

Abstract
Neurodegenerative disorders such as Alzheimer's Disease, Parkinson's Disease, fronto-temporal dementia, Huntington's Disease and Creutzfeldt-Jakob Disease (CJD) are characterized by progressive accumulation of protein aggregates in selected brain regions. Protein misfolding and templated assembly into aggregates might result from an imbalance between protein synthesis, aggregation and clearance. While protein misfolding and aggregation occur in most neurodegenerative disorders, the concept of spreading and infectivity of aggregates in the CNS has been reserved to prion diseases such as CJD and bovine spongiform encephalopathy. Emerging evidence suggests that prion-like spreading may occur in other neurodegenerative disorders, taking place with secreted proteins, such as amyloid-β,) and cytosolic proteins, such as tau, huntingtin and α-synuclein. Underlying molecular mechanisms and therapeutic implications are discussed.
Neurodegenerative disorders with accumulation of amyloidogenic proteins such as Alzheimer's disease (AD) and Parkinson's disease (PD) are major causes of morbidity in the aging population. Development of novel therapies will require understanding not only why proteins locally accumulate but how this pathology spreads throughout the brain. In fact, progressive accumulation of protein aggregates across specific brain regions is now recognized as a critical characteristic in many major neurodegenerative disorders1. For example, in AD, amyloid β (Aβ) protein and tau accumulate in extracellular and intraneuronal compartments, while PD is characterized by accumulation of the synaptic protein α-synuclein in axons and neuronal cell bodies. In Huntington's disease (HD) and other expansion diseases polyglutamine (polyQ) proteins accumulate in both, the nucleus and the cytoplasm, and in Creutzfeldt-Jakob disease (CJD) misfolded prions accumulate in the neuropil. Some of these proteins are common to more than one disease. For example, tau aggregates are also found in fronto-temporal dementia (FTD), progressive supranuclear palsy, and other disorders that are now collectively referred to as tauopathies. Mixed Aβ and α-synuclein aggregates are also found in many diseases, including AD, dementia with Lewy bodies, and multiple system atrophy.
In many of these disorders neurodegeneration is likely to initiate at the synaptic site, where discrete protein aggregates -denominated oligomers- impair neuronal transmission and functioning. While oligomers are usually the diffusible, non-fibrillar, small order aggregates, larger polymers -in the form of amyloid fibrils- comprise the inclusion bodies and extracellular deposits that characterize these disorders and that are now believed to represent a pathway for sequestration of more toxic oligomers2. Varying degrees of toxicity have been associated with different types of Aβ oligomers3. These oligomers are thought to differ in their underlying structure and may follow different assembly pathways, some of which eventually lead to fibril formation3. Intermediates along the pathway from oligomer to fibril have also been reported to form ‘pore-like’ structures that may themselves disrupt cellular ionic homeostasis and contribute to cell death4.
The mechanism(s) through which oligomers are generated and that trigger neurodegeneration are under extensive investigation and alterations in the balance of protein synthesis, folding and clearance (either due to familial mutations or post-translational modifications) have all been postulated to play important roles (Fig. 1). Evidence from prion studies suggests that protein propagation might not only contribute to the spreading and progression of the disease but also to neurodegeneration5 (Fig. 1). Recent studies suggest that such protein spreading might also be at play in AD, PD, FTD, HD and other neurodegenerative disorders.
Figure 1. Aggregate clearing activity, seed formation, and aggregate burden.
