

SUMMARY
Emerging evidence suggests that many proteins may be regulated through cysteine modification, but the extent and functions of this signaling remain largely unclear. The endoplasmic reticulum (ER) transmembrane protein IRE-1 maintains ER homeostasis by initiating the unfolded protein response (UPRER). Here we show in C. elegans and mammalian cells that IRE-1 has a distinct redox-regulated function in cytoplasmic homeostasis. Reactive oxygen species (ROS) that are generated at the ER or by mitochondria sulfenylate a cysteine within the IRE-1 kinase activation loop. This inhibits the IRE-1-mediated UPRER, and initiates the p38/SKN-1(Nrf2) antioxidant response, thereby increasing stress resistance and lifespan. Many AGC-family kinases (AKT, p70S6K, PKC, ROCK1) seem to be regulated similarly. The data reveal that IRE-1 has an ancient function as a cytoplasmic sentinel that activates p38 and SKN-1(Nrf2), and indicate that cysteine modifications induced by ROS signals can direct proteins to adopt unexpected functions, and may coordinate many cellular processes.
eTOC BLURB
We report that the transmembrane endoplasmic reticulum (ER) stress sensor IRE-1 has a redox-regulated cytoplasmic signaling function. Localized ROS sulfenylate an IRE-1 cysteine, inhibiting its ER functions and initiating the antioxidant response. IRE-1 therefore monitors cytoplasmic homeostasis through localized ROS signaling, suggesting that cysteine modifications have broad and diverse regulatory functions.
INTRODUCTION
Organisms encounter stresses that include reactive small molecules from metabolic or exogenous sources, and accumulation of misfolded or damaged proteins. To defend against these perturbations, complex stress responses have evolved that attempt to restore homeostasis and repair damage (Fulda et al., 2010). It is an intriguing question how stresses might be perceived in a rapid manner that minimizes damage, and how stress responses might be coordinated with each other and with other cellular processes.
In the endoplasmic reticulum (ER), where secretory and membrane-bound proteins are synthesized, accumulation of misfolded ER proteins (ER stress) triggers a complex unfolded protein response (UPRER) (Ron and Walter, 2007; Wang and Kaufman, 2014). This response includes activation of genes that promote ER homeostasis. The most ancestral transducer of the transcriptional UPRER is the ER transmembrane kinase/RNase Ire1 (Figure S1A)(Maly and Papa, 2014; Ron and Walter, 2007). In the presence of unfolded proteins, IRE-1 oligomerizes and is autophosphorylated by its cytoplasmic kinase (Ron and Walter, 2007; Wang and Kaufman, 2014). These events activate the Ire1 RNAse, which splices the mRNA that encodes the UPRER transcription factor XBP1 (Figure S1A). IRE-1 is important in development, immunity, and metabolism, and is dysfunctional or a potential therapeutic target in many human diseases (Fu et al., 2012; Hetz et al., 2013; Wang and Kaufman, 2014). The various biological functions of IRE-1 are thought to derive from its role as an unfolded protein sensor.
ER protein folding depends upon appropriate crosslinking of cysteines (Cys), which requires that the ER maintain an oxidizing environment that would be deleterious to the cell at large (Kakihana et al., 2012; Sevier and Kaiser, 2008). An antioxidant response that defends against reactive oxygen species (ROS) and other small molecules is orchestrated by the transcription factor Nrf2, and its C. elegans ortholog SKN-1 (Blackwell et al., 2015; O’Connell and Hayes, 2015; Suzuki and Yamamoto, 2015). This response promotes redox homeostasis, detoxifies small molecules, enhances proteostasis, regulates metabolism, and has been implicated in lifespan extension in various organisms. Nrf2 activity is increased when electrophiles interact with the Nrf2-binding ubiquitin ligase adaptor Keap1, allowing Nrf2 to escape degradation (Suzuki and Yamamoto, 2015). The paradigm of Nrf2 regulation by Keap1 has been studied in great detail, but it is still unclear how Nrf2 is regulated by some ROS-based stimuli (Jomova et al., 2011; Lau et al., 2013). Moreover, C. elegans lacks a Keap1 ortholog, implying that additional mechanisms regulate SKN-1/Nrf2 responses to stress. In C. elegans, ROS and other stresses increase SKN-1 levels in nuclei within the intestine, the digestive system counterpart (Blackwell et al., 2015). This generally requires phosphorylation of SKN-1 by the p38 mitogen-activated protein kinase (MAPK) (Figure S1B) (Inoue et al., 2005). ROS activate p38 pathway signaling in C. elegans and mammals (Inoue et al., 2005; Nadeau et al., 2007), but the primary trigger of this response is unknown.
While high levels of ROS damage proteins, a growing body of evidence suggests that ROS also have physiological signaling functions that are mediated through oxidation of specific Cys residues (D’Autreaux and Toledano, 2007; Gould et al., 2015; Holmstrom and Finkel, 2014; Paulsen et al., 2012; Tonks, 2005). Many examples have been described in which stress or exogenously provided ROS induce modifications that affect protein function (D’Autreaux and Toledano, 2007; Holmstrom and Finkel, 2014), and in some cases endogenously generated ROS have been demonstrated to regulate proteins physiologically. For example, certain growth factors induce NADPH oxidase (NOX) enzymes to generate ROS that inhibit phosphatases (Bae et al., 1997; Finkel, 2011; Sundaresan et al., 1995; Tonks, 2005), and potentiate epidermal growth factor receptor kinase activity (Paulsen et al., 2012). In brown adipocytes, cold induces production of mitochondrial ROS that oxidize uncoupling protein 1, leading to thermogenesis (Chouchani et al., 2016). Remarkably, in cultured human cells over 700 proteins are prone to Cys oxidation, in patterns that can be altered by oxidizing or growth conditions (Gould et al., 2015; Yang et al., 2014). The number of cellular processes that might be regulated physiologically through such modifications remains an open question, and it is largely unknown how versatile these modifications are with respect to their effects on protein function.
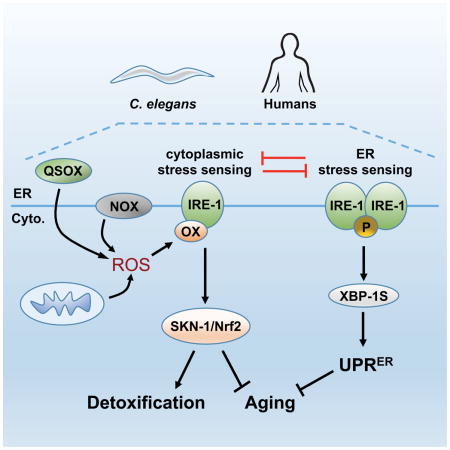
Here we show that Ire1 not only acts in the secretory pathway, but also has an evolutionarily conserved function as a sentinel that maintains cytoplasmic redox homeostasis. Localized ROS production at the ER leads to rapid oxidation of a single Cys within the Ire1 kinase active site. This modification inhibits the UPRER and directs Ire1 to adopt a distinct function in which it activates p38/SKN-1(Nrf2), and thereby promotes both stress resistance and longevity in C. elegans. This molecular switch reveals remarkable versatility in redox-based signaling, and suggests a paradigm by which physiological Cys modifications may coordinate numerous cellular processes.
