

Abstract
This paper presents our adaptation of Fryback and Thornbury’s hierarchical scheme for modeling the efficacy of diagnostic imaging systems. The original scheme was designed to evaluate new medical imaging systems but is less successful when applied to evaluate new radiopharmaceuticals. The proposed adaptation, which is specifically directed toward evaluating targeted imaging agents, has 6 levels: in vitro characterization, in vivo animal studies, initial human studies, impact on clinical care (change in management), impact on patient outcome, and societal efficacy. These levels, particularly the first four, implicitly define the sequence of studies needed to move an agent from the radiochemistry synthesis laboratory to the clinic. Completion of level 4 (impact on clinical care) should be sufficient for initial approval and reimbursement. We hope that the adapted scheme will help streamline the process and assist in bringing new targeted radiopharmaceuticals to approval over the next few years.
Keywords: efficacy, evaluation, radiopharmaceutical, FDA approval process
In 1991, Fryback and Thornbury (1) published an important paper that defined a hierarchical scheme for modeling the efficacy of diagnostic imaging systems. This 6-level scheme, which has become a widely accepted guideline for the evaluation of new diagnostic tests, has 6 levels: technical efficacy, diagnostic accuracy, diagnostic thinking, therapeutic efficacy, patient outcomes, and societal efficacy.
Fryback and Thornbury described a systematic approach to establishing the diagnostic accuracy of an imaging test, its impact on therapeutic efficacy, and, eventually, its benefit to society. About 10 years after the publication of their influential paper, the concept of molecular imaging was introduced, and during the last 15 years it has grown to become a major focus of imaging research. Although several definitions of molecular imaging probes have been proposed, an essential goal of molecular imaging is repetitive and quantitative assessment of the expression or function of molecular targets. Detecting the presence of a certain disease (diagnostic accuracy) remains an important goal of molecular imaging, but it also addresses several other clinical problems, such as assessing prognosis, predicting and monitoring response to molecularly targeted interventions, and evaluating the distribution and binding occupancy of receptors. The important concepts introduced by Fryback and Thornbury are only partially applicable to the evaluation of molecular imaging. Conversely, validation of molecular imaging requires additional steps that were not described by Fryback and Thornbury. Several modifications of the Fryback and Thornbury scheme have been published, but none has addressed the evaluation of new radiopharmaceuticals (2). An adaptation of the original scheme needs to be defined to help developers and regulators understand how to evaluate the efficacy of these agents.
The agents requiring this new scheme are those targeting a specific receptor or metabolic pathway in tissue where the imaging results can make a major difference in how a patient will be treated. Examples include PET and SPECT agents binding to cell surface receptors of malignant tumors and ligands binding to amyloid deposits in the cerebral cortex. Often, the goal is to identify tumors with high levels of receptor expression that can then be treated with a similarly targeted therapeutic agent. This concept is often referred to as theranostics. Another goal is to monitor the pharmacodynamic effects of targeted drugs in order to predict and determine whether there is a response to therapy.
Because the targeted imaging approach goes beyond the type of diagnostic imaging envisioned by Fryback and Thornbury, the hierarchical scheme needs to be significantly modified to allow for efficient and appropriately designed clinical trials of molecular imaging. In making this modification, we have retained the underlying philosophy, starting with fundamental assessment of the technical details of the test, then moving to approaches for evaluating the test in clinical practice, and finally looking at the impact of the test on patient outcome and on society.
The hierarchy suggested in this paper (Table 1) follows the temporal sequence needed to develop a targeted agent: initial evaluation in the laboratory, studies on animals, human studies to obtain regulatory approval, and application to clinical management. The suggested approach is not intended to be comprehensive but recommends the most important steps at each level and identifies testing that is inappropriate or not feasible. The approach takes into account the significant preclinical evaluation needed to ensure that a molecularly targeted imaging probe visualizes and quantifies its specific target. The new hierarchy also reduces emphasis on diagnostic accuracy studies, because the quantitative nature of molecular imaging goes beyond the binary classification of presence or absence of disease that is fundamental to the concept of diagnostic accuracy.
TABLE 1.
