

Abstract
Motile airway cilia that propel contaminants out of the lung are oriented in a common direction by planar cell polarity (PCP) signaling, which localizes PCP protein complexes to opposite cell sides throughout the epithelium to orient cytoskeletal remodeling. In airway epithelia, PCP is determined in a 2-phase process. First, cell-cell communication via PCP complexes polarizes all cells with respect to the proximal-distal tissue axis. Second, during ciliogenesis, multiciliated cells (MCCs) undergo cytoskeletal remodeling to orient their cilia in the proximal direction. The second phase not only directs cilium polarization, but also consolidates polarization across the epithelium. Here, we demonstrate that in airway epithelia, PCP depends on MCC differentiation. PCP mutant epithelia have misaligned cilia, and also display defective barrier function and regeneration, indicating that PCP regulates multiple aspects of airway epithelial homeostasis. In humans, MCCs are often sparse in chronic inflammatory diseases, and these airways exhibit PCP dysfunction. The presence of insufficient MCCs impairs mucociliary clearance in part by disrupting PCP-driven polarization of the epithelium. Consistent with defective PCP, barrier function and regeneration are also disrupted. Pharmacological stimulation of MCC differentiation restores PCP and reverses these defects, suggesting its potential for broad therapeutic benefit in chronic inflammatory disease.
Planar cell polarity signaling regulates mucociliary clearance, barrier function, and regeneration in airway epithelia, and depends on the acquisition of multiciliated cell fate.
Introduction
The mature pseudostratified epithelium of the larger conducting airways and sinonasal cavities consists of a patchwork of multiciliated cells (MCCs) and mucus-secreting cells exposed at the luminal surface and underlying basal (stem) cells. MCCs each possess 200 to 300 motile cilia that beat in a coordinated, directional manner to propel inhaled contaminants trapped by the mucus layer out of the lungs (1). This mucociliary clearance process is essential for respiratory health. Breakdown of airway clearance can precipitate and/or exacerbate acute infections and chronic inflammatory conditions such as cystic fibrosis (CF), primary ciliary dyskinesia (PCD), chronic rhinosinusitis (CRS), chronic obstructive pulmonary disease (COPD), and asthma (1).
CF is regarded as the most severe mucociliary clearance disorder (2). Mutations in the CF transmembrane conductance regulator (CFTR) lead to dehydration of the mucosal surface and accumulation of thick, abnormal mucus that both hinders airway clearance and serves as a nidus for polymicrobial infections. These events contribute to severe, chronic inflammation and to cycles of repeated injury and imperfect repair. These in turn bring about epithelial dysfunction, which includes structural and functional changes such as hyperplasia of mucus-secreting cells, decrement in MCC numbers, abnormal tissue architecture with scarring, diminished barrier function, and decreased regenerative capacity (3). CF patients march down an inevitable slope of chronic cough, dyspnea, sinusitis, recurrent antibiotic and steroid use, and oxygen dependence. Epithelial dysfunction in CF is thought to be a major factor in disease progression, ultimately resulting in lung transplantation once medical options become exhausted (4).
Effective mucociliary clearance requires that airways maintain an optimum ratio of mucus-secreting cells and MCCs (1). MCCs are terminally differentiated and arise from the basal cells or secretory cell types of the airway epithelium beginning in embryonic development and continuing as a regenerative process throughout life (5, 6). MCC differentiation starts with a Notch signaling event, in which the ligand-expressing cell is directed to the MCC fate via an MCC-specific gene expression program that drives differentiation and ultimately the production of hundreds of regulatory and structural components required for motile cilium biogenesis (7). Cilia are composed of the basal body (also known as the centriole), which anchors the cilium to the plasma membrane, and the axoneme, the motile shaft that projects from the cell surface (8). During ciliogenesis, hundreds of basal bodies are generated in the cytoplasm and then traffic to and dock with the apical plasma membrane where they elongate their axonemes. Robust mucociliary clearance requires production of cilia of the correct number, length, beat frequency and waveform, and, importantly, correct directionality along the tissue axis.
Motile cilia are physically oriented for directional motility along the proximal-distal (P-D, oral-lung) axis of the airways by the planar cell polarity (PCP) signaling pathway (9). The PCP pathway is a developmental regulator of directional cell behaviors, including cell migration, oriented cell division, and polarized cell morphologies (10). Defects in PCP signaling lead to a wide range of problems such as open neural tube, defective kidney, lung, and breast tubulogenesis, and to misoriented stereocilia, hair follicles, and primary and motile cilia. The PCP mechanism has been most intensively studied in Drosophila, in which its multiple functions include the orientation of cellular extensions called hairs on the pupal wing (11). Extensive studies have identified a core PCP signaling mechanism that polarizes neighboring wing epithelial cells with each other along the P-D (hinge-tip) axis via cell-cell communication. Signaling is mediated by 2 core PCP protein complexes, which segregate into opposing, asymmetrically localized membrane domains termed “crescents” at the proximal and distal side apical cell junctions. The Fz/Dvl crescent (contains Frizzled [Fz], Dishevelled [Dvl] and Flamingo [Celsr in mammals]) localizes to the distal side and the Vangl/Pk crescent (Van Gogh [Vangl in mammals], Prickle [Pk] and Flamingo) localizes to the proximal side. Fz/Dvl complexes interact with and recruit Vang/Pk complexes to the adjacent membrane of neighboring cells, forming intercellular complexes. While initially randomly distributed, planar polarization results from mutual exclusion of oppositely oriented intercellular complexes from a given region of cell-cell contact. Given a biasing input, this bistable feedback mechanism results in the coordinated tissue-wide segregation of Fz/Dvl complexes to one side and Vang/Pk complexes to the opposite side of cells. This feedback mechanism both amplifies molecular polarization and propagates molecular asymmetry from cell to cell. The directionality of the core PCP system is determined by global PCP signals that bias the mutual exclusion. Within individual cells, the polarized Fz/Dvl and Vang/Pk complexes serve as cues for polarized cytoskeletal remodeling to localize and orient cellular structures such as hairs.
This core PCP mechanism shows remarkable evolutionary conservation in at least some vertebrate epithelia, such as the skin, inner ear, and the multiciliated epithelia of the oviduct, ependyma, and respiratory tract (10). Mechanisms in other, especially nonepithelial directional cell behaviors, such as oriented cell division, cell migration, and intercalation are less well characterized. In some cases, core PCP components localize in a polarized manner and cytoskeletal dynamics mediate morphological changes along the axis of polarity (12–14), but how directional cues are specified and transmitted remains unknown.
We have previously demonstrated that during mouse airway epithelial development core PCP proteins segregate into asymmetric membrane domains with the Fz/Dvl crescent at the proximal and the Vangl/Pk crescent at the distal side of cells (9) (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/jci.insight.88027DS1). As in the fly, the asymmetric localization of the 2 complexes is interdependent, and they mediate cell-cell communication to coordinate polarization between cells. When motile cilia emerge in MCCs, they are physically oriented (and thus will beat) towards the proximal side via PCP-dependent microtubules that originate at the basal bodies and are captured by their plus ends at the Fz/Dvl crescent. Vangl1–/– and other PCP mutant airway epithelial cells fail to establish core PCP crescents and have misaligned cilia, which leads to impaired mucociliary clearance. In addition to mispolarized airway epithelia, PCP mutants show defective airway axis elongation and distal airway morphogenesis (15, 16). Furthermore, changes in PCP gene expression are associated with lung cancer (17) and pulmonary arterial hypertension (18). PCP signaling therefore regulates multiple processes in airway development and disease.
Here, we studied the acquisition of PCP signaling in the mammalian airway epithelium using mouse mutants and mouse and human primary airway epithelial cell cultures, which faithfully model many features of the in vivo respiratory mucosa (19, 20). We find that molecular planar polarization, as indicated by Vangl1 crescents (9), emerges first in all cells prior to MCC differentiation. It is then further strengthened selectively in MCCs as motile cilia appear. Vangl1 crescent evolution not only correlates with, but also depends on the MCC transcriptional program, although not the elaboration of motile cilia. Correspondingly, we find that human airway epithelia from patients with CF and other chronic inflammatory diseases with sparse or absent MCCs also have defective PCP. Further, we demonstrate that loss of PCP signaling not only impairs MCC polarization, predicting defective mucociliary clearance, but also correlates with decreased barrier function and regenerative capacity. This indicates that impaired PCP signaling has a wide impact on airway epithelial homeostasis and may be causative of epithelial dysfunction in airway disease. Importantly, we find that driving MCC differentiation by pharmacological suppression of Notch signaling rescues these defects, suggesting a potential therapeutic intervention for CF and other inflammatory respiratory conditions.
Results
MCCs regulate PCP signaling in the airway epithelium.
