‐bound states. To investigate the consequences of ligand binding, we extracted frames of the final 0.5 μs from each simulated system and analyzed its solvent accessible surface area (SASA; Figure 2). We found that the binding pocket of agonist‐bound systems (Figure 2 B,D) were distinctly larger than those of the antagonist‐bound states (Figure 2 A,C). Specifically, the SASA in P2Y1R*‐BPTU was about 3300 Å2, slightly smaller than that of P2Y1R‐MRS2500 with a value of 3500 Å2 (Figure 2 E). However, when P2Y1R bound the agonist molecule ADP, the SASA increased to 4050 and 3800 Å2 in agonist‐bound P2Y1R*‐ADP and P2Y1R‐ADP respectively, owing to the broken D204ECL2‐R3107.39 ionic lock.
Figure 2.
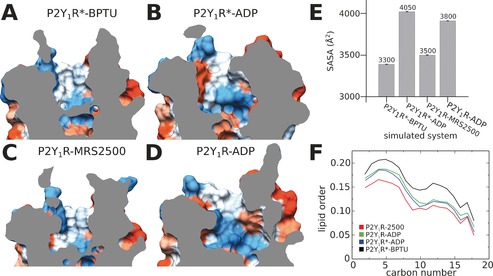
Solvent‐accessible surface area (SASA) and lipid order of P2Y1R at the end of MD simulations. The binding pocket of P2Y1R*‐BPTU (A), P2Y1R*‐ADP (B), P2Y1R‐MRS2500 (C), and P2Y1R‐ADP (D). E) SASA values of P2Y1R calculated from the final 0.5 μs MD simulations. F) Lipid order parameters averaged over the final 0.5 μs MD simulations.
Because the antagonist molecule BPTU is exposed to the lipid environment between TM2 and TM3, we investigated whether the binding of ligands affects the lipid order (Figure 2 F). Interestingly, the lipid order was as low as 0.15 when the antagonist MRS2500 bound to the classic extracellular vestibule of P2Y1R. However, when the antagonist BPTU bound to the lipid vicinity around the TM2‐TM3 bundles, the lipid order increased to 0.20. Moreover, in the two agonist‐bound systems P2Y1R*‐ADP and P2Y1R‐ADP, the lipid orders were identical, with a value of about 0.15. In addition, we found that the b‐factor in the extracellular region of P2Y1R*‐BPTU was noticeably smaller than in both MRS2500‐ and ADP‐bound P2Y1R, which correlates with our lipid order observations (Figure S4). From these combined findings, we reached the following conclusions: When the allosteric antagonist BPTU attaches to the helix bundles, it stabilizes the receptor, which leads to the increased lipid order. In contrast, when MRS2500 binds to the classic extracellular vestibule, the lipid order remains relatively low and the antagonist executes its function by occupying the binding pocket. Finally, when the ADP agonist binds to the receptor, the lipid order becomes altered owing to TM movements after activation.
Water molecules have been shown to play important roles during the GPCR activation process.6f, 9 In this work, we also observed distinct water channels inside the receptor (Figure S5). In antagonist‐bound P2Y1R*‐BPTU (Figure S5 A,E), the extracellular ligand‐binding pocket was filled with water molecules. However, in the intracellular zone next to the highly conserved Y3247.53, a hydrophobic layer with a thickness of 8 Å was observed. About 12 water molecules were found within 5 Å of Y3247.53 in P2Y1R*‐BPTU. In contrast, when the agonist ADP bound to the same structure (P2Y1R*‐ADP), it induced a continuous water channel in this region (Figure S5 B,F) and the number of water molecules increased to 24 in the corresponding space. In antagonist‐bound P2Y1R‐MRS2500, the number of water molecules stabilized at about 12 and a hydrophobic layer around 10 Å was observed. However, in the corresponding agonist system P2Y1R‐ADP, the number of water molecules increased to 21 and a continuous water channel also formed at the end of the MD simulations.
Furthermore, we noted that a highly conserved Tyr residue, Y3247.53 at the NPxxY motif, underwent a conformational switch in agonist‐bound P2Y1R (Figure S6). This occurred at 0.8–1.2 μs MD time scale. However, in both bound antagonists P2Y1R*‐BPTU and P2Y1R‐MRS2500, the conformation of Y3247.53 did not change during the entire simulations. These results agree with previous findings9 showing that Y3247.53 plays a role as a switch, forming a continuous water channel during GPCR activation.