Comparison of Schemes
| Level | Proposed scheme | Fryback and Thornbury scheme |
|---|---|---|
| 1 | In vitro characterization | Technical efficacy |
| Kon, Koff, Kd, Bmax, and IC50 Partition coefficient and binding potential |
||
| Labeling efficiency and yield | ||
| In vitro label stability | ||
| 2 | In vivo animal studies | Diagnostic accuracy |
| In vivo stability | ||
| Target vs. nontarget tissue specificity | ||
| Pharmacokinetics | ||
| Radiochemistry optimization | ||
| Dosimetry and toxicity | ||
| 3 | Initial human studies | Diagnostic thinking |
| Safety, dosimetry, and target specificity | ||
| Tracer stability in vivo | ||
| Pharmacokinetics (including metabolites) | ||
| Reproducibility | ||
| Determination of sensitivity, specificity, PPV, and NPV IND application | ||
| Chemistry, manufacturing, and controls development | ||
| 4 | Impact on clinical care (change in management) | Therapeutic efficacy |
| Diagnosis (patients with suspicion of disease) | ||
| Staging (patients with known disease) | ||
| Response to therapy (imaging before and after therapy) | ||
| Evaluation for targeted therapy | ||
| Current-good-manufacturing-practices implementation | ||
| 5 | Impact on patient outcome | Patient outcomes |
| Assessment of implementation of change in management | ||
| Assessment of correctness of change in management | ||
| Survival with and without test (Kaplan–Meier plots) | ||
| Quality-adjusted life years | ||
| 6 | Societal efficacy | Societal efficacy |
| Cost-benefit analysis | ||
| Risk-benefit analysis | ||
| Postapproval monitoring for side effects |
Kon = binding rate constant; Koff = release rate constant; Kd = dissociation constant; Bmax = maximum number of binding sites; IC50 = inhibitory concentration of 50%; NPV = negative predictive value; PPV = positive predictive value.
Another reason for deemphasizing the concept of diagnostic accuracy is that in oncologic imaging there is frequently no unbiased reference standard against which to base the presence or absence of disease. As a consequence, for many important applications of molecular imaging, diagnostic accuracy cannot be determined in an unbiased way.
Our main goal in writing this paper is to provide a clear pathway for efficient development, evaluation, and application of targeted imaging agents. The process is not a simple one, and there is a definite need to understand it better and develop better strategies toward it with the goal of accelerating the clinical application of new agents.
LEVEL 1. IN VITRO CHARACTERIZATION
In the Fryback and Thornbury hierarchy, level 1 is “technical efficacy.” It is concerned with image quality (resolution, modulation transfer function), a type of assessment that is inappropriate for imaging agents. Instead, for a new imaging agent this level is concerned with the preliminary detailed in vitro characterization essential to demonstrate that the agent is likely to bind to the target of interest. Initial studies are usually done in cell-free systems, such as columns with bound receptors. More complex studies require cell suspensions or tissue cultures. Chemical and metabolic stability can be studied by incubation in buffer, cell medium, and blood or plasma, after which the degradation products, including free radionuclide, can be detected, identified, and quantified. A recent paper by Wynendaele et al. contains a detailed description of many additional aspects of in vitro characterization of radiopharmaceuticals (3).
LEVEL 2. IN VIVO ANIMAL STUDIES
In the Fryback and Thornbury hierarchy, level 2 is “diagnostic accuracy,” with the major emphasis being on determining test accuracy in terms of sensitivity and specificity, including use of receiver-operating-characteristic curves.
The sensitivity and specificity of imaging tests have been extensively studied in the literature. Calculation of sensitivity and specificity is appropriate if there is an established gold reference standard that can be compared with the results of the imaging test. An example is the evaluation of solitary pulmonary nodules by 18F-FDG PET using histology as the reference standard. However, for whole-body tumor staging, one of the most common applications of imaging tests, histology generally cannot be used to evaluate the accuracy of all sites that are considered positive or negative by a novel imaging modality. Therefore, a common approach has been to use as a reference standard a consensus interpretation of all available imaging studies (the new imaging modality and the conventional imaging tests combined). However, histologic evaluation can be performed only for sites that are positive on at least one imaging test or perhaps a limited number of sites that are negative on imaging. Consequently, there is always a verification bias that causes a systematic overestimation of both sensitivity and specificity. Therefore, the concept of sensitivity and specificity is problematic. For example, it is a common observation that the sensitivity of novel imaging techniques systematically decreases over time. The sensitivity of 111In-octreotide imaging for neuroendocrine tumors was described to be close to 100% when the technology was introduced in the 1990s (4). However, in a more recent study comparing 111In-octreotide SPECT with 68Ga-DOTATOC PET/CT, its sensitivity was only about 50%. This striking difference is obviously due to the fact that, in the more recent paper, 111In-octreotide SPECT was compared with a more sensitive imaging modality and that therefore the denominator for calculation of sensitivity markedly increased (5).