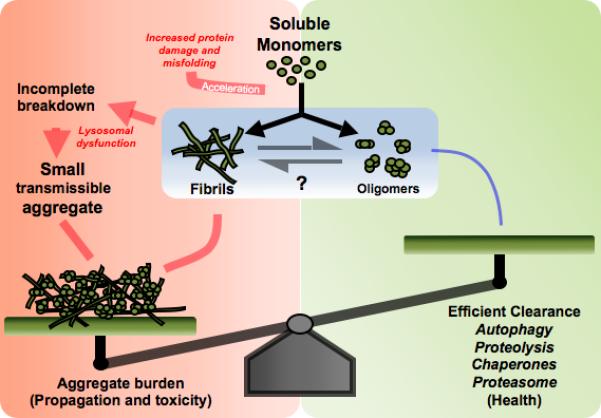
The formation of misfolded/aggregated protein is an inevitable outcome of a protein's life and is normally cleared by the cellular quality control mechanisms. In situation such as aging and disease, the combined effects of accelerated production, due to elevated oxidative stress, and the reduced ability of cells to degrade damaged proteins, increse protein aggregation, Incomplete degradation of aggregated proteins may result in the production of smaller fragments that can serve as seeds for further aggregation thereby increasing the aggregate burden. Therefore, reduced function in proteing degradation systems plays a critical role in aggregate propagation in neurodegenerative diseases.
There are contrasting views regarding the initiation of protein aggregation in neurodegenerative diseases. One hypothesis considers that formation of inclusion bodies is a multifocal event, with lesions in each brain region being independent of others. Alternatively, the pathology may initiate in a few discrete regions and then disseminate to other areas by a prion-like mechanism of propagation with spreading of protein aggregation. The staging of the pathological severity for AD6 and Lewy body disease7 suggests a predictable spreading pattern. In AD, tau aggregates first appear in the transentorhinal cortex and spreads through the hippocampal formation into broad areas of neocortex, whilst in PD, α-synuclein aggregation in the central nervous system (CNS) initiates in the lower brain stem nuclei and sequentially propagates into the midbrain, mesocortical and neocortical regions.
The main focus of this article will be to discuss a conceptual and mechanistic perspective emerging from new evidence that suggests that prion-like spreading of protein aggregates might play a role in non-prion neurodegenerative disorders.
Evidence for spreading of non-prion protein aggregates
Propagation of disease by “infectious” protein particles in the CNS has been reserved exclusively to prions for two decades. Outside of the CNS prion-like behavior has been described for systemic A-amyloidosis and ApoAII amyloidosis8,9. Emerging evidence from many neurodegenerative diseases has recently expanded the notion of spreading of protein aggregates to include several non-prion structures (Table 1). While evidence of propagation of Aβ, tau, α-synuclein and polyQ proteins have been extensively reported recently, still the mechanisms governing the spreading of non-prion aggregates remains to be elucidated.
Table 1.
Evidence for spreading of non-prion protein aggregates in the CNS
| Protein | Inoculums | Host | Propagation Effect |
|---|---|---|---|
| Amyloid β | Intracerebral injection of AD and APP tg brain homogenates | Amyloid precursor protein (APP) transgenic mice | Aβ deposition at injection site and adjacent brain structures17,18,34. |
| Tau22 | Tau fibrils | Neuronal cultured cells | Endocytic uptake of exogenous tau fibrils and induction of cytoplasmic endogenous tau proteins. Cell-to-cell transmission of uptaken tau22. |
| Intracerebral injection of brain extracts from tau transgenic mice | Transgenic mice expressing human wild type tau | Spreading of tau from site of injection to other brain structures23. | |
| α-synuclein | Aggregate producing-neuronal cell cultures | Neuronal cells | Endocytic uptake of α-synuclein aggregates36. |
| Transgenic mice overexpressing human α-synuclein | Mouse neuronal progenitor cells grafted into the mice brains | Interneuronal transmission of human α-synuclein36. | |
| Parkinson's disease patients brains | Fetal stem cells grafted into the PD brains | Interneuronal transmission of Lewy inclusions16,37-39. | |
| PolyQ proteins | In vitro generated polyQ peptide fibers | Mammalian cells in culture | Internalization of fibers with subsequent recruitment of soluble endogenous polyQ proteins and aggregate formation16. |
Molecular mechanisms of spreading
Seeded aggregation and conformational propagation
Despite the seemingly unrelated primary structures of monomer subunits, disease-linked protein aggregates have common conformational properties10, a compelling argument for the existence of a general pathogenic principle underlying different neurodegenerative disorders. In support of this hypothesis, protein aggregation processes in neurodegenerative diseases feature similar kinetics, where a long lag phase wherein nuclei are formed, precedes the aggregate growth. The addition of preformed aggregates eliminates the lag phase11, process known as “seeded” polymerization, which in combination with aggregate breakage, represents the theoretical basis for prion transmission/”infectivity”. This effect, widely investigated for polymers forming fibrils, may also exist for oligomers.