We also noted that the b‐factor of intracellular helix bundles (Figure S4 B,D) of agonist‐bound systems was much higher than that of antagonist‐bound systems (Figure S4 A,C), implying that the intracellular helices undergo considerable movement in the activated state. In agonist‐bound P2Y1R, we found that several trans‐membrane helixes underwent striking shifts including: TM3, ≈2 Å; TM5, ≈4 Å; TM6, ≈7 Å; and TM7 ≈5 Å (Figure 3). Because TM3, TM6, and TM7 directly contact the Gα protein and exert an essential role in GPCR activation,10 we plotted the distances between different pairs of helices (Figure 3 B). This strategy identified three distinct states of P2Y1R activation: the inactive, the intermediate, and the active states (Figure 3 B). In the inactive state, the average distances between each TM region were low: d(TM3‐TM6), 7 Å; d(TM6‐TM7), 5 Å; and d(TM7‐TM3), 12 Å. In the intermediate state, the average distances between each TM region increased slightly: d(TM3‐TM6), 10 Å; d(TM6‐TM7), 8 Å; and d(TM7‐TM3), 14 Å. However, in the active state, the average distances between each TM region increased: d(TM3‐TM6), 14 Å; d(TM6‐TM7), 14 Å; and d(TM7‐TM3), 16 Å.
Figure 3.
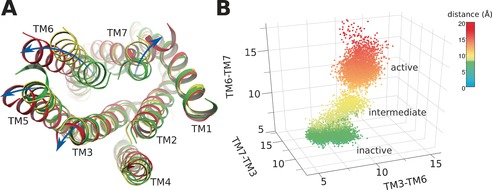
Helix movements of agonist‐bound P2Y1R. A) Helix positions of inactive (green), intermediate (yellow), and activated (red) P2Y1R. B) Distances between transmembrane helix TM3, TM6, and TM7 in inactive (green), intermediate (yellow), and activated (red) receptor.
Interestingly, the helix movements led to obvious changes in the residue interaction network (Figure S7). Analyses of the interactions between residue side chains revealed that in both antagonist‐bound complexes P2Y1R*‐BPTU and P2Y1R‐MRS2500, most of the residues inside the receptor were in contact with multiple neighbors (Figure S7 A,C). In contrast, in the agonist‐bound complexes P2Y1R‐ADP and P2Y1R*‐ADP, (Figure S7 B,D), interactions between the side chains inside the receptor were disrupted by helix shifting (Figure 3) and the accompanying water influx (Figure S5), dispersing the side chain interactions into smaller local groups indicative of larger inner void spaces. Residues without any contacts were found preferentially on surfaces or in loops of the receptor.
To aid in further interpretation and quantification of state‐specific couplings, we constructed correlation networks in which each node represents a cluster of protein residues, and each connecting edge is weighted by the correlation value between the two clusters (Figure 4). This approach has been used successfully to unravel allosteric couplings in a range of systems.11 In both antagonist‐bound P2Y1Rs (Figure 4 A,C), there were more nodes than in the agonist‐bound systems: 13 nodes were found in P2Y1R*‐BPTU, and the same number in P2Y1R‐MRS2500.
Figure 4.
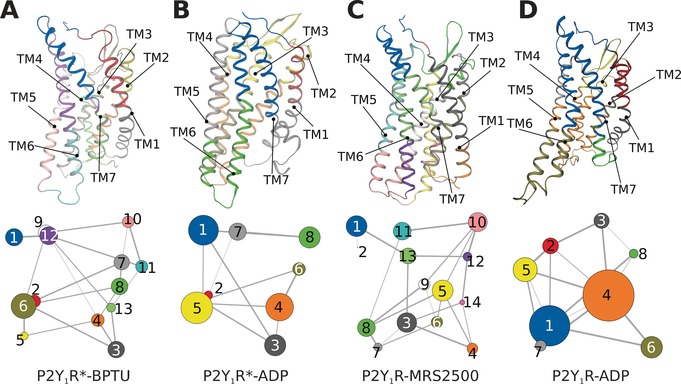
Simultaneous view of the community residue interaction network and 3D structures of A) P2Y1R*‐BPTU, B) P2Y1R*‐ADP, C) P2Y1R‐MRS2500, and D) P2Y1R‐ADP.