An additional problem of using the terms sensitivity and specificity for cancer staging is that both are dependent on the number of sites analyzed, as illustrated by many studies evaluating PET/CT for lymph node staging. Imaging findings can be correlated with the presence or absence of lymph node metastases on a whole-patient basis, on the location of lesions in relatively large regions of lymph nodes (e.g., left and right sides of the pelvis) or in smaller regions of lymph nodes (e.g., left internal iliac nodes), or on individual potentially involved nodes. When the size of the analyzed sites decreases, their total number generally increases. As a consequence, the specificity of an imaging test will be higher if smaller regions of lymph nodes are analyzed, because the number of false-positive findings will be divided by a larger total number of true-negative sites.
Even more fundamental, there can be cases that can be considered true-positive and false-negative at the same time. For example, consider a 11C-choline PET/CT scan that shows a 11C-choline–avid left internal iliac node in a patient with biochemical recurrence of prostate cancer. Let’s also assume that the patient then undergoes salvage lymphadenectomy, which finds a lymph node metastasis in the left internal and left common iliac regions. On a patient basis, the scan is true-positive for lymph node metastasis. The scan is also true-positive if lymph nodes are classified as “left and right iliac nodes,” but the study is false-negative and true-positive if the iliac lymph node regions are subdivided into external and internal iliac nodes.
In summary, determination of sensitivity requires knowledge of all true-positive sites of disease. For whole-body cancer staging, positive sites can be identified only by imaging tests, making estimates of sensitivity and specificity inherently biased. With improvements in instrumentation, sensitivity and specificity consequently change over time.
Calculation of specificity requires knowledge of all true-negative sites. Because the number of true-negative sites is most of the body, specificity becomes critically dependent on how many regions the body is divided into. It is possible to determine specificity on an individual patient basis, but like sensitivity, specificity is dependent on the capabilities of the instrumentation used as the reference and cannot be accurately measured.
In the proposed hierarchy, the emphasis at this level is not on sensitivity and specificity but on accurate characterization of the behavior of the new agent in animals before human administration. A potential pitfall with these studies is that the agent may behave quite differently in animals and in humans. Accordingly, it may be best to move rapidly to early human studies rather than investigating multiple animal species.
The essential testing done at this level is defined in the subheadings listed in Table 1: in vivo stability, target specificity, pharmacokinetics, radiochemistry optimization, radiation dosimetry, and toxicity.
In vivo stability is essential. There are numerous circulating enzymes in blood that may rapidly degrade the new agent. Slow degradation may be acceptable but has to be defined, and the behavior of the metabolites should be characterized.
Target specificity is also essential. This is very different from test performance specificity. Target specificity of a novel targeted agent is difficult to assess in vitro, and only when it is injected and imaged in an animal will it become clear if there is a high target-to-background ratio. Additional studies are also often needed to show lack of uptake in tissue that lacks the target receptor. Target specificity is usually evaluated with transplanted tumors in immunoincompetent mice. Ideally, one type of tumor has receptors and shows good uptake whereas another type of similar tumor lacking receptors shows no uptake. This type of study firmly demonstrates agent specificity and makes it unlikely that increased capillary permeability will be the primary mechanism of uptake. The other major approach for demonstrating specificity is blocking uptake using relatively high levels of a known receptor targeting ligand—often, a stable nonradioactive version of the study agent. In such studies, the high dose may have physiologic effects that can perturb the delivery of the probe molecule by effects other than simply blocking of the receptors.
Pharmacokinetics is quite important. How rapidly the agent is taken up into the target and how rapidly it is excreted will define the timing of imaging and also the limits for the acceptable half-life of the radioactive label. If the agent takes days to localize, labeling it with a short-lived isotope is not feasible. Pharmacokinetics should also include identification of metabolites, particularly those that retain the radioisotope label. Because labeled metabolites can represent a significant fraction of the detected radioactivity, their appearance and time course should be determined.