During the course of seeded polymerization, subtle conformational variations in aggregates are self-propagated by converting newly added monomers into their particular structures. This conformational propagation, characteristic of prion strains, has recently been reported for other disease-linked proteins, such as Aβ12, α-synuclein13, and tau14, raising the possibility that neurodegenerative pathologies involving protein aggregation may spread within the brain via a mechanism analogous to prion-like self-propagation.
A critical issue in this hypothesis is whether the transferred cytoplasmic aggregates are indeed amplified via seeded polymerization of the endogenous protein. These mechanisms have been reported for both tau and polyQ aggregates, where addition of exogenous aggregates into cultures induced the aggregation of endogenous, otherwise soluble cytoplasmic proteins15,16. Internalized aggregates induced the aggregation of endogenous proteins16 of their own kind, suggesting that this process is driven by the sequence-specific interaction between the exogenous nuclei and the endogenous monomers. For example, cationic liposome-mediated delivery of α-synuclein fibrils into cultured cells induced the formation of Lewy-like inclusions17, whilst hippocampal injections of brain extracts from transgenic mice with distinct plaque patterns produced Aβ deposition in the host18. This prion strain-like phenomenon is a strong indication of the seeding-dependent self-propagation of aggregates. In addition, cross seeding between dissimilar proteins that share beta-sheet structure has been described. Although the best evidence for this propagation comes from non CNS amyloidoses (like amyloid A and ApoAII)9, cross seeding of Aβ and α-synuclein4, tau and α-synuclein19, and prion protein and Aβ20 has been demonstrated. Alternative mechanisms, such as the disruption of basic cellular proteostasis by exogenous aggregates, cannot be excluded21.
Reaching neighbor's cytosol
Unlike Aβ and prion, which are naturally exposed to the extracellular space, cytosolic proteins such as tau, α-synuclein, polyQ, and other disease-linked proteins, have to cross membrane barriers at least twice before they gain access to the neighboring cell's cytosol. Therefore, mechanisms must exist by which these proteins are released from neurons and subsequently enter another neuron's cytosol. These mechanisms may include exocytosis, cell injury, exosomes, and tunneling nanotubes (Fig. 2). α-synuclein and its aggregated forms are secreted at low levels in neuronal cultures. This secretion is increased under protein misfolding stress, suggesting the existence of release mechanisms susceptible to dysregulation under disease conditions22. Consistent with this notion, both monomeric and oligomeric forms of α-synuclein and tau are present in human blood and CSF from PD and AD patients, respectively23,24 suggesting that the release of normally intracellular proteins might be a common event in neurodegenerative disorders. In addition to secretory mechanisms, neuronal protein aggregates may also be released from severely injured neurons through disrupted membranes or pore-formation4.
Figure 2. Models of cell-to-cell protein transmission.
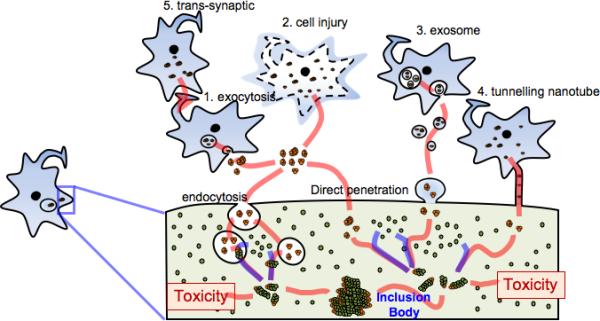
This diagram illustrates potential mechanisms by which misfolded/aggregated proteins are transmitted from one neuron to another. Proteins may be released from neurons via vesicle-mediated exocytosis (1) or simple leakage through damaged membranes (2), then internalize into neighboring neurons either via endocytosis or via direct penetration of the memebrane. Alternatively, as reported with the prion protein, proteins may be transmitted to the neighboring neurons by packaging into exosomes (3) or through tunneling nanotubes (4). Some or all of these mechanisms might work simultaneously, with a specific protein preferring a certain pathway to others. These mechanisms might act between the cell bodies, but could also occur trans-synaptically (5). Internalized aggregates (orange) might act as seeds for the aggregation of the endogenous native proteins (green). This seeded aggregation may produce toxic aggregate species during the course of dynamic aggregation process, which ultimately leads to the formation of pathological inclusion bodies.