RESULTS
Mutually exclusive UPRER and antioxidant response functions of IRE-1
Previously we observed that in C. elegans, RNA interference (RNAi) knockdown of ire-1 largely blocked activation of the p38/SKN-1 antioxidant response (Glover-Cutter et al., 2013). We investigated why this would be the case, first by asking whether ire-1 is required for p38 to be activated specifically by ROS. RNAi against ire-1 diminished acute p38 activation by the ROS generators sodium arsenite (AS) and paraquat (PQ), but not osmotic or heat stress (Figure 1A). The SKN-1 antioxidant response can be monitored in vivo with the gst-4P::GFP reporter, in which the SKN-1 target gst-4 promoter drives GFP (green fluorescent protein) expression (Glover-Cutter et al., 2013). Only AS and PQ required ire-1 to activate gst-4P::GFP (Figure 1B), and ire-1(ok799) nulls were defective in AS-induced p38 activation and sensitive to oxidative stress (Figure S1C and S1D; Table S1). Treatment with the antioxidant glutathione (GSH) inhibited AS-induced ROS production, p38 activity, and gst-4P::GFP expression (Figure S1E–H). ire-1 therefore appears to be required specifically for ROS to activate p38/SKN-1, and is important for redox homeostasis.
Figure 1. Distinct IRE-1-dependent responses to ROS and ER stress.

(A–B) IRE-1 is required for the C. elegans p38/SKN-1 response to ROS. ire-1 RNAi diminished the p38 (A) and gst-4P::GFP (B) responses to sublethal exposure to AS (30 min) or PQ (30 min), but not Sorbitol (ST, 30 min) or heat (30 min). Phosphorylation of p38 (P-p38) indicates p38 activation (Inoue et al., 2005). (C–E) Acute AS treatment does not activate the UPRER. AS (30 min) did not upregulate hsp-4P::GFP (C), with intestinal GFP scoring in (D), or promote IRE-1 and HSP-3/4 dissociation as determined by co-immunoprecipitation (co-IP) (E). (F–I) AS pre-treatment blocks IRE-1 and UPRER activation in response to ER stress. Prior AS exposure (30 min) inhibited IRE-1 phosphorylation (F), xbp-1 mRNA splicing, as indicated by quantitiative PCR assay of levels of the indicated mRNA forms (G), XBP-1S protein accumulation (H), and hsp-4P::GFP induction (I–J) by ER stress (TM; 5 hr). AS-treated samples were allowed to recover for 30 min. prior to TM exposure. (K) AS does not inhibit the PERK kinase PEK-1, as indicated by phosphorylation of its substrate eIF2-α. In the right panels PEK-1 expression was blocked by RNAi. (L–O) Pre-exposure to ER stress inhibits the antioxidant response. Exposure to TM (5 hr) prior to AS treatment (30 min.) decreased p38 activation (L), SKN-1 nuclear localization (M) and intestinal gst-4P::GFP expression (N–O). In all figures, mock incubations and zero time points are indicated by “C”. (B, D, J, M, O) GFP quantification with high (H), medium (M) or low (L) scoring. p < 0.0001****; p < 0.001***; p < 0.01**. All immunoblots in the paper are representative of at least two and in most cases three experiments. See also Figure S1.
Our current understanding of IRE-1 predicts that this requirement would be mediated through its canonical UPRER function. Prolonged ER stress may lead to cytoplasmic oxidative stress (Chaudhari et al., 2014; Kakihana et al., 2012), but we had assayed p38 activity after AS treatment for only 30 min, under conditions that required 12 hr to result in death (Figure S1D). C. elegans UPRER activation can be monitored with a transcriptional reporter for hsp-4, an ortholog of the ER chaperone BiP (Figure S1A)(Calfon et al., 2002; Henis-Korenblit et al., 2010; Taylor and Dillin, 2013). In contrast to ER stress from tunicamycin (TM), acute AS treatment did not activate hsp-4p::GFP (Figure 1C and 1D) or increase levels of the fluorescent protein Pnhx-2cpl-1W32AY35A::YFP, which misfolds and accumulates under ER stress conditions (Miedel et al., 2012) (Figure S1I). AS also did not induce disruption of the IRE-1/HSP-4(BiP) complex, a key step in UPRER activation (Figure 1E and S1A)(Ron and Walter, 2007). On the other hand, ER stress did not increase p38 activity (Figure S1J). We conclude that AS-induced ROS trigger rapid IRE-1-dependent p38/SKN-1 antioxidant signaling without activating the UPRER.
We next asked whether the IRE-1-dependent responses to ROS and ER stress might interfere with each other. Remarkably, short-term AS treatment reduced the IRE-1-mediated response to ER stress, as indicated by IRE-1 phosphorylation, xbp-1 mRNA splicing, production of the spliced form of the XBP-1 protein (XBP-1S), and hsp-4P::GFP expression (Figure 1F–J). This suggests that acute ROS exposure inhibits the UPRER function of IRE-1. The UPRER also involves IRE-1-independent mechanisms, including suppression of protein synthesis that is triggered by activation of the ER membrane kinase PERK (Ron and Walter, 2007; Wang and Kaufman, 2014). Treatment with AS increased activity of the C. elegans PERK ortholog PEK-1 (Figure 1K), suggesting that ROS exposure interferes specifically with IRE-1 and not all UPRER mechanisms. Having observed that pre-exposure to ROS antagonized IRE-1/XBP-1S signaling, we investigated whether ER stress might interfere with IRE-1-dependent p38/SKN-1 activation by ROS. Pretreatment with the ER stressor TM at a sub-lethal dose (Glover-Cutter et al., 2013) suppressed AS-induced p38 signaling, accumulation of a SKN-1::GFP fusion in intestinal nuclei, and gst-4P::GFP reporter activation (Figure 1L–O). p38 and SKN-1 target gene activation were similarly diminished when we activated the UPRER genetically by knocking down the retro-translocon regulator tfg-1 (Levi-Ferber et al., 2014) (Figure S1K–M). IRE-1 therefore mediates responses to acute ER stress and ROS through mechanisms that are distinct, and mutually exclusive.
IRE-1 kinase domain sulfenylation drives the antioxidant response
Having determined that ROS exposure acutely inhibits the canonical UPRER activity of IRE-1, we investigated whether IRE-1 might become modified by ROS. The initial step in Cys oxidation is conversion of the thiol group (SH) to sulfenic acid (SOH)(Figure 2A and Figure S2A). This modification can be detected specifically with a chemical probe (Dimedone or its derivatives)(Yang et al., 2014)(Figure 2A). Using a biotin-linked Dimedone derivative (DCP-BIO1), we discovered that C. elegans IRE-1 is readily and transiently sulfenylated (IRE-1:SOH) in vivo by AS-generated ROS, with the extent of sulfenylation peaking at around 30 min. (Figure 2B and 2C). The AS-dependence of this SOH signal eliminated the possibility that it arose through post-lysis oxidation. AS treatment did not alter the gel mobility distribution of IRE-1 (Figure S2B), suggesting that sulfenylated IRE-1 did not progress to disulfide bond formation. The time course of AS-induced p38 activity roughly paralleled that of IRE-1 sulfenylation (Figure 2D), consistent with the idea that IRE-1 sulfenylation might be involved in MAPK activation. Our finding that ER stress antagonizes the IRE-1/p38/SKN-1 response to ROS (Figure 1L–1O) suggested that IRE-1 sulfenylation might also be inhibited. Accordingly, ER stress induced by either TM or tfg-1 RNAi blocked subsequent IRE-1 sulfenylation (Figure 2E and S2C).