Both radiation dosimetry studies and toxicity studies are essential before an agent can be injected into humans. Because most of these agents are injected at or below microgram levels, it may be practically impossible to reach truly toxic levels even in small animals such as mice. Therefore, it is acceptable to show no acute toxicity in one species (usually mice) with 100 times the dose (mg/kg) likely to be used in humans and when the anticipated human dose is less than 100 μg (6,7).
LEVEL 3. INITIAL HUMAN STUDIES
In the Fryback and Thornbury hierarchy, level 3 is “diagnostic thinking efficacy,” a relatively vague and difficult-to-quantitate concept since it addresses how the test results change the thinking of the referring clinicians. In our proposed adapted hierarchy, this level is concerned with demonstrating the safety of the agent in humans and making a preliminary demonstration of efficacy. Such phase 1 studies are typically closely monitored and may be conducted on patients or healthy volunteers. These studies are designed to determine the metabolism and pharmacokinetics of the drug in humans, to uncover any side effects, and, if possible, to gain early evidence of effectiveness (8).
An important part of these studies usually consists of measuring vital signs, performing electrocardiography, and obtaining a limited panel of blood chemistries before the study and then at one or more time points afterward. It is also important to ask the subjects whether they experienced any symptoms immediately after the injection or later. It should not be necessary to continue following up the subjects for longer than about 5 half-lives of the injected agent. The follow-up period should be based on the biologic half-life of the nonradioactive agent, not the physical half-life of the radioisotope label. The selection of blood tests should reflect reasonable postulated toxicity based on the structure of the compound and on the animal toxicity study.
Human dosimetry can be performed using quantitative pharmacokinetic PET data and is necessary before proceeding to phase 2 clinical efficacy studies. Collection of data and calculation of radiation dose to various organs and to the whole body can be accomplished once quantitative PET data have been obtained at several time points after injection. Similar studies can be done with SPECT, although the methodology is more challenging and accuracy may be lower.
Target specificity for agents can be defined as the ratio of uptake in target tissue to uptake in normal tissue. Most new agents have relatively high target-to-background ratios. This ratio is particularly important if there is a companion theranostic agent, if the ligand is labeled with a β- or α-emitter, and the intention is delivery of a high radiation dose to the target while avoiding an unacceptable radiation dose to normal tissue.
Pharmacokinetic studies involve collection of blood time–activity data, derived from sampling, and of target and normal-tissue data, typically derived from sequential quantitative imaging. These data are used to calculate radiation dosimetry, to determine the optimal imaging time after injection, and to determine the appropriate injected dose for the radiopharmaceutical.
Determining the reproducibility of the quantitative behavior of a new agent is essential before attempting to use the agent for assessing response to therapy. Once reproducibility is determined, it is possible to define the degree of change in uptake that is significant and often shows improvement or progression of disease. Typically, such studies are done by repeating imaging of the same subject within a few days, with no therapy during the interval.
Although initial biodistribution studies of radiopharmaceuticals previously used in humans can be performed under Radioactive Drug Research Committee oversight, a Food and Drug Administration (FDA) Investigational New Drug Application (IND) will need to be filed and approved before clinical research can be conducted. First-in-human studies require an IND or an exploratory IND. An essential part of the IND is a section on chemistry, manufacturing, and controls. Documentation for this section should be developed at this level. The IND application needs to include any available information on safety and dosimetry; the section on chemistry, manufacturing, and controls; and a study protocol that describes the proposed trial design in detail.
LEVEL 4. IMPACT ON CLINICAL CARE (CHANGE IN MANAGEMENT)
In the Fryback and Thornbury hierarchy, level 4 is “therapeutic efficacy.” It was essentially defined as the fraction of tests that resulted in a change in management. Similarly, in the new scheme at this level the emphasis is on change in management in several specific settings.
Studies at this level are phase 2 and 3 clinical trials (8). The FDA expects diagnostic PET drugs to be produced under the rules associated with good manufacturing practices for phase 3 clinical trials and for subsequent manufacturing for marketing of the new drug.
Diagnosis
The test may be useful in patients with symptoms, laboratory findings, and imaging results that suggest the presence of the target disease. The measure of diagnostic efficacy is yield, that is, the fraction of patients studied with the new agent who are found to have the target disease, subsequently resulting in initiation of therapy.