The hallmark of PCP signaling in airway epithelia is the localization of core PCP proteins, such as Vangl1, to proximal or distal (Vangl1 is distally localized) side crescents at or near the adherens junctions (9) (Supplemental Figure 1A). Vangl1 asymmetric localization can be quantified by the Manders’ overlap coefficient (MOC) (21), which describes the extent of overlap between the Vangl1 signal and the E-cadherin (Ecad) signal marking the adherens junctions (Supplemental Figure 1B). A MOC of 1.00 indicates completely uniform Vangl1 localization, whereas maximal asymmetric localization produces a MOC of approximately 0.5 (see Methods for details). Crescent localization of core PCP proteins can also be observed in primary mouse tracheal epithelial cell (MTEC) cultures generated from isolated basal cells that have been differentiated at an air-liquid interface (ALI, Figure 1) (22, 23). Despite the absence of a tissue axis to globally organize polarization in MTECs, crescents (and cilium polarity) are coordinated among groups of neighboring cells by the activity of the core PCP signaling mechanism (9). MCCs differentiate in MTECs over approximately 2 weeks of ALI culture that begin with no MCCs (no motile cilia or nuclei positive for the MCC transcription factor Foxj1 detected) at the time of ALI creation (ALI+0 days). At ALI+4 days, many actively ciliating and some mature MCCs are present, and by ALI+14 days, all MCCs have fully matured (Supplemental Figure 1C). We observed that Vangl1 crescents evolve as MCCs appear: prior to ciliogenesis (ALI+0 days), Vangl1 crescents show asymmetric localization with approximately uniform labeling intensity in nearly all undifferentiated cells; as MCCs emerge (ALI+4 days) the crescents become more strongly labeled and more asymmetric (decreased MOC) selectively at MCC junctions; after ciliogenesis (ALI+14 days) the crescents are maximally asymmetric and strongly labeled in MCCs and only weakly detectable in other cells (Figure 1 and Supplemental Figure 1D). This observation raises the possibility that MCCs are not only polarized by PCP signaling, but also that MCCs may regulate PCP activity by sustaining and enhancing asymmetric core PCP protein localization specifically in MCCs.
Figure 1. Vangl1 expression and localization evolves during airway epithelial differentiation.
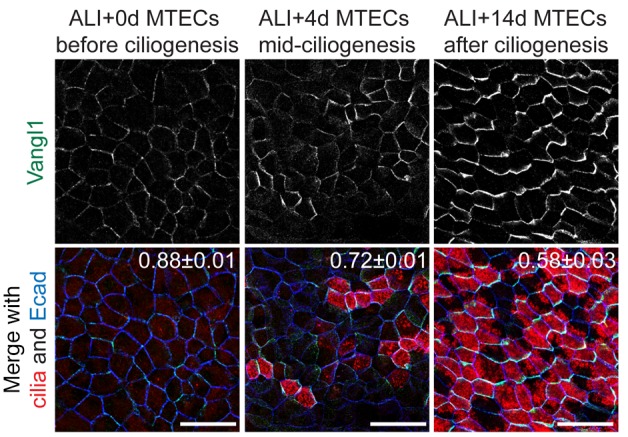
Mouse tracheal epithelial cells (MTECs) undergoing differentiation labeled with Vangl1 (green), acetylated α-tubulin (cilia, red) and Ecad (blue) antibodies show that before ciliogenesis (air-liquid interface [ALI]+0 days, left) Vangl1 signal is low and localization is weakly asymmetric in all cells. As multiciliated cells (MCCs) appear (ALI+4 days, middle) Vangl1 signal increases in nascent MCCs and becomes more asymmetric, and finally in mature, fully ciliated MTECs (ALI+14 days, right) Vangl1 crescents are robust and fully asymmetric in MCCs and weak in other cells. Manders’ overlap coefficient ± standard error indicated on merged images. Images representative of n > 10 MTEC timecourses. Scale bars: 20 μm.
Consistent with the hypothesis that MCCs regulate PCP, we observed a strong correlation between the extent of MCC differentiation and the asymmetry of Vangl1 localization. In the rare, poorly ciliated areas of mature MTEC cultures, asymmetry of Vangl1 localization is only weakly detectable in non-MCCs, whereas in more fully ciliated regions, asymmetry of Vangl1 localization is more robust (Supplemental Figure 2A). Indeed, the asymmetry of Vangl1 localization (MOC) and the fraction of MCCs are linearly related with a correlation coefficient of 0.84 (Supplemental Figure 2B). To more directly test whether the consolidation of Vangl1 crescents depends on MCCs, we examined the effects of blocking MCC differentiation or the transcriptional regulatory machinery driving MCC differentiation. MCC fate acquisition involves a Notch signaling event followed by the activation of an MCC-specific transcriptional network that turns on hundreds of ciliary genes (7). Foxj1–/– mice, which fail to complete motile ciliogenesis due to lack of Foxj1-driven MCC gene expression (24), show asymmetric Vangl1 localization early during development, but uniform Vangl1 localization with lower labeling intensity in the adults (Figure 2A). Similar effects were observed in vitro by blocking the differentiation of MCCs in MTECs at various steps in their differentiation pathway. We blocked MCC fate acquisition (marked by the presence of cilia and/or the expression of Foxj1) by culturing MTECs submerged in medium (i.e., no ALI), which has been shown to interfere with Notch signaling (25); by treating MTECs with the cytokine IL-13, which promotes the differentiation of mucus-secreting cells at the expense of MCCs (26); and by expressing a dominant-negative form of the transcriptional activator Mcidas (ΔCC-Mcidas = coiled-coil domain deletion), which blocks MCC gene expression (27) (Figure 2, B and C, and Supplemental Figure 2, C–F). In each of these cases Vangl1 localization and abundance failed to evolve as in control cultures, resulting in either decreased asymmetry and intensity of Vangl1 localization (IL-13 treatment or ΔCC-Mcidas expression) or undetectable junctional Vangl1 (submerged). The different outcomes with these conditions may reflect distinct effects of the different treatments, or might represent different kinetics of breakdown of the asymmetric Vangl1 crescents. In contrast, we found that induction of excess MCCs, either by treating MTECs with the Notch inhibitor DAPT (25) or by lentiviral expression of full-length Mcidas (27) (Supplemental Figure 2G; see Supplemental Figure 7A for DAPT treatment validation), led to exceptionally robust Vangl1 crescents in MCCs. Thus, our results indicate that PCP requires the regulatory machinery driving MCC differentiation.
Figure 2. Vangl1 expression and localization depend on multiciliated cell differentiation.

(A) Foxj1+/– and Foxj1–/– E18.5 (left) and adult (right) mouse tracheas whole-mount labeled with Vangl1 (green), acetylated α-tubulin (cilia, red), and Ecad (blue) antibodies show that Vangl1 is initially asymmetric in Foxj1–/– cells, but uniformly localized in adults. Images are representative of n = 3 Foxj1–/– mice and littermate controls. (B) Mouse tracheal epithelial cells (MTECs) cultured submerged (no air-liquid interface [ALI]) for 5+14 days (control MTECs were cultured submerged for 5, then at ALI for 14 days) or treated with IL-13 from ALI+0 to 14 days and labeled with Vangl1 (green), acetylated α-tubulin (cilia, red), and Ecad (blue) antibodies, showing that Vangl1 is absent when multiciliated cell (MCC) differentiation is blocked. (C) MTECs infected with GFP or myc-ΔCC-Mcidas lentivirus at day 4 of culture and labeled at ALI+14 days with Vangl1 (green), acetylated α-tubulin (cilia, red), and Ecad (blue) antibodies show that Vangl1 localization does not evolve when MCC differentiation is blocked. (D) MTECs infected with scrambled or Cep83 shRNA lentivirus at day 4 of culture labeled at ALI+14 days with Vangl1 (green), γ-tubulin (centrioles, red), and Ecad (blue) antibodies show that Vangl1 localization is unaffected in Cep83-depleted MCCs that fail to dock their basal bodies. (E) MTECs infected with scrambled or Ift88 shRNA lentivirus at day 4 of culture and labeled at ALI+14 days with Vangl1 (green), acetylated α-tubulin (cilia, red), and Ecad (blue) antibodies show that Vangl1 localization is unaffected in Ift88-depleted MCCs without axonemes. Acetylated α-tubulin faintly labels docked basal bodies without strongly labeling axonemes in Ift88-depleted MCCs. Manders’ overlap coefficient ± standard error indicated on merged images. Scale bars: 25 μm (A), 10 μm (B–E). Images in B–E are representative of n = 3 lentiviral infections or drug treatments of MTECs.
Next, we asked whether MCC regulation of PCP requires just the establishment of MCC cell fate and the MCC gene expression program, or if elaboration of motile cilia is required. We found that human tracheal epithelial cell cultures (HTECs) grown from donors with immotile cilia, but otherwise normal MCCs due to PCD (the presence of immotile cilia was confirmed by high-speed video microscopy, not shown) have robust Vangl1 crescents. This demonstrates that PCP acquisition, as measured by Vangl1 crescent evolution, is not dependent on ciliary motility (Supplemental Figure 2H). We then blocked motile ciliogenesis by interfering with the docking of basal bodies by lentiviral RNAi-mediated depletion of the centriolar distal appendage component Cep83 (28) and the elongation of axonemes by depleting the intraflagellar transport protein Ift88 (29) (Supplemental Figure 2I). These treatments prevent the formation of motile cilia, but not the acquisition of MCC fate, as marked by Foxj1 antibody labeling (Supplemental Figure 2J). In both cases we found that Vangl1 was still enriched and more asymmetric in MCCs at ALI+14 days (Figure 2, D and E), indicating that neither the physical presence of motile cilia nor the docking of basal bodies is required for PCP signaling.