Figure 2. IRE-1 sulfenylation within the kinase activation loop.

(A) Capture of sulfenylated (SOH) proteins with DCP-BIO1. (B) ROS induce sulfenylation of IRE-1. Lysates from animals treated with AS (30 min) in the absence or presence of GSH were incubated with DCP-BIO1, with sulfenylated IRE-1 (IRE-1:SOH) identified by immunoblot. (C–D) sulfenylation of IRE-1 parallels p38 activation. Animals treated with AS for 0–120 min were assessed for IRE-1:SOH (C) and active p38 (D) by immunoblot. (E) ER stress interferes with IRE-1 sulfenylation. Animals pretreated with TM (5 hr) show reduced IRE-1:SOH in response to AS (30 min). (F–G) Conservation of C663 within the IRE-1 kinase domain. In (F), adjacent basic amino acids are labeled in bold. A model of the IRE-1 kinase domain based upon the human Ire1α crystal structure (PDB ID: 3P23) shows C663 protruding into the ATP-binding pocket (G). (H) C663 is the site of IRE-1 sulfenylation. IRE-1:SOH was not detected in IRE-1C663S animals after AS treatment (30 min). In (H–J), results are representative of two independent transgenic lines. (I) C663 mutation does not interfere with ER stress-induced XBP-1S production. XBP-1S protein levels were determined in ire-1 (ok799) animals and animals expressing either IRE-1WT or IRE-1C663S following TM (5 hr) treatment. (J) C663 is required for ROS-induced p38 activation. IRE-1C663S animals exhibit diminished AS-induced p38 activity comparable to ire-1(ok799) null animals. (K) IRE-1C663S animals exhibit enhanced oxidative stress sensitivity compared to ire-1(ok799) that was rescued with WT IRE-1. Day-1 adult worms were exposed to 5mM AS (p < 0.01**). (L) IRE-1 sulfenylation is not compatible with IRE-1 phosphorylation. The sulfenylated IRE-1 fraction, which was isolated by Biotin (Dimedone) pull-down, does not contain any detectable phosphorylated IRE-1. Short exposure (S.E); long exposure (L.E). See also Figure S2 and Table S1.
We sought to identify the sulfenylated cysteine in IRE-1. Cys residues are differentially prone to oxidation if they are located near basic amino acids that may stabilize their thiol group as an anion (Poole, 2015). C663 of IRE-1 is widely conserved, and located adjacent to basic amino acids within the activation loop of the IRE-1 kinase domain (Figure 2F and 2G). It is also close to the Mg2+-binding DFG motif, which is critical for kinase function (Bayliss et al., 2012). We generated ire-1(ok799) nulls that transgenically expressed wild-type IRE-1 (IRE-1WT), or IRE-1 in which C663 was mutated to serine (IRE-1C663S). AS induced rapid in vivo sulfenylation of IRE-1WT but not IRE-1C663S, suggesting that IRE-1 is sulfenylated uniquely at C663 (Figure 2H). Rescue with IRE-1WT and IRE-1C663S comparably allowed xbp-1 mRNA splicing in response to ER stress, indicating that this mutation does not disrupt IRE-1 folding or its canonical UPRER functions (Figure 2I). In striking contrast, the IRE-1C663S mutation prevented AS from activating p38 above the ire-1(−) mutant background (Figure 2J). Accordingly, the IRE-1C663S transgenics and ire-1(ok799) nulls were comparably sensitive to oxidative stress (Figure 2K; Table S1). We speculated that sulfenylation of IRE-1 at C663 might inhibit its kinase activity, given the critical location of this residue. ER stress strongly increased IRE-1 phosphorylation, as predicted (Ron and Walter, 2007), but elicited only a low level of IRE-1 sulfenylation (Figure 2L). In striking contrast, the sulfenylated IRE-1 population appeared to be completely unphosphorylated (Figure 2L), consistent with evidence that AS inhibited ER stress-induced IRE-1 phosphorylation (Figure 1F). Together, the data suggest that when IRE-1 is exposed to ROS in the cytoplasm, sulfenylation at C663 inhibits its canonical kinase and UPRER activities, but is required for p38/SKN-1 pathway activation.
Localized ROS production initiates the p38 cascade at IRE-1
We next investigated how IRE-1 sulfenylation promotes p38 signaling. In C. elegans, the p38/SKN-1 antioxidant response depends largely upon the p38 MAPKKK NSY-1 (Figure S1B) (Inoue et al., 2005). We found that NSY-1 forms a complex with IRE-1, and that this was increased by AS treatment (Figure 3A). In mammalian cells, tumor necrosis factor-alpha and other peptide factors activate p38 by recruiting MAPKKK to their membrane receptor through bridging by TRAF2 (Cuadrado and Nebreda, 2010). Knockdown of C. elegans TRAF2 (trf-1) decreased IRE-1/NSY-1 complex formation, AS-induced p38 phosphorylation, SKN-1 nuclear accumulation, and gst-4P::GFP activation, and increased oxidative stress sensitivity (Figure 3B–3F and S3A; Table S1). By contrast, trf-1 RNAi did not interfere with AS-induced sulfenylation of IRE-1 (Figure 3G), indicating that TRF-1 is not required for AS-induced ROS production. Knockdown of trf-1 also did not reduce UPRER activation by ER stress, indicating that TRF-1 is not required for this aspect of IRE-1 function (Figure S3B and S3C). The data indicate that TRF-1 allows the MAPKKK NSY-1 to be recruited to IRE-1, and that this interaction is essential for activation of the p38 response to ROS.
Figure 3. A local ROS signal initiates the IRE-1/p38/SKN-1 response.

(A) AS promotes IRE-1 and NSY-1 complex formation. IRE-1 was IP’d from C. elegans that had been treated with vehicle or AS (30 min), then assayed by immunoblot for NSY-1. Non-specific IgG acted as control. (B) trf-1 promotes AS-induced IRE-1 and NSY-1 interaction. Lysates of animals fed control (EV) or trf-1 RNAi and treated with AS (30 min) were assayed by immunoblot for IRE-1/NSY-1 interaction. (C–E) trf-1 is required for the p38/SKN-1 response to AS. Knockdown of trf-1 diminished the p38 (C) and gst-4P::GFP (D) responses to AS (30 min), with GFP scoring shown in (E). (F) Reduced TRF-1 levels sensitize to oxidative stress. Day 1 adults were scored for survival in 5mM AS. (G) Sulfenylation of NSY-1 but not IRE-1 requires trf-1. Knockdown of trf-1 blocked AS-induced (30 min) sulfenylation of NSY-1. (H) AS induces NSY-1 phosphorylation comparably in control (EV) and ire-1(RNAi) animals. (I) AS induces NSY-1 sulfenylation in an ire-1-dependent manner. Animals fed either control or ire-1 RNAi were assessed for NSY-1:SOH after AS (30 min) treatment. (J–K) bli-3 (DUOX) is required for the p38/SKN-1 response to AS. bli-3 RNAi reduced intestinal gst-4P::GFP activation by AS (30 min) (J) with intestinal GFP scoring in (K). (L) AS exposure (30 min) increases BLI-3 activity. NOX-dependent superoxide production was measured by chemiluminescence. (M) AS-induced sulfenylation of IRE-1 was diminished by bli-3 RNAi. (N) bli-3 is required for the p38 response to AS. bli-3 RNAi decreased AS-induced p38 activity. (O) Interaction of IRE-1 and BLI-3 is enhanced by AS, assessed by co-IP. (E, K) GFP scored as high (H), medium (M) or low (L). p < 0.0001****; p < 0.01**; p < 0.05*. See also Figure S3.