Staging
In patients with known malignant disease, the new agent may be useful for staging, that is, accurately defining the extent and location of disease. Because of the problems with determining sensitivity and specificity in this setting, we propose that existing and new imaging tests be compared for discrepant findings that lead to a change in management. For example, if the new imaging test detects a bone metastasis that will change treatment from curative to palliative, a biopsy of this metastasis should be performed and used as the reference for comparison of the two tests. Findings that are concordantly positive or have no impact on patient management do not need to undergo further evaluation. The discrepantly positive and negative findings of the new and existing imaging tests can be checked for statistical significance by the McNemar test, the result of which depends only on the discrepant cases. Therefore, no reference standard is needed for sites that are concordantly positive or negative. The McNemar test for lesions that are positive on histology but discrepant on the two imaging modalities will reveal whether the new modality is significantly more sensitive than the existing modality. Conversely, the McNemar test for all lesions that are negative on histology will reveal whether the new modality is more specific than the existing modality. Thus, improvements in sensitivity over existing imaging technologies can be determined even if the absolute sensitivity and specificity are unknown.
Demonstration of accurate identification of abnormal tissue is often sufficient to show that imaging with a new agent will result in a change in management. A recent paper written by FDA personnel (9) stated: “The FDA imaging product guidance recognizes how the clinical usefulness of some imaging information may be obvious in certain clinical settings, such as the staging of cancer or the detection of clinically important pathology.” The paper additionally states: “In such situations, imaging drug developers are not expected to perform clinical studies that demonstrate again the clinical benefit of the imaging information.”
Response to Therapy
A significant advantage of PET imaging is that uptake can be determined quantitatively. This capability lends itself to assessing response to therapy by quantitative comparison of uptake before therapy to uptake at some time afterward. The optimum timing of the follow-up studies and the criteria for response have to be determined in appropriate clinical trials. The major rationale for such studies is that if the studies show lack of response to therapy, another treatment regimen can be implemented. Early identification of lack of response may benefit patients by limiting the duration they are exposed to ineffective but potentially toxic drugs. The reference standard in this setting is often change in tumor size, as seen with anatomic imaging using RECIST (10). In addition, survival of patients classified as responders or nonresponders can be compared.
Monitoring tumor response to therapy is related to assessing the pharmacodynamic effects of targeted drugs. For example, blocking of androgen receptors by antiandrogens can be imaged with 18F-labeled dihydrotestosterone. The reference standard in this setting can be biopsies showing downregulation of target-dependent signaling pathways that correlate with a decrease in uptake of the imaging agent.
Evaluation for Targeted Therapy
New diagnostic targeted agents are often developed in conjunction with a companion therapeutic agent that differs only in the radioisotope label, for example, 68Ga-DOTATATE and 177Lu-DOTATATE for diagnosis and treatment of neuroendocrine tumors. Diagnostic imaging with the targeted agent is essential before administration of the therapeutic companion to ensure high uptake into the target and acceptable uptake into normal tissue. In this setting, accurate quantitation of radiotracer uptake and calculation of radiation dose with a clinically feasible imaging protocol is the key outcome parameter, not the sensitivity and specificity for detection of metastases.
LEVEL 5. IMPACT ON PATIENT OUTCOME
In the Fryback and Thornbury hierarchy, level 5 is “patient outcome efficacy.” This includes the fraction of patients whose outcome improved because of the test (compared with without the test), the fraction of patients in whom morbidity was avoided by undergoing the test, the change in quality-adjusted life years, and the cost per quality-adjusted life year saved (cost effectiveness). In the proposed new hierarchy, the goals are similar.
Assessment of Implementation of Change in Management
At level 4, the major goal is assessment of the frequency with which clinical management is changed in response to information obtained from the new test. Usually, this assessment is done by requiring the treating physician to record the treatment plan before the results of the new test are available and then to record the new plan once the results are available. This assessment is really looking at the change in intended management. At level 5, a more rigorous criterion is required, confirmation that the changes were actually implemented and were appropriate.
Assessment of Correctness of Change in Management
The correctness of a change in management is not easily determined. It requires either a panel of experts to assess the situation and determine the appropriateness of the change or follow-up to see how well the patient does after the change. Both approaches have inherent weaknesses. The experts may not have sufficient knowledge or information to accurately determine appropriateness in all settings, and follow-up cannot reveal what would have happened if the test had not been done. In addition, subsequent testing and changes in management may occur that are not related to the original diagnostic test.