Based on these results, we propose that PCP acquisition in the airway epithelium is a 2-phase process (Supplemental Figure 1D). In phase I, all cells participate in the intercellular communication of polarity information via asymmetrically localized PCP crescents. However, in the subsequent MCC-dependent phase II, PCP crescents are consolidated selectively in MCCs, while weakening in non-MCCs. We suggest that phase II may enable the alignment of basal bodies and therefore cilia with respect to the PCP crescents in MCCs.
Prickle2 expression is controlled by the MCC transcriptional machinery.
While other core PCP proteins, including Vangl1, are present in all cells and are asymmetrically localized prior to the emergence of MCCs (Figure 1 and data not shown), Prickle2, a putative binding partner of Vangl1 (30), is expressed only in MCCs (9), where it colocalizes in the same crescent with Vangl1. Like the phase II consolidation of Vangl1, we found that Prickle2 expression depends on MCC specification. Prickle2 antibody labeling showed that while interfering with cilium biogenesis itself had no effect on Prickle2 crescents (Figure 3A), Prickle2 was not present in MTECs in which MCC fate acquisition and the MCC gene expression program were blocked (Figure 3B). Consistently, Foxj1–/– mice lacked Prickle2 crescents (Figure 3C). Thus, Prickle2 expression not only correlates with, but depends on MCC differentiation.
Figure 3. Prickle2 expression and localization depends on multiciliated cell differentiation.

(A) Mouse tracheal epithelial cells (MTECs) infected with lentivirus containing scrambled sequence or Cep83 shRNA at day 4 of culture and labeled at air-liquid interface (ALI)+14 days with Prickle2 (green), γ-tubulin (cilia, red), and Ecad (blue) antibodies show that Prickle2 localization is unaffected in Cep83-depleted multiciliated cells (MCCs) that fail to dock their basal bodies. (B) MTECs cultured submerged for 5+14 days (control MTECs were cultured submerged for 5, then at ALI for 14 days) and labeled with Prickle2 (green), acetylated α-tubulin (cilia, red), and Ecad (blue) antibodies show that Prickle2 is absent when MCC differentiation is blocked. (C) Foxj1+/– and Foxj1–/– adult mouse tracheas whole-mount labeled with Prickle2 (green), acetylated α-tubulin (cilia, red), and Ecad (blue) show that Prickle2 is absent in Foxj1–/– cells. Images are representative of n = 3 Foxj1–/– mice and littermate controls. (D) Luciferase reporter assay using the human PRICKLE2 promoter (left) in 293T/17 cells shows that it is responsive to MCC transcriptional regulators compared to vector-only control. Luciferase reporter assay using a human PRICKLE2 genomic fragment (right) with predicted MYB and RFX3 binding sites shows that it is responsive to MCC transcriptional regulators compared with vector-only control. Assays were performed in triplicate with n = 3 transfections per reporter construct. Box and whisker plots show the minimum, lower quartile, median, upper quartile, and maximum values. Two-tailed Student’s t test for each transcriptional regulator compared with vector-only transfection; *P < 0.05, **P < 0.01. Scale bars: 10 μm (A and B), 25 μm (C). Images in A and B are representative of n = 3 lentiviral infections or drug treatments of MTECs.
We sought to examine whether Prickle2 expression in MCCs is driven by known MCC transcriptional regulators. In addition to Foxj1, the transcription factors Myb, Rfx3, and E2F4 coupled with Mcidas are known activators of the MCC gene expression program, and many target genes are activated by more than one transcription factor (7). We found that these MCC transcriptional activators can drive luciferase reporter gene expression from a human genomic region predicted to contain the PRICKLE2 promoter and a highly conserved internal PRICKLE2 genomic region predicted to contain MYB and RFX3 binding sites (31, 32) coupled with a minimal promoter (Figure 3D and Supplemental Figure 3A). Our data demonstrate that Prickle2 expression depends on MCC differentiation, and the luciferase assays suggest that it might be a direct target of the MCC gene expression program.
Prickle2 in MCCs may serve to link asymmetric PCP protein localization to downstream regulators of basal body orientation. This possibility is consistent with the mild cilium misorientation observed in Prickle2–/– mutant tracheas (Supplemental Figure 3B). Alternatively, or in addition, the observation that the asymmetry of Vang (and other core PCP proteins) in Drosophila depends on Pk (33, 34) suggested the possibility that Prickle2 might be required for phase II of PCP acquisition in MCCs. However, we found that Prickle2–/– mutant epithelia acquire and fully differentiate MCCs and that Vangl1 (and other core PCP proteins) localizes normally (Supplemental Figure 3C and data not shown), indicating that Vangl1 crescent evolution does not require Prickle2. Nonetheless, the finding that Prickle1, Prickle3, and Prickle4, the last 2 being thus far uncharacterized mammalian PCP components, are also expressed and localize asymmetrically in mouse airway epithelial cells (Supplemental Figure 3D) allows for the possibility of redundant function. Similarly to Prickle2, we found that Prickle4 expression is also restricted to MCCs (Supplemental Table 1), which suggests that Prickle2 and Prickle4 may act together in MCCs to regulate PCP.
Asymmetric VANGL1 localization is lost in diseased human airway epithelia.
It has long been known that the MCCs of the in vivo respiratory epithelium are highly vulnerable to external damage and diseases, such as cigarette smoke, viral infections, asthma, and CF (1). Physical injury and structural changes brought on by chronic inflammation result in decreased MCC numbers, disrupted MCC differentiation, and damage to MCCs and cilia (1). Because PCP depends on MCCs, this led us to ask whether PCP signaling is misregulated in diseased airways, and whether this has any consequence for cilium orientation for mucociliary clearance or other aspects of epithelial function.
We obtained healthy and diseased human sinonasal epithelia from patients undergoing surgical procedures at Stanford Hospital and tested VANGL1 localization by whole-mount antibody labeling. We found that in healthy human tissues, similar to wild-type MTECs or mouse trachea (Figures 1 and 2A), VANGL1 was robustly asymmetric and more abundant in MCCs than in other cells (Figure 4A). In contrast, in CF (due to homozygous F508del CFTR mutation) and CRS tissues obtained from grossly affected regions that lacked MCCs, VANGL1 localization was disrupted. CF tissues had robust, but completely uniform VANGL1 signal, whereas CRS samples had weak VANGL1 signal that ranged from uniform to partially asymmetric. Some samples also had increased cytoplasmic VANGL1 (Figure 4A). This points to a possible dysfunction in the recruitment or maintenance of VANGL1 in crescents in these diseased tissues. Similar to MTECs (Figure 2, A–C), the differences in VANGL1 mislocalization in the CF and CRS samples may derive from unrelated defects or represent discrete steps in a common pathway. We examined distinct regions of intact excised turbinates from CRS patients, and found that the degree of VANGL1 disruption depended on the extent of ciliation: regions lacking MCCs (area 1 and parts of area 2) had absent or weak VANGL1 localization, whereas better-ciliated regions (areas 3 and 4) had a mix of uniform and asymmetric localizations (Figure 4B). In contrast, a well-ciliated nasal polyp had nearly normal Vangl1 crescents (Figure 4A). The disruption of VANGL1 localization is likely not due to the disruption of epithelial apical junctions, as only areas with robust and uniform junctional ECAD labeling were analyzed. As CF can arise due to a variety of CFTR mutations and CRS is a heterogeneous disease, sinonasal tissue from multiple individuals with CRS or with different CFTR mutations was examined, and similar alterations in PCP were found (data not shown). Based on our results in MTECs, we hypothesize that the disruption of VANGL1 crescents seen here is due to the decreased number of MCCs present in the diseased tissues.
Figure 4. VANGL1 localization is abnormal in human cystic fibrosis (CF) and chronic rhinosinusitis (CRS) sinonasal epithelia.

(A) Human sinonasal epithelia from healthy donors and donors with CF, CRS, and nasal polyps whole-mount labeled with VANGL1 (green), acetylated α-tubulin (cilia, red), and ECAD (blue) antibodies show that VANGL1 is no longer asymmetric in diseased tissues that lack multiciliated cells (MCCs). Images are representative of samples from n = 5 CF and n = 3 CRS and nasal polyp donors. (B) CRS tissue areas with varying degrees of ciliation labeled with VANGL1 (green) and acetylated α-tubulin (cilia, red) antibodies show that VANGL1 crescent formation depends on the presence of MCCs. Images are representative of samples from n = 3 CRS donors. (C) CF and healthy human nasal epithelial cells (HNECs) labeled with VANGL1 (green), ECAD (red), and acetylated α-tubulin (cilia, blue) antibodies treated with inflammatory cytokines show that MCCs are not present and VANGL1 crescents do not form. Images are representative of n = 3 drug treatments of CF HNECs from n = 5 donors. Manders’ overlap coefficient ± standard error indicated on merged images. Scale bars: 50 μm (A), 25 μm (B), 10 μm (C).
