

Abstract
The brains of Alzheimer’s disease (AD) patients contain abundant amyloid plaques composed of Aβ peptides. It is generally assumed that amyloid plaques and soluble Aβ oligomers induce neuronal pathology in AD. The mechanism of amyloid-mediated pathological effects is not clearly understood. Recent in vivo calcium (Ca2+) imaging studies with AD mouse models provide novel insights into changes in brain function resulting from accumulation of amyloid plaques. The unexpected lesson from these studies is that amyloid plaques result in both localized and global changes in brain function. The amyloid-induced effects include “short-range” changes in neuronal Ca2+ levels, “medium-range” changes in neuronal activity and ‘long-range” changes in astrocytic Ca2+ signaling and induction of intracellular Ca2+ waves spreading via astrocytic network. These results have potential implications for understanding synaptic and neuronal network dysfunction in AD brains.
The dominant idea in the AD field is the “amyloid hypothesis” which states that increased production of amylogenic Aβ42 peptide (or an increase in Aβ42/40 ratio) is a major cause of neuronal and synaptic loss in AD [1]. The experimental support for the “amyloid hypothesis” comes from (i) accumulation of amyloid plaques in the brains of AD patients; (ii) the familial AD (FAD) cases resulting from missense mutations in Aβ-precursor APP protein; (iii) the FAD cases resulting from missense mutations in presenilins (PS), which form a catalytic subunit of APP-cleaving enzyme γ-secretase. It is postulated that formation of soluble Aβ oligomers and insoluble amyloid plaques drive the pathological processes in the AD brains. Based on these assumptions the “amyloid-targeting” therapies have been the main focus of the current AD drug development [2]. The key unanswered question about the “amyloid hypothesis” is how exactly amyloid impairs brain function? Several recent in vivo Ca2+ imaging studies performed in APP-PS1 mouse models provide first look at changes in brain function resulting from accumulation of amyloid plaques. The unexpected lesson from these studies is that amyloid plaques result in both localized and global changes in brain function.
Usually pathogenic actions of Aβ oligomers are assumed to be highly localized - at the level of individual synapses or spines. Local effects of Aβ oligomers on AMPA receptors internalization [3,4] and NMDAR trafficking and function [5–7] have been described. Aβ oligomers are also able to generate Ca2+-permeable pores in plasma membrane directly [8]. The ability of Aβ oligomers to form Ca2+ permeable channels in neuronal plasma membrane is consistent with the recent in vivo Ca2+ imaging experiments performed with APP (APPsw) and APP-PS1 (APPsw, PS1-ΔE9) transgenic mice [9]. These authors found that resting Ca2+ levels were significantly elevated in approximately 35% of neurites located in an immediate vicinity (< 25 μm) from the Aβ plaques. The most likely explanation to these results is that high local concentration of Aβ oligomers in the area surrounding amyloid plaques causes formation of Ca2+-permeable ion channels in the neuronal plasma membrane. The neurites with elevated Ca2+ levels lacked spines and displayed abnormal morphology [9]. These studies provide strong support to “local” (< 25 μm) pathogenic effects of amyloid, which result in destabilization of intracellular neuronal Ca2+ signaling and impaired synaptic function (Fig 1).
Fig. 1.
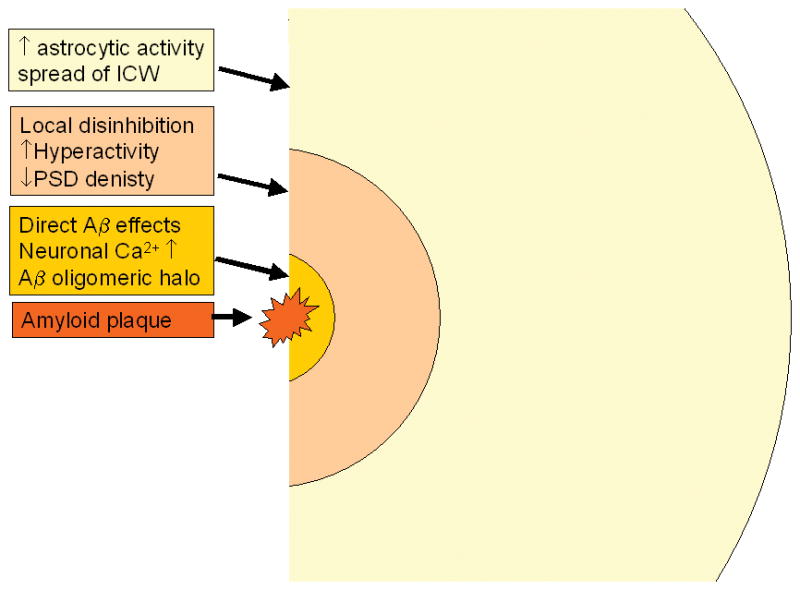
“Short-range” (< 25 μm), “medium-range” (< 60 μm) and “long-range” (< 200 μm) effects of an amyloid plaque are shown. The presence of an amyloid plaque in brains of APP-PS1 mice causes increase in neuronal resting Ca2+ levels at the short range [9], increases local neuronal spontaneous activity in the medium range [10] and causes spread of intracellular Ca2+ waves via astrocytic network in the long-range [12]. The resulting changes in neuronal and astrocytic properties are likely to lead to dysfunction of neuronal network in the proximity of the amyloid plaques in AD brains.