It would be ideal to determine outcome (relapse-free survival, overall survival) in a randomized trial with patients who did and did not undergo the new test. The practical problems with implementation of this approach are lack of clinical equipoise and lack of control of subsequent treatment decisions. Many of these new agents show high target-to-background ratios, and after inspection of a few examples, it is often intuitively compelling that the new agent is superior to prior approaches, making it difficult to recruit subjects for a randomized clinical trial (RCT). Even if recruited, many subjects will attempt to have the new test done outside the scope of the trial, making a rigorous survival trial impossible. Another factor that affects the feasibility and meaningfulness of a survival trial is lack of control over subsequent treatment decisions. The only setting in which subsequent treatment is well controlled is a therapeutic clinical trial, but in such trials it would be problematic to introduce an experimental diagnostic agent that might bring about a change in management.
Although it would be ideal to be able to calculate cost per quality-adjusted life year, similar problems are encountered in that there is often a lack of uniformity in the treatment of patients during the months and years after they undergo imaging with the new agent. Thus, survival and quality of life may be only loosely related to the test results.
An important limitation of using survival as an endpoint is the large number of patients needed to demonstrate differences in outcome. An alternative treatment is unlikely to improve survival in all patients who do not respond to the first treatment. The fraction of patients who can potentially improve the outcome of the whole patient population therefore becomes small. For example, if 50% of the patients are classified as nonresponders by the new test and an alternative treatment improves survival in 20% of these patients, only 10% of the patients will ultimately benefit from the use of the new test to assess response. Studies with sufficient statistical power to detect an improvement in overall survival in such a setting will generally require randomization of several hundred patients. In addition, the results are likely to be confounded by patients in the control group who are identified as nonresponders by conventional imaging at a later time. These patients are likely to receive alternative treatments as well, and some of these patients will likely benefit from the alternative therapy.
In practice, the only feasible way to make meaningful conclusions about survival and cost effectiveness is to model the probability of subsequent events after the test, given knowledge of the fraction of patients for whom there was a change in management and knowledge (or assumptions) about appropriate subsequent therapy decisions and outcomes (Fig. 1) (11).
FIGURE 1.
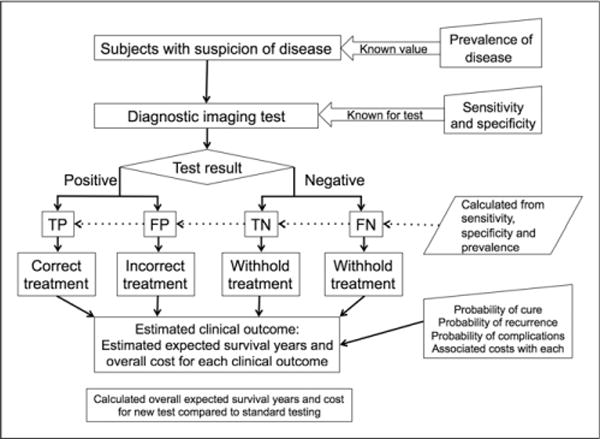
Scheme for modeling the outcomes expected for a new medical test. Although the process of defining disease incidence and test sensitivity and specificity is straightforward, it is more challenging to define treatment impact (probability of cure and of complications) and to estimate survival and costs for each test-result pathway. FN = false-negative; FP = false-positive; TN = true-negative; TP = true-positive.
LEVEL 6. SOCIETAL EFFICACY
It is certainly desirable and important to determine whether a new test is valuable at the societal level, particularly in extending useful life-span and in lowering overall health-care costs. Explicit determination of these measures is essentially impossible but may be feasible with appropriate modeling.
DISCUSSION
The goal of this paper is to present an organized consensus view of a logical and efficient approach for evaluation of the efficacy of new targeted radiopharmaceuticals. It is built on the framework of a 1991 publication, “The Efficacy of Diagnostic Imaging,” by Dennis G. Fryback and John R. Thornbury (1). The levels of the approach represent the sequence of studies necessary to move a new agent from the radiochemistry synthesis laboratory to the clinic. Completion of level 4 (impact on clinical care) should be sufficient for initial approval and reimbursement. Levels 5 (impact on patient outcome) and 6 (societal efficacy) should be addressed once a new agent becomes widely available.