We also noted that where present in diseased epithelia, VANGL1 crescents were often not localized along the same side of cells (i.e., showing only local, not global, alignment), indicating that these samples have defective tissue-wide coordination of PCP (Supplemental Figure 4A), which would be expected to produce misaligned cilia and diminished directional mucociliary clearance. We hypothesize that these are regions that have undergone previous wounding and re-epithelialization in which orientation of MCCs was incorrectly restored.
Chronic inflammation, a predominant feature of both CF and CRS, is known to favor the differentiation of mucus-secreting cells at the expense of MCCs (35), so we set out to test whether it may play a role in the PCP defect we observed in the patient samples. We first tested whether the treatment of healthy ALI-cultured human nasal epithelial cells (HNECs) with inflammatory cytokines, which models some of the changes that take place in the respiratory epithelium during chronic inflammation (3), will affect PCP acquisition. We found that healthy HNECs treated during differentiation with IL-1β, IL-13, or TNF-α, inflammatory cytokines commonly detected in CF epithelia (36–38), have no or greatly decreased numbers of MCCs, and VANGL1 shows little or no asymmetry (Figures 4C and 2B for IL-13–treated MTECs). This correlation, taken together with the observation that Vangl1 asymmetry depends on MCC differentiation in MTECs, suggests that the lack of VANGL1 crescents in the differentiating HNECs is due to the failure of MCC differentiation. In contrast, when already differentiated healthy HNECs were treated with the cytokines, areas with numerous MCCs always had robust VANGL1 crescents (Supplemental Figure 4B), indicating that the lack of VANGL1 crescents in the differentiating HNECs is not directly due to the cytokine treatment.
We next examined HNECs derived from CF (homozygous F508del) sinonasal tissue. Although generated from equal numbers of basal cells, similar to the in vivo CF donor tissue, CF HNECs had fewer MCCs and defective VANGL1 crescents compared with healthy HNECs (Figure 4C). Furthermore, the degree of inflammation in the donor tissue was proportional to the decrease in MCCs and loss of VANGL1 asymmetry in the cultures. HNECs were derived from different regions of the same CF donor tissue, which were designated as either mildly or severely inflamed (mildly inflamed tissues showed only slight erythema, whereas severely inflamed tissues had extensive erythema and edema, purulent secretions, and crusting; data not shown), and MCCs and VANGL1 localization were assessed and compared with HNECs from healthy donors. In all cases, HNECs grown from severely inflamed regions had fewer MCCs and more abnormal VANGL1 localization compared with those from mildly inflamed regions, which, in turn, were more affected compared with those from healthy donors (Supplemental Figure 4C). Thus, our data are consistent with inflammation being a factor in the loss of MCCs, which in turn leads to PCP dysfunction. Furthermore, the effect of inflammation is retained in the basal cells, giving rise to commensurate defects in the HNEC cultures.
PCP defects impact multiple aspects of airway epithelial homeostasis.
Our results support a model in which inflammation in diseased human sinonasal epithelia leads to the decrement of MCCs, and loss of MCCs impairs PCP. PCP is required for the planar polarized motility of cilia that drives directional mucociliary clearance (9). Airway clearance is known to be disrupted in CF, CRS, and many other diseased airway epithelia (1), so we assessed motile cilium polarity by transmission electron microscopy (TEM). The orientation of the basal foot ciliary appendage points toward ciliary beat direction, and a line drawn through the axonemal central microtubule doublet is perpendicular to ciliary beat direction (23) (Supplemental Figure 5A). We obtained healthy, CF, and CRS sinonasal tissues as above, and first assessed VANGL1 localization (see Figure 4A) and the presence and morphology of MCCs by antibody labeling and scanning electron microscopy (SEM) for normal and CF (Supplemental Figure 5B, CRS not shown). If intact MCCs were detected, we processed a part of the tissue for TEM. SEM of CF sinonasal tissue confirmed the variability in the extent of ciliation in different regions, ranging from near normal to virtually absent, and revealed that cilia were poorly organized even in better-ciliated areas (Supplemental Figure 5B). TEM showed that, in contrast to healthy tissue, where basal feet on cilia always pointed in a common direction, CF and CRS tissues with disrupted VANGL1 crescents had many misaligned cilia (Figure 5), indicating that mucociliary clearance over that area would be disrupted.
Figure 5. Human cystic fibrosis (CF) and chronic rhinosinusitis (CRS) sinonasal epithelia have misaligned cilia.

Ciliary planar polarization was evaluated by transmission electron microscopy (TEM) for the orientation of basal feet (top) and central axonemal microtubule (MT) doublets (bottom). Green lines are drawn through basal feet in 2 cells in each image with boxed area shown magnified in each cell. Red lines are drawn through the central MT doublets in some axonemes. The green and red lines are shown again underneath each TEM image to clearly demonstrate directionality. Both structures indicate that cilia are aligned in a common orientation in healthy tissues, but not in CF or CRS. Scale bars: 2 μm (top), 0.5 μm (bottom). Images are representative of samples from n = 3 donors.
Diseased airway tissues show a range of epithelial structural and functional abnormalities (3). We therefore examined whether the disruption of PCP signaling per se can affect epithelial function and if this may account for or contribute to disease phenotypes. PCP mediates a wide range of cell behaviors involved in development and disease, including migration, oriented division, and morphological polarization (10). Airway tissues frequently rely on robust regeneration to repair injuries caused by disease or mechanical agents (5, 39). Repair may involve PCP-driven directional migration for wound healing followed by re-epithelialization. Thus, we asked whether loss of PCP in the airway epithelium disrupts wound healing. We induced a scratch wound in mature MTECs using a Pasteur pipet and observed wound closure during regeneration (Supplemental Figure 6A). While wild-type MTECs repaired the wound robustly, we found that MTECs generated from mice with a germline deletion of the transmembrane domains in exon 4 of Vangl1 (Vangl1CKO–/–; see ref. 9) or Prickle2–/– mice failed to close the wound (Figure 6A and Supplemental Figure 6B). It is known that diseased or chronically injured tissues such as those in the CF airways have diminished repair capability (40); thus, we propose that PCP dysfunction may be a contributing factor.
Figure 6. Planar cell polarity–mutant mouse tracheal epithelial cells (MTECs) have defective wound healing capacity and decreased barrier function.

(A) Wild-type, Vangl1CKO–/–, and Prickle2–/– MTECs scratch wounded at air-liquid interface (ALI)+14 days of culture show that planar cell polarity (PCP)-mutant cultures have defective wound healing response. Missing 48-hour data point for Prickle2–/– (n/a) is due to cell death. Wound healing was measured in n = 3 cultures in triplicate, wild-type and Prickle2–/–, and wild-type and Vangl1CKO–/– values were compared using a 2-way ANOVA test and were found to be significantly different at P < 0.0001. (B) Transepithelial electrical resistance (TEER) measurements at ALI+14 days for Vangl1CKO–/– and Prickle2–/– MTECs show that PCP-mutant cultures have lower TEER than wild-type MTECs. Rt indicates transepithelial resistance. TEER was measured in n = 3 cultures in triplicate. Two-tailed Student’s t test; *P < 0.05, **P < 0.001. Box and whisker plots show the minimum, lower quartile, median, upper quartile, and maximum values.
Finally, since Vangl1 and other core PCP proteins act at the apical cell junctions, we hypothesized that they may also contribute to junctional integrity or function, and disrupted PCP may result in compromised barrier function. Consistent with this, we found that, compared with wild-type MTECs, cultures generated from Vangl1CKO–/– or Prickle2–/– mice have lower transepithelial electrical resistance (TEER), and their junctions disassemble more readily in the presence of Ca++-chelating agents (Figure 6B and Supplemental Figure 6C). Decreased barrier function is a cardinal feature of chronically inflamed respiratory epithelia, including CF (41). Our results raise the possibility that defective core PCP complexes at the adherens junctions may play a role in weakened junctional integrity in CF airway epithelia.
In summary, we propose that in CF and CRS sinonasal epithelia, chronic inflammation and/or other changes during disease progression leads to decrease or loss of MCCs that in turn causes loss of VANGL1 asymmetry. This defect in PCP signaling may contribute to disease phenotypes via decreased mucociliary clearance, disrupted barrier function in intact epithelia, and a diminished capacity for repair in case of injury. Our results suggest that PCP is a factor in multiple aspects of airway epithelial homeostasis, and MCC loss or damage may be a more detrimental part of the disease process than previously appreciated.
Notch pathway inhibition restores MCCs and PCP signaling in diseased epithelia in vitro.