Another in vivo Ca2+ imaging study with APP-PS1 (APPsw, PS1-G384A) transgenic mice reported disturbances in neuronal excitability in an area surrounding amyloid plaques [10]. It has been observed that in the area close to the plaques the fraction of “hyperactive” neurons was increased by 16-fold and the fraction of “silent” neurons was increased by 3-fold when compared to the wild type mice [10]. These “medium-range” effects of amyloid (< 60 μm) have been linked to impaired local synaptic inhibition, which resulted in hyperactivity of “disinhibited” neurons [10] (Fig 1). Similar hyperactivity has been previously described in electrophysiological studies of APP transgenic mice [11]. It has been speculated that the loss of inhibition is due to local anatomical remodeling of synaptic contacts in the vicinity of amyloid plaque, which in turn occurs as a response to amyloid-induced activation of microglia.
The latest in vivo Ca2+ imaging study [12] indicates that pathological effects of amyloid can spread even farther in the brain. These authors studied Ca2+ dynamics in astrocytes from APP-PS1 transgenic mice (APPsw, PS1-ΔE9). They found that resting Ca2+ levels were globally elevated in the astrocytic network in the APP-PS1 mice. They also found significant (4-fold) increase in a fraction of spontaneously active astrocytes in the brains of APP-PS1 mice. The spontaneous Ca2+ signals in APP-PS1 astrocytes were also 50% higher in amplitude than the Ca2+ signals in wild type mice. The effects on resting Ca2+ levels and spontaneous activity of astrocytes were observed globally and did not depend on proximity to the plaques. The increase in astrocyte Ca2+ activity was not blocked by TTX and appear to result from direct interactions of Aβ oligomers with the astrocyte network and not mediated by neuronal activity. In addition to these effects, the authors also observed increased correlation between astrocytic Ca2+ signals and frequent occurrence of intracellular Ca2+ waves (ICW) spreading via astrocytic network [12]. The correlation in astrocyte activity and propagation of ICW was observed at distances reaching as far as 200 μm from the plaques. Although some of the effects observed by the authors were reported to be global, the study was performed in 6–8 months old mice with dense plaques (mean astrocyte-to-plaque distance was 50 μm [12]). Thus, it is clear that presence of the plaques affects astrocytic Ca2+ signals at least as far as 200 μm (Fig 1). Additional imaging studies with “sparse” plaques brains (at earlier age of the mice) will be needed to determine upper limit for long-ranging effects of amyloid plaques on astrocytic Ca2+ signals. The mechanism involved in plaque effects on the astrocytic Ca2+ signals appear to involve “local” stimulation of initiator astrocytes (within 25 μm from the plaque) followed by long-range ICW propagation via astrocytic network [12].
The results of in vivo Ca2+ imaging studies in APP-PS1 mice [9,10,12] indicate that amyloid plaques have “short-range” (< 25 μm), “medium-range” (< 60 μm) and “long-range” (< 200 μm) effects on Ca2+ signaling and excitability (Fig 1). What is an importance of these findings for understanding AD pathogenesis? AD has been proposed to result from “synaptic failure” due to local attack by Aβ oligomers on synapses and dendritic spines [13]. On another hand, the manifestation of AD symptoms have been proposed to result from a neuronal network dysfunction [14,15]. It has been reasoned that the localized synaptic loss and activation of microglia and astrocytes may affect properties of the neuronal network as a whole [14,15]. The results from the in vivo Ca2+ imaging studies discussed here indicate that presence of amyloid plaques may affect properties of neuronal networks by acting both locally and at the distance. For example, long-range changes in astrocytic Ca2+ signals may affect glutamate clearance mechanisms, which in turn may affect neuronal network behavior. It will be very interesting to determine if inhibition of ICW in astrocytes of APP-PS1 mice can result in improvement in behavioral performance of these mice in memory tasks. If it is, it would suggest that block of astrocytic Ca2+ waves may have a therapeutic potential for treating AD or at least for alleviating some of the symptoms in human AD patients.
Acknowledgments
IB is a holder of Carla Cocke Francis Professorship in Alzheimer’s Research and supported by the McKnight Neuroscience of Brain Disorders Award and the NINDS grant R01NS056224.
References
- 1.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer’s disease. Science. 2006;314:781–4. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 4.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder EM, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 7.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 8.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. 1993;90:567–71. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–25. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busche MA, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–9. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 11.Palop JJ, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science. 2009 doi: 10.1126/science.1169096. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 14.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–73. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 15.Small DH. Network dysfunction in Alzheimer’s disease: does synaptic scaling drive disease progression? Trends Mol Med. 2008;14:103–8. doi: 10.1016/j.molmed.2007.12.006. [DOI] [PubMed] [Google Scholar]