Others have considered the issue of the optimal strategy for approval of diagnostic imaging agents. A major issue in prior discussions has been the question of the need for RCTs. This issue was considered at length by Valk in 2000 (12) and more recently by Hicks et al. in 2012 (13) in a critique of a paper on a review of RCTs in PET (14). Both papers (Valk’s and Hicks’) clearly make the point that RCTs are not necessary, feasible, or effective in the assessment of new radiopharmaceuticals.
Vach et al. (15) have also addressed this question and have considered the problem of “generating evidence for clinical benefit of PET/CT in diagnosing cancer patients.” They considered two different RCT designs but concluded that practical issues of clinical equipoise, time to conduct a trial, and the need for multiple trials to address all possible scenarios make the RCT approach frequently impractical. They proposed that decision modeling after determination of an actual change in management is an efficient way to generate evidence of clinical benefit. This approach depends on making reasonable assumptions about the management changes that were correct, as well as the expected benefit or detriment of a change in therapy for both correct and incorrect changes. If consensus can be reached between the medical researchers and the regulatory authorities on the validity of the assumptions, then it should be feasible to move forward to approval and reimbursement.
Although there is not complete agreement, there is consensus that appropriate observational studies, carefully done, can be sufficient to establish the safety and efficacy of a new agent. A Medical Imaging and Technology Alliance conference addressed this question, although with a more specific focus on research endpoints appropriate for Medicare coverage of new PET radio-pharmaceuticals (16). At the outset of the conference, there was general agreement on specific issues presented by Louis Jacques, who was then head of the Centers for Medicare and Medicaid Services Coverage and Analysis Group. A key principle from the conference was that the potential benefits of diagnostic tests relate to their providing information to optimize treatment plans and, thereby, improve clinical care and health outcomes. A key take-home point was that coverage of new PET radiopharmaceuticals should depend on clinical evidence of effect on intermediate endpoints, such as a beneficial change in clinical management (i.e., change in subsequent therapeutic or diagnostic interventions) that can be linked to improved health outcomes. These same principles should be applied to approval by the FDA, as well as coverage by the Centers for Medicare and Medicaid Services. Although the link to improved health outcomes could conceivably be made with RCTs or long-term observational studies, the only practical way to make the link is with well-designed decision modeling studies.
The design of trials to study changes in management depends on how the new agent is likely to be used in clinical practice. For example, in applications for cancer staging, an imaging agent may be used in any of 4 ways: to detect previously unknown disease and thus allow for treatment, to detect unsuspected distant disease and thus avoid a futile operation, to confirm that a patient is negative for disease and thus avoid unnecessary treatment, or to determine whether disease is so clinically insignificant as to not require treatment, such as would be the case for stable prostate or thyroid cancer. In addition, the study design needs to consider whether the test is replacing or being added to an existing test, the potential consequences of both positive and negative results, and whether intended changes were actually implemented. Although change in management is a potential powerful tool for assessing the efficacy of and need for a new agent, its measurement is not trivial and must be approached with care (17).
In addition to the class of molecular imaging agents intended to guide clinical management, there is a class of agents intended to assess the pharmacokinetic and pharmacodynamic behavior of novel therapeutic drugs during early development (18). This characterization can have a major effect on decision making about subsequent development of the new drug. These agents often include a radiolabeled version of the therapeutic drug or may be targeted at a specific metabolic pathway presumed to be blocked or stimulated by the therapeutic drug. Often, these agents are never intended for clinical use but are essential in the initial characterization of a new therapeutic drug. They need to be characterized carefully at levels 1–3 to demonstrate the validity of their behavior. Some may be specific for the study drug (e.g., showing the biodistribution and tumor uptake of a certain antibody), whereas others may be more generic (e.g., showing the expression of a target for multiple drugs, such as the density of free estrogen or androgen receptors).
CONCLUSION
Currently, several targeted radiopharmaceuticals are being developed by multiple academic and commercial groups throughout the United States and the world. Many of these agents have significant potential to make a real difference in how medicine is practiced in the future and are likely to be a major part of true personalized medicine. However, because of uncertainty and inconsistency regarding the optimum pathway from discovery to clinical application, the development of these agents is less efficient, more expensive, and slower than it should be. We hope that the suggestions presented in this paper will help streamline the process and assist in bringing many of these agents to approval over the next few years.
Acknowledgments
We thank the members of the SNMMI FDA Task Force for numerous comments and critiques during the formulation and writing of the manuscript. This paper has been formally endorsed by the SNMMI.
Footnotes
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
References
- 1.Fryback DG, Thornbury JR. The efficacy of diagnostic imaging. Med Decis Making. 1991;11:88–94. doi: 10.1177/0272989X9101100203. [DOI] [PubMed] [Google Scholar]
- 2.Lijmer JG, Leeflang M, Bossuyt PM. Proposals for a phased evaluation of medical tests. Med Decis Making. 2009;29:E13–E21. doi: 10.1177/0272989X09336144. [DOI] [PubMed] [Google Scholar]
- 3.Wynendaele E, Bracke N, Stalmans S, De Spiegeleer B. Development of peptide and protein based radiopharmaceuticals. Curr Pharm Des. 2014;20:2250–2267. doi: 10.2174/13816128113196660663. [DOI] [PubMed] [Google Scholar]
- 4.Shi W, Johnston CF, Buchanan KD, et al. Localization of neuroendocrine tumours with (111In) DTPA-octreotide scintigraphy (Octreoscan): a comparative study with CT and MR imaging. QJM. 1998;91:295–301. doi: 10.1093/qjmed/91.4.295. [DOI] [PubMed] [Google Scholar]
- 5.Gabriel M, Decristoforo C, Kendler D, et al. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nucl Med. 2007;48:508–518. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]
- 6.Guidance for industry, investigators, and reviewers: exploratory IND studies. U.S. Food and Drug Administration website. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078933.pdf. Published January 2006. Revised April 1, 2015. Accessed January 11, 2016.
- 7.Position paper on the non-clinical safety studies to support clinical trials with a single micro dose (CPMP/SWP/2599/02Rev 1) European Medicines Agency website. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000400.jsp. Published June 2004. Accessed January 11, 2016.
- 8.Code of Federal Regulations: title 21, volume 5, section 312.21. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.21. Accessed January 11, 2016.
- 9.Gorovets A, Marzella L, Rieves D, Yang L. Efficacy considerations for U.S. Food and Drug Administration approval of diagnostic radiopharmaceuticals. J Nucl Med. 2013;54:1479–1484. doi: 10.2967/jnumed.112.117804. [DOI] [PubMed] [Google Scholar]
- 10.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Gambhir SS. Decision analysis in nuclear medicine. J Nucl Med. 1999;40:1570–1581. [PubMed] [Google Scholar]
- 12.Valk PE. Randomized controlled trials are not appropriate for imaging technology evaluation. J Nucl Med. 2000;41:1125–1126. [PubMed] [Google Scholar]
- 13.Hicks RJ, Hofman MS, Ware RE. Not-so-random errors: randomized controlled trials are not the only evidence of the value of PET. J Nucl Med. 2012;53:1820–1822. doi: 10.2967/jnumed.112.111351. [DOI] [PubMed] [Google Scholar]
- 14.Scheibler F, Zumbe P, Janssen I, et al. Randomized controlled trials on PET: a systematic review of topics, design, and quality. J Nucl Med. 2012;53:1016–1025. doi: 10.2967/jnumed.111.101089. [DOI] [PubMed] [Google Scholar]
- 15.Vach W, Høilund-Carlsen PF, Gerke O, Weber WA. Generating evidence for clinical benefit of PET/CT in diagnosing cancer patients. J Nucl Med. 2011;52(suppl 2):77S–85S. doi: 10.2967/jnumed.110.085704. [DOI] [PubMed] [Google Scholar]
- 16.Hillman BJ, Frank RA, Abraham BC. The Medical Imaging & Technology Alliance conference on research endpoints appropriate for Medicare coverage of new PET radiopharmaceuticals. J Nucl Med. 2013;54:1675–1679. doi: 10.2967/jnumed.113.127886. [DOI] [PubMed] [Google Scholar]
- 17.Staub LP, Lord SJ, Simes RJ, et al. Using patient management as a surrogate for patient health outcomes in diagnostic test evaluation. BMC Med Res Methodol. 2012;12 doi: 10.1186/1471-2288-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthews PM, Rabiner EA, Passchier J, Gunn RN. Positron emission tomography molecular imaging for drug development. Br J Clin Pharmacol. 2012;73:175–186. doi: 10.1111/j.1365-2125.2011.04085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]