We found that, as in vivo, HNECs generated from CF sinonasal tissue have very few MCCs and absent or decreased VANGL1 crescents (Figures 4C and 7A). This likely reflects a state of severe and chronic inflammation that persists even after the progenitor basal cells are isolated and placed into culture (40, 42, 43). To test our hypothesis that the loss of MCCs leads to VANGL1 localization defects, and to ask whether increasing the fraction of MCCs can correct the PCP defect in diseased tissues, we treated CF HNECs with the Notch pathway inhibitor DAPT, which increases the fraction of MCCs 2-fold in both MTECs (25) and HNECs (Supplemental Figure 7A). DAPT treatment of CF HNECs restores normal MCC numbers, and we observed that VANGL1 crescents are robust and asymmetrically localized (Figure 7, A and B). Next, we asked whether restoring MCC differentiation also improves epithelial function. Consistent with previous reports, we found that CF HNECs have decreased TEER and are unable to or are slower to repair an epithelial scratch wound (3, 40) (effect variable among donors, Figure 7, C and D, and Supplemental Figure 7B). In contrast, DAPT-treated CF HNECs from all donors tested were able to robustly repair a scratch wound (Figure 7C). Furthermore, treatment of CF HNECs with DAPT resulted in increased TEER compared to untreated CF cultures, indicating improved barrier function (Figure 7D). Thus, reinstating or improving MCC differentiation restores multiple structural and functional characteristics of the CF airway epithelium in vitro. We propose that this is in part due to the restoration of PCP signaling by inducing the proper differentiation of airway epithelial cell types.
Figure 7. Notch inhibition restores planar cell polarity and improves epithelial function in cystic fibrosis (CF) human nasal epithelial cells (HNECs).

(A) Healthy and CF HNECs were labeled with VANGL1 (green), acetylated α-tubulin (cilia, red), and ECAD (blue) antibodies at air-liquid interface (ALI)+10 (early) and ALI+30 (late) days. VANGL1 crescents become robust in well-ciliated healthy HNECs, but fail to do so in poorly ciliated CF HNECs. DAPT treatment of CF HNECs from ALI+0 to +30 days restores multiciliated cell (MCC) differentiation and robust VANGL1 crescent formation. Manders’ overlap coefficient ± standard error indicated on merged images. Scale bars: 10 μm. Images are representative of n = 3 drug treatments of CF HNECs from n = 5 donors. (B) Quantification of the fraction of MCCs in healthy HNECs and untreated and DAPT-treated CF HNECs; n > 1,000 cells were evaluated in triplicate. One-way ANOVA with post-hoc Dunnett’s multiple comparison test; *P < 0.0001. (C) Healthy HNECs and untreated and DAPT-treated CF HNECs were injured in a scratch wound assay at ALI+30 days and allowed to regenerate. Depending on donor, CF HNECs mostly failed to repair the wound; however, DAPT-treated CF HNECs were able to robustly close the wound (see Supplemental Figure 7B). Wound healing was measured in n = 3 cultures in triplicate, CF HNEC and CF+DAPT HNEC values were compared using a 2-way ANOVA and were found to be significantly different at P < 0.0001. (D) Transepithelial electrical resistance (TEER) measurements at ALI+30 days for IL-13– or DAPT-treated healthy HNECs and untreated and DAPT-treated CF HNECs show that IL-13–treated healthy HNECs and untreated CF HNECs have lower TEER than healthy HNECs. TEER can be increased in both healthy and CF cultures by DAPT treatment. TEER was measured in n = 3 cultures in triplicate. One-way ANOVA with post-hoc Bonferroni’s multiple comparison test; *P < 0.05, **P < 0.0001, ***P < 0.00001. Box and whisker plots show the minimum, lower quartile, median, upper quartile, and maximum values.
Discussion
MCCs are required for a 2-phase core PCP mechanism in the airway epithelium.
We have previously shown that motile cilia are oriented by PCP signaling, and that localization of Vangl1 and other core signaling components to the P-D side apical junctions of airway epithelial cells reports the activity of this pathway (9, 23). We examined the kinetics of core PCP protein (Vangl1) localization in the mouse airway epithelium, and found that PCP emerges in a 2-phase process. In phase I, prior to ciliogenesis, Vangl1 crescents of equivalent intensity form in every cell, and in phase II, after ciliogenesis, Vangl1 crescents evolve such that their intensity is strengthened in MCCs and weakened in non-MCCs. We show that phase II not only correlates with but also depends on the differentiation of MCCs. In the Foxj1–/– mouse trachea, which lacks complete MCC differentiation (24), or in ALI cultures in which MCC differentiation is blocked, Vangl1 crescents are initially present (phase I), but dissipate rather than evolve (phase II). These results demonstrate that in addition to mediating mucociliary clearance, MCCs also control PCP. Further results show that PCP regulates additional key features of airway epithelial homeostasis.
While PCP does not rely on the physical presence of motile cilia, it requires the MCC gene expression program. This program is launched upon MCC fate acquisition to turn on the hundreds of regulatory and structural motile ciliogenesis genes (7). We propose that the MCC gene expression program also induces the expression of factors that control phase II of PCP acquisition. One such candidate factor is Prickle2. We considered 2 potential functions for Prickle2. First, Prickle2 may link PCP crescents to the cellular cytoskeletal machinery that polarizes MCCs. Second, it is possible that Prickle2 contributes to the creation or maintenance of the crescents, a function analogous to that of Drosophila Pk (33, 34). Both possibilities are consistent with the mild polarity disruption observed in Prickle2–/– mutant tracheas. Although Vangl1 crescents evolved normally in Prickle2–/– MCCs, the presence of other Prickle proteins in MCCs leaves open the possibility that Prickle2 may act redundantly with other Prickle family members in one or both of these functions.
The 2-phase mechanism of respiratory epithelial PCP acquisition may reflect the differing cellular responses to PCP in this tissue, in which MCCs require polarity cues to orient cellular structures, while neighboring secretory cells display no overt planar polarized morphology. We suggest that in phase I, all cells participate in the communication of PCP cues to establish molecular planar polarity throughout the tissue. Then, in phase II, MCCs reinforce and use the polarity information to orient their cilia. During phase II, crescents are still present, although only weakly detectable, in non-MCCs. This may reflect a requirement for the maintenance of PCP crescents in MCCs, which would involve the interaction of opposing complexes across adjacent cell boundaries (10). It may also reflect the requirement that new cells born into the epithelium acquire correct polarity with respect to their neighbors. It is noteworthy that while PCP crescents are transient in other tissues (44), they are retained in airway epithelial cells for the life of the organism. While not yet known, a similar 2-step mechanism may also be active in the cochlear epithelium of the inner ear, where hair cells with planar polarized stereociliary bundles and asymmetric Prickle2 crescents are interspersed with supporting cells that lack planar polarized structures (45).
PCP regulates development and epithelial homeostasis in the respiratory system.
Our experiments with MTECs mutant for PCP function demonstrate that PCP in airway epithelia is required not only for the alignment of motile cilia, but also for the integrity of apical epithelial junctions and for an effective wound healing response. This suggests that PCP signaling is not only critical for the 2 major functions of the airway epithelium — mediating mucociliary clearance and acting as a physical barrier — but also for its regeneration in case of injury. Although we are beginning to understand how PCP orients motile cilia (9, 44, 46), the mechanisms underlying its other roles remain unclear. The core PCP cassette assembles and functions at apical cell junctions, and we find that the junctions are defective in PCP mutant airway epithelial cells. The core PCP proteins Vangl2 and Celsr1 were previously shown to interact with and control the localization of Ecad in kidney epithelial cells (47) and lymphatic endothelial cells (48), respectively, indicating that PCP can regulate the content and functionality of apical junctional complexes. Diminished barrier function in airway epithelial cells may reflect a similar dependence on core PCP proteins to maintain functional cell-cell junctions.
Directional cell migration defects are likely responsible for the diminished wound closure response of PCP mutant airway epithelial cells in the scratch wound assay. In other systems, core PCP proteins have been shown to localize to the leading or lagging edges of migrating cells, in some cases asymmetrically, and to control cytoskeletal dynamics for locomotion (18, 49, 50). The airways may incur large-scale injuries with epithelial denudation (as modeled by the scratch wound assay) due to viral infections or mechanical damage during intubation or transplant surgery. Small-scale injuries or damage limited to small numbers of cells are likely to be more frequent, and the healing of these may not require directional cell migration. Nonetheless, the PCP pathway must still play a role in reestablishing a planar polarized airway epithelium in these cases. Future studies using in vitro and in vivo injury models will be needed to clarify the role of PCP in the regenerative response.
At the organ level, PCP is known to control airway axis elongation, pulmonary branching, and distal lung morphogenesis (15, 16). Taking these results into consideration together with our data, we conclude that PCP is vital for both pulmonary development and epithelial homeostasis. Given that PCP controls a wide range of directional cell behaviors, these diverse processes likely represent multiple distinct PCP mechanisms under the control of a common set of regulators. Delineating these mechanisms in the respiratory system using the established mouse and primary cell culture models will lead to a better understanding of airway biology and also inform studies in other, less tractable vertebrate tissues and organ systems where PCP operates.