We examined how NSY-1 becomes activated when bound to IRE-1. Phosphorylation within the kinase activation loop is critical for MAPKKK activation (Raman et al., 2007). Importantly, AS treatment induced activating phosphorylation of NSY-1 independently of ire-1 (Figure 3H), indicating that IRE-1 must be required for a different NSY-1 activation step. NSY-1 lacks a Cys at DFG+2 (not shown), but was sulfenylated robustly in response to AS treatment (Figure 3G and 3I). This NSY-1 sulfenylation required both ire-1 and trf-1, in contrast to NSY-1 phosphorylation (Figure 3G–3I). Thus, while IRE-1 sulfenylation at C663 inhibits its kinase, sulfenylation of NSY-1 at a different position correlates with its interaction with IRE-1, and p38 cascade activation (Figure S3D). Together, the data suggest that AS-induced activation of the p38 cascade depends upon TRF-1-dependent recruitment of phosphorylated NSY-1 to an oxidizing environment at IRE-1, and the cytoplasmic surface of the ER membrane (Figure S3D).
Cellular antioxidant mechanisms limit the extent of ROS diffusion, making it necessary for ROS signals to be produced locally with respect to the target (Winterbourn, 2008). How might AS induce production of a localized pool of ROS in the vicinity of IRE-1? AS can damage proteins and lipids directly, but it is not known how AS exposure induces ROS production (Hughes et al., 2011). NOX enzymes represent a potential source of inducible ROS production. These enzymes generate superoxide anions by reducing NADPH, are produced in the ER, and are present in membrane structures (Lambeth and Neish, 2014; Laurindo et al., 2014). They have well-described roles in pathogen defense and have been implicated in signaling by some growth factors, but less is known about their other functions. The two C. elegans NOX enzymes (BLI-3 and DUOX-2) are dual oxidases (DUOXs), which may directly convert superoxide to hydrogen peroxide. Knockdown of bli-3 but not duox-2 dramatically attenuated the gst-4p::GFP response to AS (Figure 3J, 3K and S3E). AS treatment increased BLI-3 activity, leading to ROS production that was blocked by a NOX inhibitor (Figure 3L). Accordingly, bli-3 RNAi diminished AS-induced IRE-1 sulfenylation and p38 activation (Figure 3M and 3N). IRE-1 and BLI-3 interacted physically, and their association was increased by AS treatment (Figure 3O). We conclude that AS causes stress that leads to activation of BLI-3 that is at or near the ER, leading to ROS production. These ROS signal locally through IRE-1 to activate the p38/SKN-1 antioxidant response.
p38 activation and longevity induced by ROS at the ER
Our model predicts that any stimulus that generates ROS in the vicinity of the ER could activate the IRE-1/p38/SKN-1 pathway. Mitochondria are a major source of cellular ROS, and perturbations in mitochondrial respiration can increase ROS production to levels that are devastating. The ER interacts physically with mitochondria (Kornmann, 2013), raising the question of whether mitochondrially-produced ROS might signal through IRE-1. PQ induces mitochondrial ROS production by inhibiting complex 1 of the electron transport chain (Yang and Hekimi, 2010). Accordingly, PQ but not AS activated the mitochondrial UPR (Pellegrino et al., 2014), a biomarker of mitochondrial ROS production (Figure S4A). PQ rapidly induced IRE-1 sulfenylation (Figure 4A), as expected from the ire-1-dependence of the p38/SKN-1 response to PQ (Figure 1A and 1B). PQ-induced appearance of IRE-1/p38/SKN-1 response markers was diminished by the antioxidant Mito-Tempo (MT), which scavenges mitochondrial ROS specifically (Figure 4A–4D). Importantly, and in contrast to AS, PQ activated this response independently of the NOX enzyme bli-3 (Figure S4B) (Hoeven et al., 2011). We conclude that the p38 antioxidant response is initiated at IRE-1 in response to localized ROS that can be generated either by NOX activation, or mitochondria.
Figure 4. ER-associated ROS activate the IRE-1/p38/SKN-1 pathway to increase lifespan.

(A) IRE-1 is sulfenylated in response to mitochondrial ROS production. IRE-1 sulfenylation (SOH) that is induced by PQ (30 min) was inhibited by pre-treatment of C. elegans with the mitochondrial ROS scavenger mito-tempo (MT) (1 hr). (B–D) Mitochondrial ROS activate the p38/SKN-1 response. Mito-tempo (MT) (1 hr) reduced the p38 response to PQ (30 min) (B) and diminished PQ-induced gst-4P::GFP expression (C), with GFP scoring shown in (D). GFP was quantified as high (H), medium (M) or low (L). (E–I) ero-1 RNAi induces ROS production (E), IRE-1:SOH (F), and p38 signaling (G) to extend lifespan (H, I). The MAPKK SEK-1 phosphorylates p38 in response to ROS and is required for p38 activity (Inoue et al., 2005). Lifespan extension from ero-1 RNAi depended upon ire-1- and sek-1 (H), and in a presumed null skn-1 mutant ero-1 RNAi shortened lifespan (I). Additional lifespan experiments and statistics are described in Table S2. p < 0.0001****; p < 0.05*. See also Figure S4 and Table S2.
We also investigated whether the IRE-1/p38/SKN-1 pathway could be activated by ROS that are produced by the ER itself, through the process of secretory protein folding. ER oxidoreducin 1 (ERO1) drives disulfide bond formation within the ER lumen by participating in a redox relay that generates hydrogen peroxide (Kakihana et al., 2012; Sevier and Kaiser, 2008). Surprisingly, in mice deletion of both ERO1 genes only subtly impaired ER disulfide formation, implying that an alternative mechanism can drive Cys thiol oxidation (Zito et al., 2012). These mutant mice exhibited increased sulfenylation of membrane-associated proteins, depletion of the antioxidant Ascorbic Acid, and a greater proportion of oxidized glutathione (Zito et al., 2012), suggesting that ROS levels were increased. As these mouse studies predict, knockdown of ero-1 in adult C. elegans increased ROS levels, IRE-1 sulfenylation, ire-1-dependent p38 phosphorylation, and SKN-1 target gene activity (Figure 4E–4G, S4C, and S4D). This p38 activation was abolished by GSH, suggesting that it is ROS-driven (Figure S4E). Knockdown of ero-1 therefore resulted in ROS production that activated the IRE-1/p38/SKN-1 antioxidant response. Consistent with our evidence that this response and the UPRER are mutually exclusive, ero-1 RNAi also interfered with TM-induced production of the spliced form of XBP-1 (Figure S4F).