Once released, protein aggregates, such as α-synuclein and tau15,25, are internalized through endocytosis and packaged into endocytic vesicles, therefore, another mechanism is required to gain access to the host cytosol (Fig. 2). It is worth noting that protein aggregates can disrupt lipid bilayers, like endocytic vesicle membranes, a mechanism that grants them access into the cytosolic space. Endocytosis and subsequent delivery to late endosomes may concentrate the internalized proteins, forming aggregates that might seed endogenous protein aggregation26. Direct membrane penetration is another plausible mechanism of entry, since internalized polyQ aggregates do not seem to be enclosed within vesicles16.
In the case of prions, intercellular transmission by means of exosomes27 or tunneling nanotubes28 has been suggested, and it is possible that similar mechanisms may play a role in the transmission of other aggregated proteins (Fig. 2).
Spreading through neural connections
Considering that α-synuclein, as well as other proteins involved in neurodegeneration, is enriched in the synaptic sites, the aggregates transferred from the postsynaptic cells would have ample source of native proteins to convert in the presynaptic terminal (Fig. 2). Cellular prions are transported via both fast anterograde and retrograde axonal transport and internalization and movement of the scrapie form of prion along neuronal processes has also been reported29. It is likely that the routes of aggregate transmission are determined by the unique cell biological characteristics of particular proteins, including where in the neuron does protein aggregation occur, what is the mechanism of intraneuronal transport and how are the aggregates released. In addition, synaptic activity may influence the release and transmission of cytosolic proteins, since synaptic stimulation has been shown to induce secretion of Aβ30, promote APP transport and to regulate synaptic localization of α-synuclein31, although precise mechanisms remain to be addressed.
Molecular species responsible for propagation and neurotoxicity
It is important to address which molecular species are responsible for aggregate propagation and whether these species are responsible for neurotoxicity. If the neurotoxic species are generated during the course of the seeding-dependent aggregate propagation process, are they generated as the final product or as relatively stable intermediates? Whether there is a common structural motif in these molecular species, regardless of the constituent subunits, is another important question. These considerations are highly relevant to diagnostic and therapeutic drug development.
A related issue is whether or not the aggregate propagation mechanism involves co-factors cooperating with the aggregate itself. Consistent with the involvement of co-factors, injection of pure protein aggregates of prion and Aβ showed either no transmission or very low transmission rates18,32, whereas injection of aggregate-containing tissue extracts showed efficient transmission processes. The cellular form of prion protein seems to have various functions and to interact with a long list of molecules (reviewed in33), suggesting that the pathogenic conversion of this protein may not occur as a free protein. Although the aggregate inducing abilities of pure protein aggregates of tau and polyQ proteins have been shown in tissue culture systems, they remain to be validated in animal models.
Distinct propagation properties of non-prion proteins
Although protein aggregation and spreading now appears to be a common feature of several neurodegenerative disorders, it remains unclear as to whether these disorders qualify as “infectious” diseases in a similar way to prion diseases. β-amyloidosis was not induced by oral, intravenous, intraocular, or intranasal inoculations in APP transgenic mice; however direct contact of the host brain with Aβ-contaminated wires triggered β-amyloidosis34. Differences in transmission may be attributed to the presence or absence of a system that transports seeding particles from the periphery to the CNS. The peripheral and systemic transmission of α-synuclein, tau, and polyQ proteins is yet unknown.
Aggregate propagation in prions can occur at a relatively fast and efficient pace. This propagation is accelerated when the aggregate breaks into smaller seeds that serve to recruit additional monomers (Fig. 1). Prion aggregate stability correlates with incubation period to terminal disease suggesting that a more fragile aggregate may rapidly break and produce seeds accelerating propagation29. Therefore, prions may be more easily fragmented in vivo producing small seeds than other protein aggregates. The stability of amyloid fibrils can vary dramatically and its effects on propagation remains to be determined.