Cilia and PCP in disease.
MCC loss or damage resulting in impaired mucociliary clearance, and defective barrier function and regeneration are well documented in chronic inflammatory diseases (1). We report that well-defined core PCP crescents are absent in human airway epithelia from CF and CRS donors that lack MCCs. HNECs generated from basal cells derived from such CF donors also produce few or no MCCs and show defective PCP, along with defective apical junctions and wound healing response. Thus, we propose that PCP dysfunction contributes to disease phenotypes in chronic inflammatory conditions. While the evidence linking defective PCP signaling to impaired barrier function and regenerative capacity in human disease tissues is correlative, the direct evidence of causality in the mouse system strongly suggests that PCP contributes to these phenotypes in human tissue.
Similar to mouse MCCs with blocked differentiation (observed in Foxj1–/– mice or upon submerged culture or IL-13 treatment of MTECs), we find that in human disease tissue lacking MCCs, PCP proteins show diminished asymmetric or completely uniform distribution, suggesting that phase I of the core mechanism may still be at least partially operational, but phase II does not occur in the absence of MCCs. Although we do not directly demonstrate that PCP dysfunction in the patient tissues is due to the loss of MCCs, this hypothesis is supported by the observation that PCP crescents are present in ciliated areas of diseased tissues, and the degree of ciliation and crescent asymmetry are positively correlated. Furthermore, we show that CF HNECs, which model many aspects of the diseased in vivo epithelium including diminished MCCs, have defective PCP signaling. We can rescue these defects by inducing MCC differentiation with Notch inhibitor treatment, strongly implying that the defective PCP observed in vivo also results from loss of MCCs.
MCC loss or damage in chronic airway diseases like CF is thought to be caused by infections that target MCCs or indirectly by inflammation (51). Damage is localized sporadically throughout the airways, and may be reversible, although chronic injury to the epithelial surface eventually gives rise to epithelial remodeling with irreversible structural changes (3). Robust mucociliary clearance requires an optimal ratio of MCCs and mucus-secreting cells, and proper planar polarization of cilia. We find that the PCP signaling that confers that polarity also depends on a sufficient ratio of MCCs to mucus-secreting cells. Mucous cell hyperplasia caused by inflammation disrupts this relationship, and this may result in the lack of PCP crescents we observed in the diseased human airway specimens (see Supplemental Figure 5B). Because we can reproduce these effects with inflammatory cytokines in culture, we propose that defective PCP signaling may be a universal feature of chronically inflamed airways, and that it may contribute to the epithelial dysfunction that drives disease progression.
In addition to impaired mucociliary clearance, deficient wound healing and barrier function are prominent phenotypes in CF airways (40, 52). These phenotypes are reproduced in HNECs derived from these patients. Our results do not directly demonstrate that deficient wound healing and barrier function can be attributed to the disruption of PCP signaling in human airway disease tissues, but the observation that PCP-mutant MTECs show the same defects strongly implies that disrupted PCP signaling contributes to, if not accounts for, these defects.
We do not wish to suggest that defective PCP signaling is a primary defect in CF or other chronic inflammatory conditions. Rather, we propose that these conditions diminish MCC differentiation, that loss of MCCs impairs PCP signaling, and that impaired PCP signaling contributes to phenotypes that exacerbate chronic inflammation, thereby functioning in a feedback loop. Interventions that restore MCC differentiation could interrupt this feedback loop by rescuing PCP function and associated phenotypes. Experiments in cultures suggest this may be the case. We found that HNECs generated from basal cells isolated from CF patients continue to exhibit epithelial dysfunction, including fewer MCCs, decreased or absent PCP crescents, diminished barrier function, and reduced wound healing response. This is consistent with previous reports that basal cells may incur long-term injury in CF airways (3, 40, 42, 43). Importantly, we were able to restore all of these structural and functional deficits by treatment of cultures with a Notch inhibitor, which induces the differentiation of MCCs (25). Although we cannot rule out other Notch-dependent mechanisms, this raises the intriguing possibility of using biologic or small-molecule inhibitors of the Notch pathway to restore MCCs and PCP signaling in the treatment of chronic inflammatory airway diseases. Restoring MCCs may be especially valuable in treating CF, as other therapies are aimed at correcting or increasing the function of the CFTR protein, which is expressed in MCCs (53). Future studies using in vivo disease models will be necessary to validate and further advance these concepts.
Methods
Mouse husbandry and MTEC culture.
Vangl1CKO–/– (9), Prickle2–/– (54) (gift from Naoto Ueno, NIBB, Okazaki, Japan and Alex Bassuk, University of Iowa, Iowa City, Iowa, USA), Foxj1–/– (24) (gift from Mark Krasnow, Stanford University School of Medicine), and Foxj1/EGFP (55) (gift from Larry Ostrowski, UNC Chapel Hill, Chapel Hill, North Carolina, USA) mice have been previously described. C57BL/6J mice were obtained from JAX. MTEC culture and lentiviral infection were performed as previously described (22). In short, tracheas were excised and incubated overnight in protease solution to release epithelial cells. Cells were seeded on porous Transwell filters (Corning) and cultured submerged in proliferation medium until confluence. An ALI was created by adding differentiation medium only to the bottom compartment of the culture vessel to promote MCC differentiation. MTECs reach maturity with 60%–80% MCCs by ALI+14 days. Cells were treated with 1 μM DAPT (Abcam) or mouse IL-13 (Peprotech) in differentiation medium for various lengths of time. For submerged culture, MTECs were cultured in differentiation medium supplied from the bottom and top compartments of the culture vessel.
Lentiviral constructs and lentivirus preparation and infection.
Lentiviral vectors containing mouse myc-Mcidas and myc-ΔCC-Mcidas cDNA (27, 56), mouse GFP-Prickle1 and GFP-Prickle2, or GFP alone cDNA (9), and mouse Cep83 (28) and Ift88 (20) shRNA have been previously described. The GFP-Prickle3 and GFP-Prickle4 constructs were generated by PCR amplifying and inserting the Prickle open reading frames (Dharmacon) into the BsrGI site of the pRRL.sin-18.PPT.PGK.GFP.pre lentiviral vector (57) to create an N-terminal GFP fusion. Lentivirus was prepared according to published methods using the psPAX2 and pMD2.G helper plasmids (Addgene) in the 293T/17 cell line (ATCC). MTECs were infected with lentivirus on day 4 of culture using spin infection following EGTA treatment to temporarily disrupt epithelial junctions (22).
Human tissues and HTEC/HNEC culture.
HTECs were generated from human tracheal epithelial brushings obtained from patients undergoing bronchoscopy at the Stanford Pulmonary and Cystic Fibrosis Center. HNECs were generated from human sinonasal epithelial brushings or from tissue obtained from patients undergoing endoscopic sinus surgery at Stanford Hospital. Cells from brushings were dissociated by incubation in a 1:1 mixture of 0.5% trypsin (Thermo Fisher) and Nonenzymatic Cell Dissociation Solution (Sigma-Aldrich) for 5 minutes at 37°C. Cells from tissue pieces were dissociated by overnight digestion in 1.5 mg/ml pronase (Roche) at 4°C. The isolated epithelial cells were cultured on Transwell filters (Corning) following the MTEC protocol (22). Y-27632 ROCK inhibitor (Abcam) was added to the proliferation medium at 10 μM for cultures derived from limited material (58). Healthy HNECs and HTECs reach maturity with 75%–90% MCCs with variable timing due to donor differences, but all cultures complete MCC differentiation by ALI+30 days. Cells were treated with human IL-1β, human IL-13, human TNF-α (all from Peprotech), or 1 μM DAPT in differentiation medium for various lengths of time.
Immunofluorescence.
Cell cultures and tissues were fixed in –20°C methanol or 4% paraformaldehyde for 10 minutes, blocked in 10% normal horse serum and 0.1% Triton X-100 in PBS, and incubated with primary antibodies for 1–2 hours, then with secondary antibodies for 30 minutes at room temperature. Transwell filters were mounted in Mowiol mounting medium containing 2% N-propyl gallate (Sigma-Aldrich). For whole-mount labeling, tissues were pinned luminal side up onto Sylgard-184 elastomer (Ellsworth Adhesives) slabs. Tissues were mounted within a gasket to support the cover slip. Trachea images are oriented proximal side up. Specimens were imaged with Leica LAS X software on a Leica SP5 or SP8 confocal microscope. For antibodies and fixation conditions, see Supplemental Table 2.
Electron microscopy.