Quiescin-Sulfhydryl Oxidase (QSOX) proteins are produced in the ER and catalyze disulfide formation, thereby generating peroxide, but their contributions to ER protein folding in vivo are unknown (Kodali and Thorpe, 2010; Sevier, 2012). Strikingly, ero-1 RNAi failed to generate ROS and activate p38 when C. elegans QSOX genes were knocked down (Figure S4G and S4H). This suggests that when ERO-1 activity is low, QSOX generates high levels of ROS and might play a compensatory role in the ER. Consistent with this idea, a strong genetic interaction between ero1 and qsox1 was previously detected in Drosophila (Tien et al., 2008). We conclude that when ER redox homeostasis is perturbed sufficiently to increase cytoplasmic ROS levels, the IRE-1/p38/SKN-1 antioxidant response becomes activated.
ROS can damage cellular macromolecules, and have long been presumed to promote aging. However, it is now known that C. elegans lifespan can be increased when mitochondrial ROS production is modestly elevated, apparently because protective mechanisms are mobilized (Lee et al., 2010; Ristow, 2014; Yang and Hekimi, 2010). ero-1 is required for C. elegans development (not shown), but was detected in a screen that identified genes for which RNAi knockdown during adulthood increases lifespan (Curran and Ruvkun, 2007). We confirmed that lifespan is increased substantially when ero-1 is knocked down in adults (18–48% extension of mean lifespan). Across multiple trials, this increased longevity largely or completely depended upon both ire-1 and p38 signaling (Figure 4H and Table S2). Moreover, in skn-1 mutants ero-1 RNAi consistently decreased lifespan (Figure 4I and Table S2). Taken together, our data suggest that ER-derived ROS that are generated upon ero-1 knockdown increase lifespan by activating the ire-1/p38/SKN-1 pathway. Knockdown of ero-1 was notably deleterious in the complete absence of the SKN-1-mediated antioxidant response, illustrating the importance of this protective response for defending against ER-derived ROS. The lifespan extension enjoyed by ero-1(RNAi) animals (Figure 4H and 4I) also suggests that the ire-1/p38/SKN-1 response promotes both homeostasis and health.
Conservation of the IRE-1 antioxidant response function
The DFG+2 Cys in Ire1 is evolutionarily conserved (Figure 2F) suggesting that Ire1 might initiate the p38 antioxidant response in mammals. Accordingly, AS treatment rapidly induced Ire1α sulfenylation and p38 activation in human (HepG2) cells (Figure 5A and 5B). AS also activated the stress-response kinase JNK, but only p38 activation was blocked by Ire1α knockdown (Figure 5B). Similar to C. elegans results with bli-3 RNAi, the mammalian p38 response to AS was attenuated by a NOX inhibitor, indicating that it was initiated by enzymatically produced ROS (Figure 5C). The human Ire1α responses to acute ROS and ER stress were also mutually exclusive: short-term AS preconditioning blocked ER stress-induced XBP-1S induction, and pre-treatment with TM inhibited AS-induced p38 activation (Figure 5D and 5E). Human Ire1α therefore initiates an acute p38 response to ROS analogously to its C. elegans counterpart. In C. elegans, stress responses can be influenced profoundly by signals between tissues (Prahlad and Morimoto, 2011; Taylor and Dillin, 2013), but our detection of this response in cultured cells demonstrates that it occurs cell-autonomously.
Figure 5. Evolutionary conservation of the IRE-1 antioxidant response function.

(A) Human Ire1α senses ROS. Ire1α sulfenylation was assessed in HepG2 cells following AS (30 min) treatment. (B) Ire1α is required for the p38 and Nrf2 response to AS-induced ROS. Levels of Nrf2 accumulation, p38 phosphorylation and activation were assayed in HepG2 cells treated with either control siRNA (si-SCR) or Ire1α siRNA (si-Ire1α) following AS (30 min) treatment. (C) NOX-derived ROS activate the p38/Nrf2 pathway. Levels of Nrf2 and p38 activation were determined in HepG2 cells treated with AS (30 min) in the absence or presence of the pan-NOX inhibitor VAS2870 (NOX-i)(1 hr). (D–E) Ire1α senses ROS and ER stress in a mutually exclusive manner. HepG2 cells were treated with AS (30 min) prior to TM (5 hr) treatment, with Xbp1S protein assayed by immunoblot (D). Levels of active p38 were determined in HepG2 cells following treatment with AS (30 min) alone, TM (5 hr) alone or pretreatment with TM prior to acute AS exposure (E). (F) Nrf2 activation by AS requires p38. HepG2 cells were treated with vehicle control or the p38 inhibitor SB203580 (p38-i) (2 hr) prior to AS (30 min). (G) Ire1α regulates Nrf2 independently of Keap1. HepG2 cells treated with either control siRNA (si-SCR) or Ire1α siRNA (si-Ire1α) were exposed to either the Keap1 inhibitor sulforaphane (SF) (30 min) or AS (30 min). (H) Activation of the p38/Nrf2 response by ER-derived ROS. Treatment of HepG2 cells with Ero1 inhibitor II (2 hr) activated p38 and Nrf2 comparably to AS. See also Figure S5.
In mammalian cells, AS treatment appears to activate Nrf2 independently of the canonical Nrf2 inhibitor Keap1 (Wang et al., 2008). Earlier work suggested a possible role for p38 signaling in the Nrf2 antioxidant response (Zipper and Mulcahy, 2000), but this has not been elucidated. Nrf2 upregulates its own expression and becomes activated through stabilization, making Nrf2 protein levels a reliable indicator of the antioxidant response (O’Connell and Hayes, 2015; Suzuki and Yamamoto, 2015). Remarkably, the Nrf2 response to AS was dramatically attenuated by treatment with Ire1α siRNA, a NOX inhibitor, or a p38 inhibitor (Figure 5B, 5C, and 5F), as would be predicted from our C. elegans data. By contrast, the classical Keap1 inhibitor sulforaphane increased Nrf2 levels independently of Ire1 (Figure 5G), suggesting that the Ire1α-regulated antioxidant response is parallel to and distinct from the Keap1-Nrf2 axis. Treatment with an Ero1 inhibitor activated p38 signaling and Nrf2 in human cells (Figure 5H), indicating conservation of this response to ER-derived ROS. Together, our C. elegans and mammalian data demonstrate the existence of a conserved ancestral Ire1α/p38 pathway that activates Nrf2 when ROS accumulate at the ER (Figure S5).
Sulfenylation of related kinases by NOX-generated ROS
Oxidative stress affects numerous cellular functions, including protein synthesis and proliferation (Martindale and Holbrook, 2002), but the mechanisms involved are largely not understood. Interestingly, a conserved Cys is present at the same DFG+2 position as IRE-1 in approximately 8% of human kinases, including most members of the AGC family (Figure 6A) (Liu et al., 2013). In many cases, this Cys is predicted to be redox-reactive (not shown), suggesting that these kinases might be modified similarly to IRE-1 when NOX enzymes produce ROS in response to stress. To test this idea, we examined AGC kinases in which the DFG+2 Cys is predicted to be reactive (Rock1, Akt, Pkc-δ and p70S6K), or non-reactive (Pak1) (Figure 6A). Using available antibodies, we found that AS rapidly induced sulfenylation of Rock1 (LET-502) and p70S6K (RSKS-1) orthologs in C. elegans (Figure 6B). In human cells, AS similarly induced sulfenylation of each AGC kinase tested except for Pak1 (Figure 6C). Remarkably, NOX inhibition blocked sulfenylation of these kinases (Figure 6C), suggesting that many kinases with a DFG+2 Cys may be susceptible to conserved modification and regulation by NOX-produced ROS signals.
Figure 6. Sulfenylation of AGC kinases by NOX-derived ROS.

(A) Alignment of the DFG+2 Cys of selected human AGC kinases. Basic amino acids are underlined. (B) The C. elegans Rock1 ortholog (LET-502) and p70S6K (RSKS-1) are sulfenylated in vivo. Lysates from animals treated with AS (30 min) were assessed for levels of Rock1:SOH and RSKS-1:SOH. (C) NOX-derived ROS sulfenylate multiple AGC kinases. HepG2 cells were treated with AS (30 min) in the absence or presence of the NOX inhibitor (NOX-i) VAS2870 (1 hr).
Discussion
A conserved IRE-1 function that is distinct from the UPRER
Over 23 years of study have yielded deep insights into the canonical IRE-1 function of responding to unfolded proteins within the ER (Maly and Papa, 2014; Ron and Walter, 2007). Here we describe a mechanistically distinct IRE-1 function, in which its cytoplasmic kinase transduces ROS signals from the ER, NOX enzymes, and mitochondria (Figure 7A). IRE-1 assumes this unexpected function through a conserved molecular switch, in which a Cys within its kinase activation loop becomes sulfenylated when exposed to a ROS signal. This addition of a single oxygen atom appears to inhibit the IRE-1 kinase and RNAse, and promotes recruitment and activation of p38 signaling at IRE-1. Our data provide a striking example of a well-described protein having an unexpected function, and suggest that Cys-based signaling can regulate proteins in diverse ways.
Figure 7. A sulfenylation-mediated switch between IRE-1 functions.

(A) Model of the UPRER and antioxidant response functions of IRE-1 (see text). (B) NOX-generated ROS sulfenylate multiple AGC kinases, resulting in IRE-1 adopting a distinct function.
IRE-1 initiates this antioxidant response upon exposure to ROS that are generated nearby, and need not be dispersed throughout the cytoplasm. Stress-induced phosphorylation of the MAPKKK NSY-1 was not sufficient to activate p38 signaling, which depended upon NSY-1 also being physically associated with IRE-1 within an oxidative environment, and becoming sulfenylated (Figure 3). At the ER, IRE-1 is in an opportune location to sense ROS, and stimuli that trigger ROS signals. The ER lumen maintains oxidizing conditions (Kakihana et al., 2012; Sevier and Kaiser, 2008) apparently by balancing ERO-1 and QSOX activities (Figure S4G). IRE-1 is ideally poised to detect ER-derived ROS that appear in the cytoplasm, and is well placed to detect mitochondrial ROS (Figure 4A–4D) because of extensive ER-mitochondrial communication (Kornmann, 2013). In addition, NOX enzymes that interact with IRE-1 transduce AS stress into a ROS signal (Figure 7A). This suggests that certain stresses, possibly lipid or protein damage at the ER (Laurindo et al., 2014), send an adaptive signal through IRE-1 to activate p38. By sensing these perturbations at the ER, IRE-1 acts as a sentinel that may limit their deleterious effects on the cell at large.
The requirement for IRE-1 sulfenylation would prevent ROS from activating p38 signaling without a “go ahead” signal from the ER environment. In C. elegans, SKN-1 and XBP-1 regulate each other’s expression, suggesting underlying cooperation between their respective stress responses (Glover-Cutter et al., 2013). However, the UPRER and ROS signaling functions of IRE-1 are mutually exclusive (Figure 1F–1J, Figure 1L–1O, Figure 5D, and Figure 5E). Under ER stress conditions it could be advantageous to suppress antioxidant response activation, because this response could impair oxidative protein folding by increasing GSH levels. In the other direction, when the antioxidant response is needed it may be advantageous to “tap the brakes” on the IRE-1-initiated UPRER and oxidative protein folding, until redox homeostasis is restored. Accordingly, when the ER becomes overly oxidizing BiP undergoes oxidation that inhibits its chaperone function, and allows it to stabilize misfolded proteins by acting as a holdase (Wang et al., 2014). The mutual exclusivity between the these two IRE-1 functions could explain evidence that the ER and cytoplasm are mutually antagonistic with respect to redox status, so that proteostasis perturbations that make the cytoplasm more oxidizing (and thus should inhibit the IRE-1 UPRER) render the ER more reducing (Kirstein et al., 2015).
It will be important to elucidate how post-translational modification of the IRE-1 kinase discriminates between these two distinct IRE-1 functions (Figure 7A), and influences the UPRER and antioxidant responses over time. For example, a considerable body of evidence indicates that the IRE-1 kinase is important for xbp-1 mRNA splicing (Prischi et al., 2014; Ron and Walter, 2007), a model consistent with our results, but in yeast the Ire1 kinase limits the duration of the UPRER (Chawla et al., 2011; Rubio et al., 2011). One interesting question is whether the sulfenylated IRE-1 Cys progresses to further oxidation steps (Figure S2A), or can be “reset” to a thiol so that this IRE-1 molecule can reassume its UPRER function. Another is how UPRER activation might inhibit IRE-1 from responding to ROS (Figure 7A). Perhaps oligomerized IRE-1 is resistant to sulfenylation within the kinase loop. Formation of a signalosome that initiates p38 signaling might require a subtle interaction with sulfenylated IRE-1, or it might be sufficient simply to inhibit the IRE-1 kinase and oxidatively modify the p38 MAPKKK. In mammalian cells, exogenous ROS administration induces the p38 MAPKKK ASK1 to form an internal disulfide that promotes signalosome assembly (Nadeau et al., 2007). Our finding that Ire1α is required for ROS-induced mammalian p38 activation suggests that ASK1 oxidation is not the initiating signaling event, but might play a similar role to NSY-1 sulfenylation in C. elegans. In earlier cell culture experiments, sustained ER stress from TM led to JNK activation that was prevented by mutational Ire1 kinase inactivation (Urano et al., 2000). It is possible that p38 and JNK signaling are activated at IRE-1 through distinct mechanisms, with only the former involving sulfenylated kinase-inactive IRE-1 (Figure 7A). In HepG2 cells we observed robust AS-induced JNK activation that did not require Ire1α (Figure 5B) but failed to detect TM-induced JNK activation (not shown), suggesting that the latter pathway might be context-dependent.
A surprising aspect of our findings was that IRE-1 functions as a direct ROS sensor for the Nrf2/SKN-1 antioxidant pathway. In mammals, analyses of Nrf2 responses to stress have been focused almost entirely around the solidly established Keap1 inhibitory paradigm (Figure S5)(Suzuki and Yamamoto, 2015). p38 phosphorylates Nrf2 and has been implicated in its regulation in mammalian cells (Sun et al., 2009; Zipper and Mulcahy, 2000), but the functional significance of these findings has remained unclear. We determined that AS activates Nrf2 by stimulating the conserved NOX/Ire1α/p38 pathway, which apparently functions independently of Keap1 (Figure 5 and S5). Similarly, inhibition of mammalian Ero1 activated p38 and Nrf2 (Figure 5H), indicating that this is pathway is a conserved response to the presence of ROS at the ER. The presence of this IRE-1/p38 pathway in C. elegans, which lacks a Keap1 ortholog, suggests that it may represent the ancestral mode through which ROS and other stresses activate the antioxidant response. Perhaps Keap1 arose in higher organisms to broaden or speed responsiveness to diverse electrophilic compounds. Nrf2 is either pathogenic or protective in certain cancers, diabetes, and other diseases (Murakami and Motohashi, 2015; O’Connell and Hayes, 2015; Uruno et al., 2013). It will be important to elucidate the extent to which the normal and disease-related functions of Nrf2 involve its regulation by Ire1α, and ER-associated ROS.
IRE-1 redox regulation in aging and disease
Ire1 is required for normal development, and is dysfunctional or a potential therapeutic target in diseases that include cancer, diabetes, and neurodegeneration (Fu et al., 2012; Hetz et al., 2013; Wang and Kaufman, 2014). Our results indicate that functional requirements for Ire1 are likely to involve its regulation of p38/SKN-1(Nrf2), as well as its UPRER functions. In mice, obesity can lead to nitric oxide modifying Ire1α within its RNase, thereby perturbing Xbp1 splicing and glucose homeostasis (Yang et al., 2015). Overall protein oxidation patterns change with age, possibly reflecting damage or maladaptive modifications (Brandes et al., 2013; Stadtman, 2006). Age- or disease-related oxidation of residues that have physiological regulatory functions, like IRE-1 C663, could have devastating effects.
Ire1 also appears to play a positive role in promoting longevity, as indicated by its being required for some genetic or dietary interventions to increase lifespan in C. elegans (Chen et al., 2009; Henis-Korenblit et al., 2010). This requirement is likely to involve not only the canonical UPRER function of IRE-1, but also activation of p38/SKN-1, a pathway linked to longevity across species (Blackwell et al., 2015). The observation that mitochondrially-produced ROS can extend lifespan has generated considerable interest, because it shows that ROS-based signals can promote longevity (Lee et al., 2010; Ristow, 2014; Yang and Hekimi, 2010). Some studies indicate that SKN-1 is essential for this beneficial effect of mitochondrial ROS (Blackwell et al., 2015). Here we determined that ER-derived ROS extend lifespan by activating the IRE-1/p38/SKN-1 pathway, but are harmful when the SKN-1 antioxidant response is completely ablated (Figure 4H and 4I). The data emphasize the importance of SKN-1-regulated mechanisms as potent drivers of longevity, identify the ER as another source of ROS that can extend lifespan, and suggest that physiological ROS-mediated signaling may profoundly affect longevity and health.
Importance of localized redox signaling for homeostasis
A conserved Cys is present at the DFG+2 position in numerous kinases (Figure 6A) (Liu et al., 2013). In each case in which this Cys was predicted to be redox-reactive, these kinases became sulfenylated in response to AS treatment and NOX activation (Figure 6C). The precedent of IRE-1 suggests that sulfenylation might not simply inhibit their kinase activity, but also could induce unexpected functions (Figure 7B). It is not understood how oxidative stress affects so many cellular processes, including protein synthesis and proliferation (Martindale and Holbrook, 2002). The sulfenylated kinases we identified are involved in cell polarity (Rock1), cell growth and survival (Akt, Pkc), and protein synthesis (p70S6K), providing a possible explanation. ROS-scavenging antioxidants have largely failed as disease therapies, possibly because they may suppress protective mechanisms (Ristow, 2014). The idea that ROS have many physiological regulatory functions provides another note of caution concerning such treatments. Great interest has arisen in designing covalent inhibitors that target Cys residues, which are present in over 200 human kinase domains (Liu et al., 2013), and recently an Ire1 inhibitor has been designed that binds its DFG+2 Cys (Waller et al., 2016). Our results suggest that many of these potential Cys targets are likely to be physiological regulatory sites, and that pharmacological targeting of such residues might allow specific activities to be modulated in multifunctional proteins like IRE-1.
We suspect that many more targets and processes are regulated physiologically by post-translational Cys modifications than is appreciated currently. At least 700 proteins are sulfenylated in mammalian cells, in patterns that are altered by ROS or growth factor treatment (Yang et al., 2014). The paradigm of IRE-1 and NSY-1 regulation we have described suggests that many of these modified Cys residues are likely to be play regulatory roles, and that this Cys-based regulation is remarkably versatile: sulfenylation inhibited the IRE-1 kinase and UPRER and induced IRE-1 to adopt a distinct function, whereas p38 signaling was activated through sulfenylation of NSY-1 that is associated with IRE-1. We propose that Cys modification by ROS from NOX enzymes or other sources allows various perturbations and signals to be transduced into a language that can be interpreted by kinases and transcription factors, or other regulators. Elucidating these regulatory interactions and the stimuli that trigger them, and determining how they influence their protein targets, is likely to yield profound insights into physiology, development, aging, and disease.
Experimental Procedures
ROS detection
Synchronized C. elegans were incubated with the ROS sensitive fluorescent probe CM-H2DCFDA (Life Technologies), with ROS levels indicated relative to total protein concentration.
GFP scoring
Expression or nuclear accumulation of transgenically expressed GFP proteins was scored as ‘low’, ‘medium’ or ‘high’ as described previously (Glover-Cutter et al., 2013).
Sulfenylation Assay
Sulfenylated proteins were isolated from C. elegans and HepG2 cells using the DCP-Bio1 probe (KeraFast, Boston), prior to immunoblotting with antibodies.
Details and other methods are in the Supplemental Experimental Procedures.
Supplementary Material
HIGHLIGHTS.
The ER stress sensor IRE-1 has a distinct function in cytoplasmic homeostasis
Local redox signals block IRE-1 ER signaling by sulfenylating a kinase cysteine
This functional switch initiates the p38/SKN-1(Nrf2) antioxidant response at IRE-1
The IRE-1 paradigm implies broad and versatile functions for signaling at cysteines
Acknowledgments
We thank Blackwell lab members and Eric Greer for helpful discussions, Sonja Bertram for experimental contributions, and Elizabeth Veal, Kunihiro Matsumoto, and Sivan Henis-Korenblit for reagents. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). The work was supported by funding from the NIH to TKB (R01GM094398) and a Diabetes Center Research Award from the NIDDK (P30DK036836). L.P.F.C is a doctoral student from Programa de Doctorado en Ciencias Bioquímicas, UNAM and received a fellowship from CONACyT and the PAEP program. The authors declare no conflicts of interest.
Footnotes
AUTHOR CONTRIBUTIONS
J.M.H and T.K.B designed the experiments and wrote the paper. J.M.H, L.E.M.M, and L.P.F.C. conducted the experiments, organized and interpreted the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- Bayliss R, Fry A, Haq T, Yeoh S. On the molecular mechanisms of mitotic kinase activation. Open Biol. 2012;2:120136. doi: 10.1098/rsob.120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, Isik M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic Biol Med. 2015;88:290–301. doi: 10.1016/j.freeradbiomed.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes N, Tienson H, Lindemann A, Vitvitsky V, Reichmann D, Banerjee R, Jakob U. Time line of redox events in aging postmitotic cells. eLife. 2013;2:e00306. doi: 10.7554/eLife.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213. doi: 10.3389/fncel.2014.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Chakrabarti S, Ghosh G, Niwa M. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol. 2011;193:41–50. doi: 10.1083/jcb.201008071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000486. doi: 10.1371/journal.pgen.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016;532:112–116. doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter KM, Lin S, Blackwell TK. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013;9:e1003701. doi: 10.1371/journal.pgen.1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould NS, Evans P, Martinez-Acedo P, Marino SM, Gladyshev VN, Carroll KS, Ischiropoulos H. Site-Specific Proteomic Mapping Identifies Selectively Modified Regulatory Cysteine Residues in Functionally Distinct Protein Networks. Chemistry & biology. 2015;22:965–975. doi: 10.1016/j.chembiol.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee SJ, Cary M, Kenyon C. Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc Natl Acad Sci U S A. 2010;107:9730–9735. doi: 10.1073/pnas.1002575107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Hoeven R, McCallum KC, Cruz MR, Garsin DA. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog. 2011;7:e1002453. doi: 10.1371/journal.ppat.1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305–332. doi: 10.1093/toxsci/kfr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, Matsumoto K. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 2005;19:2278–2283. doi: 10.1101/gad.1324805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol. 2011;31:95–107. doi: 10.1002/jat.1649. [DOI] [PubMed] [Google Scholar]
- Kakihana T, Nagata K, Sitia R. Peroxides and peroxidases in the endoplasmic reticulum: integrating redox homeostasis and oxidative folding. Antioxid Redox Signal. 2012;16:763–771. doi: 10.1089/ars.2011.4238. [DOI] [PubMed] [Google Scholar]
- Kirstein J, Morito D, Kakihana T, Sugihara M, Minnen A, Hipp MS, Nussbaum-Krammer C, Kasturi P, Hartl FU, Nagata K, et al. Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 2015;34:2334–2349. doi: 10.15252/embj.201591711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali VK, Thorpe C. Oxidative protein folding and the Quiescin-sulfhydryl oxidase family of flavoproteins. Antioxid Redox Signal. 2010;13:1217–1230. doi: 10.1089/ars.2010.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B. The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol. 2013;25:443–448. doi: 10.1016/j.ceb.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annual review of pathology. 2014;9:119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol. 2013;33:2436–2446. doi: 10.1128/MCB.01748-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurindo FR, Araujo TL, Abrahao TB. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal. 2014;20:2755–2775. doi: 10.1089/ars.2013.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Ferber M, Salzberg Y, Safra M, Haviv-Chesner A, Bulow HE, Henis-Korenblit S. It’s all in your mind: determining germ cell fate by neuronal IRE-1 in C. elegans. PLoS Genet. 2014;10:e1004747. doi: 10.1371/journal.pgen.1004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chemistry & biology. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly DJ, Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014;10:892–901. doi: 10.1038/nchembio.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Miedel MT, Graf NJ, Stephen KE, Long OS, Pak SC, Perlmutter DH, Silverman GA, Luke CJ. A pro-cathepsin L mutant is a luminal substrate for endoplasmic-reticulum-associated degradation in C. elegans. PLoS One. 2012;7:e40145. doi: 10.1371/journal.pone.0040145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Motohashi H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med. 2015;88:168–178. doi: 10.1016/j.freeradbiomed.2015.06.030. [DOI] [PubMed] [Google Scholar]
- Nadeau PJ, Charette SJ, Toledano MB, Landry J. Disulfide Bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H(2)O(2)-induced c-Jun NH(2)-terminal kinase activation and apoptosis. Mol Biol Cell. 2007;18:3903–3913. doi: 10.1091/mbc.E07-05-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MA, Hayes JD. The Keap1/Nrf2 pathway in health and disease: from the bench to the clinic. Biochem Soc Trans. 2015;43:687–689. doi: 10.1042/BST20150069. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014;516:414–417. doi: 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med. 2015;80:148–157. doi: 10.1016/j.freeradbiomed.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Morimoto RI. Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc Natl Acad Sci U S A. 2011;108:14204–14209. doi: 10.1073/pnas.1106557108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prischi F, Nowak PR, Carrara M, Ali MM. Phosphoregulation of Ire1 RNase splicing activity. Nature communications. 2014;5:3554. doi: 10.1038/ncomms4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nature medicine. 2014;20:709–711. doi: 10.1038/nm.3624. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rubio C, Pincus D, Korennykh A, Schuck S, El-Samad H, Walter P. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol. 2011;193:171–184. doi: 10.1083/jcb.201007077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS. Erv2 and quiescin sulfhydryl oxidases: Erv-domain enzymes associated with the secretory pathway. Antioxid Redox Signal. 2012;16:800–808. doi: 10.1089/ars.2011.4450. [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta. 2008;1783:549–556. doi: 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation and aging. Free Radic Res. 2006;40:1250–1258. doi: 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- Sun Z, Huang Z, Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One. 2009;4:e6588. doi: 10.1371/journal.pone.0006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien AC, Rajan A, Schulze KL, Ryoo HD, Acar M, Steller H, Bellen HJ. Ero1L, a thiol oxidase, is required for Notch signaling through cysteine bridge formation of the Lin12-Notch repeats in Drosophila melanogaster. J Cell Biol. 2008;182:1113–1125. doi: 10.1083/jcb.200805001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW, Yamamoto M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol. 2013;33:2996–3010. doi: 10.1128/MCB.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller DD, Jansen G, Golizeh M, Martel-Lorion C, Dejgaard K, Shiao TC, Mancuso J, Tsantrizos YS, Roy R, Sebag M, et al. A Covalent Cysteine-Targeting Kinase Inhibitor of Ire1 Permits Allosteric Control of Endoribonuclease Activity. Chembiochem: a European journal of chemical biology. 2016;17:843–851. doi: 10.1002/cbic.201500485. [DOI] [PubMed] [Google Scholar]
- Wang J, Pareja KA, Kaiser CA, Sevier CS. Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. eLife. 2014;3:e03496. doi: 10.7554/eLife.03496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Chen W, Li Y, Villeneuve NF, Zhang DD. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: enhanced Keap1-Cul3 interaction. Toxicol Appl Pharmacol. 2008;230:383–389. doi: 10.1016/j.taap.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nature communications. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, Fu S, Hotamisligil GS. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. 2015;349:500–506. doi: 10.1126/science.aaa0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–492. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- Zito E, Hansen HG, Yeo GS, Fujii J, Ron D. Endoplasmic reticulum thiol oxidase deficiency leads to ascorbic acid depletion and noncanonical scurvy in mice. Mol Cell. 2012;48:39–51. doi: 10.1016/j.molcel.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.