However, in both agonist systems P2Y1R*‐ADP and P2Y1R‐ADP, only 8 nodes were observed. Moreover, there were fewer large nodes in antagonist systems than in agonist systems because several smaller nodes had merged after agonist binding. Specifically, the separate nodes in intracellular TM6‐TM7 were grouped into a single node in the agonist‐bound systems. Similar phenomena also occurred at both the extracellular section of TM6‐TM7 and the intracellular section of TM2‐TM4. These observations are consistent with the MD simulation results showing that intracellular TM6 and TM7 undergo the same collective motions.
In summary, on the basis of a total of 20 μs all‐atom long‐timescale MD simulations (Figure 5), we found that both antagonist molecules can stabilize an ionic lock between K461.46 and R195ECL2 inside the receptor via different mechanisms: BPTU acts as an antagonist by stabilizing the extracellular helix bundles to increase the lipid order, whereas MRS2500 blocks signaling by occupying the ligand‐binding site. The SASA of both antagonist‐bound systems were smaller than the comparable agonist‐bound systems. When the ADP agonist bound to the P2Y1R, it induced breakage of the ionic lock and increased the SASA, inducing a bulk water influx into the binding pocket. Consequently, the rotamer of highly conserved Y3247.53 underwent a molecular switch and a continuous water channel formed inside the receptor. Finally, TM3, TM6, and TM7 shifts in the cytoplasmic region created a large void for G protein binding with subsequent activation of the receptor. These findings provide new insights into the molecular changes and actions of GPCR allosteric ligands. Such insights are applicable to innovative drug discovery.
Figure 5.
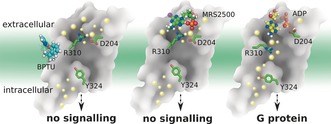
Molecular mechanism of action inferred from the P2Y1R structure. The big dots represent water molecules close to the front on the Figure, whereas the smaller dots indicate water molecules further away. Left panel: Antagonist molecule BPTU binds to P2Y1R and stabilizes the ionic lock between K461.46 and R195ECL2 inducing a decrease in the lipid order. No continuous internal water channel is formed. Middle panel: Antagonist molecule MRS2500 binds to P2Y1R and stabilizes the ionic lock between K461.46 and R195ECL2. No continuous internal water channel is formed. Right panel: Agonist molecule ADP binds to P2Y1R and disrupts the ionic lock between K461.46 and R195ECL2. Y3247.53 undergoes a molecular switch that results in the formation of a continuous water channel inside the receptor.
Experimental Section
MD simulations. All membrane systems were built with the membrane building tool in Schrodinger Maestro12 software with each receptor structure pre‐aligned in the OPM (Orientations of Proteins in Membranes) database.13 Pre‐equilibrated 128 POPC lipids coupled with 9,800 TIP3P water molecules in a periodic box of 70 Å×70 Å×96 Å were used to build the protein/membrane system. Proteins, lipids, water molecules and ions were modelled with the CHARMM36 force field14 parameter set, and the ligands were modelled with the CHARMM CGenFF small molecule force field.15 All of the ligands were submitted to the GAUSSIAN 09 program16 for structure optimization at the B3LYP/6‐31G* level prior to force field parameter generation. All of the bond lengths to hydrogen atoms in each protein/membrane system were constrained with M‐SHAKE.17 Van der Waals and short‐range electrostatic interactions were cut off at 10 Å. Results obtained from the MD simulations were analyzed in Gromacs18 and VMD.19
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Interdisciplinary Centre for Mathematical and Computational Modelling in Warsaw (grant G07‐13), providing the major computing part of this project (grant BIO120006P). Part of the work was performed at the Pittsburgh Supercomputing Center in the USA for use of the Anton supercomputer system (NIH grant R01GM116961). S.F. received funding from the National Center of Science, Poland, grant no. 2013/08/M/ST6/00788. Research in H.V.′s group was supported by the European Community (project SynSignal, grant no. FP7‐KBBE‐2013‐613879), and internal funds of the EPFL. K.P. was supported by the Arnold and Mabel Beckman Foundation. H.V. and S.F. participate in the European COST Action CM1207 (GLISTEN). We also thank the Canadian Institute for Advanced Research (CIFAR). K.P. is the John H. Hord Professor of Pharmacology.