For TEM, tissues were fixed in 2% glutaraldehyde, 4% paraformaldehyde in 0.1 M Na cacodylate buffer, pH 7.4 at 4°C overnight. Samples were osmicated, stained with uranyl acetate, and then dehydrated with a graded ethanol series and infiltrated with EMbed-812 (Electron Microscopy Sciences). Sections (80–100 nm) were mounted onto copper grids and analyzed with a JEOL JEM-1400 microscope using a Gatan Orius camera. For SEM, tissues were fixed as for TEM, osmicated, dehydrated, dried with a Tousimis Autosamdri-815 critical point dryer, and then samples were mounted luminal side up, sputter coated with a 100-Å layer of Au/Pd, and analyzed with a Hitachi S-3400N VP-SEM microscope operated at 10–15 kV, with a working distance of 7–10 mm and using secondary electron detection. Our human tissue procurement procedure does not easily permit the tracking of the global orientation of the isolated tissue during the immunofluorescence and EM preps, but we are still able to assess ciliary orientation among neighboring cells and within MCCs.
Measurement of Vangl1 asymmetric localization.
Vangl1 asymmetric localization at the apical junctions was quantified by calculating the MOC, which describes the extent of overlap between the Vangl1 and Ecad channels of the same image using the Coloc 2 function in the Fiji software (NIH). MOC is defined as the Pearson’s correlation coefficient of the pixel-by-pixel covariance in the signal levels of 2 images (21). A MOC of 1.00 indicates completely symmetric Vangl1 localization (perfect overlap with Ecad); values less than 1.00 indicate asymmetric localization. The MOC ± standard error (SE, based on measurements from n ≥ 3 representative images) is indicated on the merged image where applicable.
Luciferase reporter assays.
For the luciferase reporter assay, human PRICKLE2 promoter and enhancer genomic DNA fragments (see Supplemental Figure 3A) were cloned into pGL4.20 and pGL4.23 (Promega) firefly luciferase expression vectors, respectively, and transfected into 293T/17 cells (ATCC) using FuGENE6 (Roche) along with plasmids containing human FOXJ1 (56), MYB (56), E2F4 (gift from Tim Stearns, Stanford University) and/or MCIDAS (56) cDNA and pRL-TK (Promega) Renilla luciferase expression vector. Reporter activity was assessed using the Dual-Glo Luciferase Assay System (Promega) with a FLUOStar Omega (BMG Labtech) luminescence plate reader. Relative reporter activity was calculated by normalization to the vector-only transfection control in triplicate.
Quantitative RT-PCR.
For quantitative RT-PCR analysis of Prickle1–4 expression, cDNA was prepared from MCCs (EGFP+) and non-MCCs (EGFP–) obtained by FACS from Foxj1/EGFP mouse trachea or MTECs as previously described (20). qPCR was performed in triplicate with Power SYBRGreen MasterMix (Thermo Fisher) in a StepOnePlus Real-Time PCR System (Thermo Fisher), and gene expression was evaluated using the ΔΔCt method. For primer sequences, see Supplemental Table 3.
Measurement of basal body orientation.
Basal body orientation for 3 Prickle2–/– mice and 3 wild-type littermate controls were evaluated at 2 months of age as previously described (9). In short, the angle of basal foot orientation with respect to the proximal airway direction was measured in TEM images (see Supplemental Figure 5A for example) using ImageJ software (NIH). The mean vector of basal body orientation per cell (αcell) and the length of the mean vector (rcell = 1 – circular variance), representing the dispersion of basal body directions in each cell, were calculated using the Oriana v3.0 circular statistics software (Kovach Computing Services). The degree of intracellular orientation (rcell) was reported as mean ± SE and compared among conditions using a 2-tailed, unpaired Student’s t test. To describe intercellular orientation across the trachea, the tracheal mean vector (αtrachea) was determined by averaging αcell values per condition, and the length of the mean tracheal vector (rtrachea) was calculated to show the dispersion of cell directions. Data are represented on circular plots with the proximal direction set at 0°, and each arrow describes the orientation of a single MCC with the angle of the arrow showing the mean vector of basal body orientation (αcell) and the length of the mean vector representing the coherence of orientation within that cell (rcell).
MTEC/HNEC wound healing assay.
Mature cultures were wounded in triplicate by drawing a Pasteur pipet gently over the surface, then rinsing with PBS. Cultures were fixed with 4% paraformaldehyde at t = 0 and at multiple subsequent timepoints during wound closure and stained with DAPI (Thermo Fisher) to mark nuclei. DAPI signal was used to visualize the wound edge using SPOT Advanced v4.6 software (SPOT Imaging) on a Nikon Eclipse TE2000-U inverted fluorescence microscope. Wound healing was assessed by measuring the distance between wound edges at multiple sites along the wound (see Supplemental Figure 6A).
MTEC/HNEC epithelial integrity assays.
TEER was measured in mature cultures in triplicate using a Millicell-ERS2 Volt-Ohm Meter (EMD Millipore). Differentiation medium was added to the top compartment of ALI cultures 30 minutes prior to measurement. For the EGTA sensitivity assay, MTECs were rinsed twice in PBS, and then 10 mM EGTA in 12 mM HEPES (pH 7.4) was added to the top compartment and fresh differentiation medium was added to the bottom compartment. Cells were incubated at 37°C, 5% CO2 for 10–30 min, and then fixed for immunofluorescence.
Statistics.
Prism7 software (GraphPad Software) was used to generate graphs and perform statistical analyses in all cases except the measurement of basal body orientation (see above for circular statistics). Data are graphically represented in the box and whisker format showing the minimum, lower quartile, median, upper quartile, and maximum values. For the wound healing assays, data sets were compared using a 2-way ANOVA test. Pearson correlation was used to test the relationship between Vangl1 asymmetry (MOC) and MCC number. Pairwise comparisons were made with a 2-tailed Student’s t test or an ANOVA test. For multiple comparisons, a follow-up test (Dunnett’s or Bonferroni’s) was applied to correct for multiple hypothesis testing. For all cases, a P value less than 0.05 was considered significant.
Study approval.
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Stanford University School of Medicine in accordance with established guidelines for animal care. Human airway cells and tissues were acquired and used for research with informed consent under Institutional Review Board approval from Stanford University School of Medicine (IRB-18981, IRB-22155). Protocol and informed consent procedures were provided according to Declaration of Helsinki guidelines.
Author contributions
EKV designed and performed the experiments, analyzed the data and wrote the manuscript. JVN and CEM provided human tissue and advised on the design and analysis of the experiments. JDA supervised the design and analysis of the experiments. All authors edited the manuscript.
Supplementary Material
Acknowledgments
We thank Alan Nguyen and Dawn Bravo (Stanford) for help with human tissue procurement; Klara Fekete for help with animal husbandry; John Perrino and Lydia Joubert (Stanford) for help with electron microscopy; Jeff Wine, Nam Soo Joo and Zachary Sellers (Stanford) for advice and help with HNEC assays; and members of the Axelrod lab for helpful discussions. Work was supported by R01 GM098582 (to J.D.A.), internal funds provided by the Stanford Department of Otolaryngology – Head and Neck Surgery (to J.V.N.), and National Institutes of Health Shared Instrumentation grant 1S10RR02678001 to the Stanford University Cell Sciences Imaging Facility.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(13):e88027. doi:10.1172/jci.insight.88027.
References
- 1.Tilley AE, Walters MS, Shaykhiev R, Crystal RG. Cilia dysfunction in lung disease. Annu Rev Physiol. 2015;77:379–406. doi: 10.1146/annurev-physiol-021014-071931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruscia EM, Bonfield TL. Innate and adaptive immunity in cystic fibrosis. Clin Chest Med. 2016;37(1):17–29. doi: 10.1016/j.ccm.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Adam D, et al. Cystic fibrosis airway epithelium remodelling: involvement of inflammation. J Pathol. 2015;235(3):408–419. doi: 10.1002/path.4471. [DOI] [PubMed] [Google Scholar]
- 4.Regamey N, Jeffery PK, Alton EW, Bush A, Davies JC. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax. 2011;66(7):624–629. doi: 10.1136/thx.2009.134106. [DOI] [PubMed] [Google Scholar]
- 5.Hogan BL, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15(2):123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rock JR, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106(31):12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choksi SP, Lauter G, Swoboda P, Roy S. Switching on cilia: transcriptional networks regulating ciliogenesis. Development. 2014;141(7):1427–1441. doi: 10.1242/dev.074666. [DOI] [PubMed] [Google Scholar]
- 8.Brooks ER, Wallingford JB. Multiciliated cells. Curr Biol. 2014;24(19):R973–R982. doi: 10.1016/j.cub.2014.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vladar EK, Bayly RD, Sangoram AM, Scott MP, Axelrod JD. Microtubules enable the planar cell polarity of airway cilia. Curr Biol. 2012;22(23):2203–2212. doi: 10.1016/j.cub.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vladar EK, Antic D, Axelrod JD. Planar cell polarity signaling: the developing cell’s compass. Cold Spring Harb Perspect Biol. 2009;1(3):e88027. doi: 10.1101/cshperspect.a002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carvajal-Gonzalez JM, Mlodzik M. Mechanisms of planar cell polarity establishment in Drosophila. F1000Prime Rep. 2014;6:e88027. doi: 10.12703/P6-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin C, Kiskowski M, Pouille PA, Farge E, Solnica-Krezel L. Cooperation of polarized cell intercalations drives convergence and extension of presomitic mesoderm during zebrafish gastrulation. J Cell Biol. 2008;180(1):221–232. doi: 10.1083/jcb.200704150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luga V, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151(7):1542–1556. doi: 10.1016/j.cell.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 14.Kaucká M, et al. Asymmetry of VANGL2 in migrating lymphocytes as a tool to monitor activity of the mammalian WNT/planar cell polarity pathway. Cell Commun Signal. 2015;13:e88027. doi: 10.1186/s12964-014-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yates LL, et al. The PCP genes Celsr1 and Vangl2 are required for normal lung branching morphogenesis. Hum Mol Genet. 2010;19(11):2251–2267. doi: 10.1093/hmg/ddq104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Xiao J, Hormi K, Borok Z, Minoo P. Wnt5a participates in distal lung morphogenesis. Dev Biol. 2002;248(1):68–81. doi: 10.1006/dbio.2002.0729. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Galkin AV, Wu C, Reddy V, Su AI. CAFET algorithm reveals Wnt/PCP signature in lung squamous cell carcinoma. PLoS One. 2011;6(10):e88027. doi: 10.1371/journal.pone.0025807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan K, et al. Activation of the Wnt/planar cell polarity pathway is required for pericyte recruitment during pulmonary angiogenesis. Am J Pathol. 2015;185(1):69–84. doi: 10.1016/j.ajpath.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- 20.Vladar EK, Stearns T. Molecular characterization of centriole assembly in ciliated epithelial cells. J Cell Biol. 2007;178(1):31–42. doi: 10.1083/jcb.200703064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol, Cell Physiol. 2011;300(4):C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vladar EK, Brody SL. Analysis of ciliogenesis in primary culture mouse tracheal epithelial cells. Meth Enzymol. 2013;525:285–309. doi: 10.1016/B978-0-12-397944-5.00014-6. [DOI] [PubMed] [Google Scholar]
- 23.Vladar EK, Lee YL, Stearns T, Axelrod JD. Observing planar cell polarity in multiciliated mouse airway epithelial cells. Methods Cell Biol. 2015;127:37–54. doi: 10.1016/bs.mcb.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brody SL, Yan XH, Wuerffel MK, Song SK, Shapiro SD. Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice. Am J Respir Cell Mol Biol. 2000;23(1):45–51. doi: 10.1165/ajrcmb.23.1.4070. [DOI] [PubMed] [Google Scholar]
- 25.Gerovac BJ, Valencia M, Baumlin N, Salathe M, Conner GE, Fregien NL. Submersion and hypoxia inhibit ciliated cell differentiation in a notch-dependent manner. Am J Respir Cell Mol Biol. 2014;51(4):516–525. doi: 10.1165/rcmb.2013-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laoukili J, et al. IL-13 alters mucociliary differentiation and ciliary beating of human respiratory epithelial cells. J Clin Invest. 2001;108(12):1817–1824. doi: 10.1172/JCI13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stubbs JL, Vladar EK, Axelrod JD, Kintner C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat Cell Biol. 2012;14(2):140–147. doi: 10.1038/ncb2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanos BE, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27(2):163–168. doi: 10.1101/gad.207043.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12(3):589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bastock R, Strutt H, Strutt D. Strabismus is asymmetrically localised and binds to Prickle and Dishevelled during Drosophila planar polarity patterning. Development. 2003;130(13):3007–3014. doi: 10.1242/dev.00526. [DOI] [PubMed] [Google Scholar]
- 31.Zhao L, et al. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res. 2011;39(11):4664–4679. doi: 10.1093/nar/gkr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jolma A, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20(6):861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho B, Pierre-Louis G, Sagner A, Eaton S, Axelrod JD. Clustering and negative feedback by endocytosis in planar cell polarity signaling is modulated by ubiquitinylation of prickle. PLoS Genet. 2015;11(5):e88027. doi: 10.1371/journal.pgen.1005259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tree DR, Shulman JM, Rousset R, Scott MP, Gubb D, Axelrod JD. Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell. 2002;109(3):371–381. doi: 10.1016/S0092-8674(02)00715-8. [DOI] [PubMed] [Google Scholar]
- 35.Voynow JA, Gendler SJ, Rose MC. Regulation of mucin genes in chronic inflammatory airway diseases. Am J Respir Cell Mol Biol. 2006;34(6):661–665. doi: 10.1165/rcmb.2006-0035SF. [DOI] [PubMed] [Google Scholar]
- 36.Wojnarowski C, et al. Cytokine expression in bronchial biopsies of cystic fibrosis patients with and without acute exacerbation. Eur Respir J. 1999;14(5):1136–1144. doi: 10.1183/09031936.99.14511369. [DOI] [PubMed] [Google Scholar]
- 37.Bonfield TL, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152(6 Pt 1):2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 38.Hauber HP, Gholami D, Koppermann G, Heuer HE, Meyer A, Pforte A. Increased expression of Interleukin-13 but not Interleukin-4 in cystic fibrosis patients. J Cyst Fibros. 2003;2(4):189–194. doi: 10.1016/S1569-1993(03)00091-2. [DOI] [PubMed] [Google Scholar]
- 39.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L715–L731. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hajj R, Lesimple P, Nawrocki-Raby B, Birembaut P, Puchelle E, Coraux C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J Pathol. 2007;211(3):340–350. doi: 10.1002/path.2118. [DOI] [PubMed] [Google Scholar]
- 41.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13(9):3218–3234. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aldallal N, et al. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am J Respir Crit Care Med. 2002;166(9):1248–1256. doi: 10.1164/rccm.200206-627OC. [DOI] [PubMed] [Google Scholar]
- 43.Ribeiro CM, et al. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem. 2005;280(18):17798–17806. doi: 10.1074/jbc.M410618200. [DOI] [PubMed] [Google Scholar]
- 44.Guirao B, et al. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat Cell Biol. 2010;12(4):341–350. doi: 10.1038/ncb2040. [DOI] [PubMed] [Google Scholar]
- 45.Deans MR, Antic D, Suyama K, Scott MP, Axelrod JD, Goodrich LV. Asymmetric distribution of prickle-like 2 reveals an early underlying polarization of vestibular sensory epithelia in the inner ear. J Neurosci. 2007;27(12):3139–3147. doi: 10.1523/JNEUROSCI.5151-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitchell B, Stubbs JL, Huisman F, Taborek P, Yu C, Kintner C. The PCP pathway instructs the planar orientation of ciliated cells in the Xenopus larval skin. Curr Biol. 2009;19(11):924–929. doi: 10.1016/j.cub.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagaoka T, Inutsuka A, Begum K, Bin hafiz Km, Kishi M. Vangl2 regulates E-cadherin in epithelial cells. Sci Rep. 2014;4:e88027. doi: 10.1038/srep06940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tatin F, et al. Planar cell polarity protein Celsr1 regulates endothelial adherens junctions and directed cell rearrangements during valve morphogenesis. Dev Cell. 2013;26(1):31–44. doi: 10.1016/j.devcel.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lamonica K, Grabel L. The planar cell polarity pathway and parietal endoderm cell migration. Methods Mol Biol. 2012;839:187–200. doi: 10.1007/978-1-61779-510-7_15. [DOI] [PubMed] [Google Scholar]
- 50.Cirone P, Lin S, Griesbach HL, Zhang Y, Slusarski DC, Crews CM. A role for planar cell polarity signaling in angiogenesis. Angiogenesis. 2008;11(4):347–360. doi: 10.1007/s10456-008-9116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heijerman H. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros. 2005;4 Suppl 2:3–5. doi: 10.1016/j.jcf.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 52.LeSimple P, Liao J, Robert R, Gruenert DC, Hanrahan JW. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J Physiol (Lond) 2010;588(Pt 8):1195–1209. doi: 10.1113/jphysiol.2009.182246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kreda SM, et al. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol Biol Cell. 2005;16(5):2154–2167. doi: 10.1091/mbc.E04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tao H, et al. Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet. 2011;88(2):138–149. doi: 10.1016/j.ajhg.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ostrowski LE, Hutchins JR, Zakel K, O’Neal WK. Targeting expression of a transgene to the airway surface epithelium using a ciliated cell-specific promoter. Mol Ther. 2003;8(4):637–645. doi: 10.1016/S1525-0016(03)00221-1. [DOI] [PubMed] [Google Scholar]
- 56.Tan FE, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140(20):4277–4286. doi: 10.1242/dev.094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Follenzi A, Ailles LE, Bakovic S, Geuna M, Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet. 2000;25(2):217–222. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- 58.Horani A, Nath A, Wasserman MG, Huang T, Brody SL. Rho-associated protein kinase inhibition enhances airway epithelial Basal-cell proliferation and lentivirus transduction. Am J Respir Cell Mol Biol. 2013;49(3):341–347. doi: 10.1165/rcmb.2013-0046TE. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.