The specific localization of soluble monomers or “seeds” may also play a role in the speed of protein recruitment. The prion protein is GPI-anchored in lipid rafts at the plasma membrane33, this high local concentration of protein may lead to more efficient conversion and spread at the cell surface than in the case of cytoplasmic aggregates.
Cellular clearance mechanisms counterbalance aggregate amplification (Fig. 1). The ease of protein clearance may vary with the aggregate conformation, size, stability and abundance; with the intracellular or extracellular trafficking patterns and with the cell type producing the aggregates. Any deficit in the cellular clearance mechanisms would amplify the aggregate formation by increasing the likelihood of interactions between the “seed” and the soluble/unaggregated form of the protein.
Effect of aggregate spreading
In a number of recent studies, experimental aggregate propagation of tau35 or Aβ plaques18 in murine models was often not accompanied by neuronal loss. There are three possible scenarios to explain this observation: 1) aggregate deposition may simply precede neuronal loss and other pathological changes; 2) aggregate propagation may be a critical factor but not in itself sufficient for neurodegeneration (in this case transmitted aggregates could cooperate with unknown factors, which are limited in the mouse models) and 3) the progression of protein aggregation and neurodegeneration may be entirely unrelated and independent events.
In this regard, it is interesting to note different responses related to the nature of the host brain. Mouse neuronal progenitor cells grafted into the brains of α-synuclein transgenic mice showed increased caspase 3 activation accompanying α-synuclein uptake36. In human graft studies, while there was no evidence for neuronal loss in the transplanted tissues37-39 -in addition to Lewy body-like inclusions- a reduction in dopamine transporter and tyrosine hydroxylase was observed in grafted dopamine neurons37, implicating a functional decline in the long-term. These results support the notion that the propagation of protein aggregates is linked to neurotoxicity. In human PD cases, spreading of Lewy bodies does not always appear to be associated with neuronal dysfunction and degeneration. One approach to resolve this issue would be to identify the molecular species of proteins that are responsible for aggregate spreading and to assess their abilities to induce pathology. Elucidation of the role of aggregate spreading in neurodegeneration not only increases our understanding of the basic mechanisms of disease progression but also provides crucial information needed to enhance our current approach to cell replacement therapy.
Therapeutic implications
Potential therapeutic opportunities targeting aggregate transmission should be explored. Passive and active immunizations have already been proposed as a promising new direction by studies showing successful reduction in protein aggregate loads in transgenic mouse models for AD (Aβ and tau)40, synucleinopathies and prion diseases. Although some approaches apparently have safety and clinical effectiveness issues, immunization targeting the transmitting aggregate proteins may still be a viable approach for the treatment. The identification of specific molecular species capable of spreading the aggregates will allow refinement of the immunization approach toward pathogen-specific therapy.
Another beneficial avenue may involve enhancing the endolysosomal pathway in general and antagonizing the specific receptor for internalization, although this bears the risk of undesirable side effects. If direct transmission were indeed the crucial mechanism of disease progression, the most attractive and specific therapeutic approach would be targeting the extracellular pathogenic protein aggregates. Specific binding agents to the aggregates might prevent the uptake and seeding in neurons. These agents may be chemical compounds, peptides, or ribonucleic acids and may function as blocking agents preventing disease progression and neurodegeneration or perhaps as tracers for presymptomatic diagnosis. Again, identification of the molecular species responsible for transmission of aggregates and neurotoxicity will refine the drug targets. If one can target a common conformational element that many pathogenic molecular species share, development of drugs treating for multiple neurodegenerative diseases might be possible.
Enhancing glia-mediated clearance of extracellular aggregates may be another possibility. Extracellular aggregates of Aβ and α-synuclein have been shown to activate and be cleared by both astrocytes and microglia41 with lysosomes being responsible for the degradation of internalized proteins in both neuronal and glial cells25. Several studies have shown that autophagy plays an important role in the degradation of aggregated proteins in the cytoplasm42, therefore it would be informative to investigate whether this mechanism is involved in the clearance of transmitted protein aggregates. Aging and age-related disorders are generally associated with functional decline of lysosomes43, and genetic depletion of lysosomal hydrolases causes many human neurodegenerative diseases. Stimulation of lysosomal degradation activity may therefore be another promising approach to therapeutics.
CONCLUSIONS
Prion-like spreading of aggregates may be a general principle underlying progressive neurodegeneration associated with protein misfolding, and has significant implications in cell replacement therapies. Understanding the mechanisms of pathological spreading is crucial to develop diagnostic techniques and novel therapies for protein misfolding-associated neurodegenerative diseases.
BOX 1. Definitions.
Several recent studies have reported the propagation of non-prion protein aggregates in many neurodegenerative diseases. While the idea of considering traditional neurodegenerative diseases as prion diseases is under debate, a clear definition of terminology will shed light on setting common features and particularities of the propagation mechanisms of both types of protein entities. For that purpose, we propose in this perspective to reserve the term infectivity to refer specifically to the conveyance of prion proteins from animal to animal, while the term transmission would be extended to include the conveyance of non-prion proteins. The terms propagation, spreading and dissemination, on the other hand, might be used interchangeably to refer to the dispersal of the protein over a larger area and from cell to cell.
ACKNOWLEDGMENTS
We thank Changyoun Kim for the assistance in illustrations. This work was supported by NIH grants AG18440, AG022074, AG11385 and AG10435, by the Disease Network Research Program (20090084180) from the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology, Republic of Korea and by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (20090083737).
REFERENCES
- 1.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 2.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 3.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsigelny IF, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS One. 2008;3:e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 8.Solomon A, et al. Amyloidogenic potential of foie gras. Proc Natl Acad Sci U S A. 2007;104:10998–11001. doi: 10.1073/pnas.0700848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sponarova J, Nystrom SN, Westermark GT. AA-amyloidosis can be transferred by peripheral blood monocytes. PLoS One. 2008;3:e3308. doi: 10.1371/journal.pone.0003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 11.Jarrett JT, Lansbury PT., Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 12.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 13.Yonetani M, et al. Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem. 2009;284:7940–7950. doi: 10.1074/jbc.M807482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem. 2009;284:3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren PH, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luk KC, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 19.Kotzbauer PT, et al. Fibrillization of alpha-synuclein and tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation. Exp Neurol. 2004;187:279–288. doi: 10.1016/j.expneurol.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Morales R, et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J Neurosci. 2010;30:4528–4535. doi: 10.1523/JNEUROSCI.5924-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 22.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Agnaf OM, et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson's disease. FASEB J. 2006;20:419–425. doi: 10.1096/fj.03-1449com. [DOI] [PubMed] [Google Scholar]
- 24.Vandermeeren M, et al. Detection of tau proteins in normal and Alzheimer's disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem. 1993;61:1828–1834. doi: 10.1111/j.1471-4159.1993.tb09823.x. [DOI] [PubMed] [Google Scholar]
- 25.Lee HJ, et al. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Hu X, et al. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fevrier B, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–9688. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gousset K, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol11. 2009:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 29.Legname G, Nguyen H-OB, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cirrito JR, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 31.Fortin DL, et al. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Legname G, et al. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 33.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 34.Eisele YS, et al. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009;106:12926–12931. doi: 10.1073/pnas.0903200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paula Desplats H-JL. Eun-Jin Bae, Christina Patrick, Edward Rockenstein, Leslie Crews, Brian Spencer, Eliezer Masliah and Seung-Jae Lee. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci U S A106. 2009:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 38.Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 39.Mendez I, et al. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat Med. 2008;14:507–509. doi: 10.1038/nm1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wyss-Coray T, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 43.Bahr BA, Bendiske J. The neuropathogenic contributions of lysosomal dysfunction. J Neurochem. 2002;83:481–489. doi: 10.1046/j.1471-4159.2002.01192.x. [DOI] [PubMed] [Google Scholar]