S. Yuan, H. C. S. Chan, H. Vogel, S. Filipek, R. C. Stevens, K. Palczewski, Angew. Chem. Int. Ed. 2016, 55, 10331.
Contributor Information
Dr. Shuguang Yuan, Email: shuguang.yuan@gmail.com.
Prof. Raymond C. Stevens, Email: stevens@usc.edu
Prof. Krzysztof Palczewski, Email: kxp65@case.edu
References
- 1.
- 1a. Wootten D., et al., Nat. Rev. Drug Discovery 2013, 12, 630; [DOI] [PubMed] [Google Scholar]
- 1b. Li J., et al., J. Am. Chem. Soc. 2013, 135, 8749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Rose A. S., et al., PLoS One 2015, 10, e0143399. [Google Scholar]
- 2. Zhang D., et al., Nature 2015, 520, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gachet C., Thromb. Haemostasis 2008, 99, 466. [DOI] [PubMed] [Google Scholar]
- 4. Hechler B., et al., J. Pharmacol. Exp. Ther. 2006, 316, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chao H., et al., J. Med. Chem. 2013, 56, 1704. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Lakkaraju S. K., et al., J. Comput. Chem. 2016, 37, 416; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Schneider M., et al., FEBS Lett. 2011, 585, 3587; [DOI] [PubMed] [Google Scholar]
- 6c. Rose A. S., et al., J. Am. Chem. Soc. 2014, 136, 11244; [DOI] [PubMed] [Google Scholar]
- 6d. Nygaard R., et al., Cell 2013, 152, 532; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Yuan S., et al., Angew. Chem. Int. Ed. 2015, 54, 556; [Google Scholar]; Angew. Chem. 2015, 127, 566; [Google Scholar]
- 6f. Yuan S., et al., Nat. Commun. 2014, 5, 4733. [DOI] [PubMed] [Google Scholar]
- 7. Moro S., et al., Biochemistry 1999, 38, 3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Guo D., et al., Drug Dev. Res. 2002, 57, 173; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Moro S., et al., J. Med. Chem. 1998, 41, 1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Yuan S., et al., Angew. Chem. Int. Ed. 2015, 54, 7560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7670; [Google Scholar]
- 9b. Yuan S., et al., Angew. Chem. Int. Ed. 2013, 52, 10112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10299; [Google Scholar]
- 9c. Liu W., et al., Science 2012, 337, 232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Angel T. E., et al., Proc. Natl. Acad. Sci. USA 2009, 106, 14367;19706523 [Google Scholar]
- 9e. Garczarek F., Gerwert K., Nature 2006, 439, 109; [DOI] [PubMed] [Google Scholar]
- 9f. Yuan S., et al., Angew. Chem. Int. Ed. 2016, 55, 8661; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8803. [Google Scholar]
- 10.
- 10a. Trzaskowski B., et al., Curr. Med. Chem. 2012, 19, 1090; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Flock T., et al., Nature 2015, 524, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Karami Y., et al., BMC Bioinf. 2016, 17, 13; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Skjaerven L., et al., BMC Bioinf. 2014, 15, 399; [Google Scholar]
- 11c. Yuan S., et al., Structure 2016, 24, 816. [DOI] [PubMed] [Google Scholar]
- 12.Schrödinger, Schrödinger, LLC, 2015.
- 13. Lomize M. A., et al., Bioinformatics 2006, 22, 623. [DOI] [PubMed] [Google Scholar]
- 14. Klauda J. B., et al., J. Phys. Chem. B 2010, 114, 7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vanommeslaeghe K., et al., J. Chem. Inf. Model. 2012, 52, 3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.M. J. Frisch, et al., Gaussian, Inc., Wallingford, CT, USA, 2009.
- 17. Schmauder R., et al., ChemBioChem 2011, 12, 2431. [DOI] [PubMed] [Google Scholar]
- 18. Pronk S., et al., Bioinformatics 2013, 29, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Humphrey W., et al., J. Mol. Graphics Modell. 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary