

SUMMARY
Current approaches to cancer immunotherapy aim to engage the natural T cell response against tumors. One limitation is the elimination of self-antigen specific T cells from the immune repertoire. Using a system in which precursor frequency can be manipulated in a murine melanoma model, we demonstrate that the clonal abundance of CD4+ T cells specific for self-tumor antigen positively correlated with antitumor efficacy. At elevated precursor frequencies, intraclonal competition impaired initial activation and overall expansion of the tumor specific CD4+ T cell population. However, through clonally derived help, this population acquired a polyfunctional effector phenotype and antitumor immunity was enhanced. Conversely, development of effector function was attenuated at low precursor frequencies due to irreversible T cell exhaustion. Our findings assert that the differential effects of T cell clonal abundance on phenotypic outcome should be considered during the design of adoptive T cell therapies, including use of engineered T cells.
INTRODUCTION
Beginning when Burnet first postulated the theory of clonal selection in 1957, it has become a central tenet of immunology that the immune system has evolved to promote repertoire diversity while limiting self reactivity (Burnet, 1957, 1959). Balance is achieved by maintaining a varied repertoire of adaptive immune cells of unique specificity, which then expand upon encounter with cognate antigen through clonal expansion. Self-reactivity is prevented by eliminating high affinity clones that recognize self from the immune repertoire early in development through negative selection and peripheral tolerance. In the time since Burnet, many groups have shown that T cells specific for epitopes of common antigens can be maintained in the repertoire at precursor frequencies that range from only a few clones to pools numbering in the thousands (Blattman et al., 2002; Jenkins and Moon, 2012; Rizzuto et al., 2009; Whitmire et al., 2006). Variance in the endogenous precursor frequency of foreign antigen specific T cells impacts the magnitude of the response to pathogen (Jenkins and Moon, 2012; Moon et al., 2007). Although heterogeneity in the size of precursor populations exists, frequency is maintained within a relatively narrow physiologic range. When T cells exceed this range, their survival and ability to expand in response to antigen are impaired through intraclonal competition (Hataye et al., 2006).
While the exact mechanism of intraclonal competition has yet to be completely elucidated, it is widely believed that competition for antigen during engagement with antigen presenting cells is at least partly responsible (Kedl et al., 2000; Quiel et al., 2011; Smith et al., 2000; Willis et al., 2006). For T cells present at high precursor frequencies, this competition results in a decreased initial proliferative burst and impaired overall expansion, as well as deficiencies in the induction of effector function and generation of memory (Badovinac et al., 2007; Blair and Lefrançois, 2007; Marzo et al., 2005). However, in models where antigen may not be a limited resource, such as when the cognate antigen is a ubiquitously expressed self-molecule as in cancer, it is less well understood to what extent competition influences immunity.
It is increasingly apparent that mechanisms of central tolerance are not infallible; auto-reactive clones can escape negative selection and initiate destruction of healthy tissue (Zehn and Bevan, 2006). The first tumor rejection antigens were characterized due to aberrant responses against self and tumor and took the form of differentiation antigens, as well as cancer-testis antigens (Houghton, 1994). Our group has estimated the clonal abundance of tumor/self antigen specific CD8+ T cells to be over an order of magnitude lower than that of T cells specific for a foreign antigen, which is low enough to preclude an immune response without therapeutic intervention (Rizzuto et al., 2009). It was determined that bringing the frequency of the T cells within or above the normal physiologic range favored the proliferation and generation of polyfunctional effector T cells and potent anti-tumor immunity, while dramatically exceeding this threshold resulted in intraclonal competition and an impaired immune response.
In this report, we show that clonal abundance dictated the development of CD4+ T cell mediated anti-tumor immunity as well. Tumor specific CD4+ T cells operate within the constraints imposed by intraclonal competition despite abundant expression of cognate antigen. Unlike CD8+ T cells, the observed defects in proliferation are uncoupled from the development of effector function. Physiological precursor frequencies of self-antigen specific T cells support the rapid expansion of the population at the expense of the generation of effector function due to the onset of irreversible T cell exhaustion. Despite decreased expansion at high precursor frequencies, tumor specific CD4+ T cells accumulate in greater numbers. Through a mechanism of population-induced positive feedback involving paracrine IFN-γ sharing and traditional T cell help, we observe intraclonal cooperation resulting in strong Th1 cell differentiation and potent anti-tumor responses.
RESULTS
At high precursor frequencies, tumor-specific CD4+ T cells experience impaired expansion and activation
To investigate the effect of clonal abundance on the response of tumor specific CD4+ T cells in a model of implantable B16 melanoma, we made use of TCR transgenic CD4+ T cells specific for the melanoma differentiation antigen tyrosinase related protein 1 (TRP-1) (Muranski et al., 2008). One unique feature of this model is that anti-TRP-1 TCR transgenic T cells are negatively selected in mice expressing TRP-1, such that these TCR transgenic mice must also be maintained on a TRP-1 deficient background. These conditions imply that this T cell clone is strongly selected against and likely present at very low quantities, if at all, in the normal immune repertoire. Taking this into account, we examined how the relative paucity of this clone affects its ability to mount a successful response to B16 melanoma.
We used a previously established adoptive transfer model, in which clonal frequency in the immune repertoire can be manipulated in tumor bearing mice (Rizzuto et al., 2009). Mice bearing large (approximately 1 cm) implantable B16 melanoma tumors received sub-lethal irradiation to eliminate their existing immune repertoire. These animals are then reconstituted via adoptive transfer of naïve polyclonal splenocytes supplemented with varied frequencies of the TRP-1 specific CD4+ T cell clone of interest. The total cell transfer quantity is fixed to 30 million cells, ensuring that the only manipulated variable is the number of TRP-1 CD4+ T cells.
We first determined how precursor frequency impacted the initial proliferative burst of the tumor specific CD4+ T cells. TRP-1 CD4+ T cells were labeled with CFSE and co-transferred at titrated quantities of 103, 105 or 106 cells, with naïve splenocytes of normal repertoire diversity. At days 5 and 7, proliferation was assessed in the tumor draining lymph nodes (LN) and spleens of the treated animals (Figure 1A). At all frequencies, proliferation was observed, as expected after adoptive transfer into a lymphopenic host. However, only at the lowest frequency (103), did almost all TRP-1 CD4+ T cells undergo robust proliferation (Figure 1B,C). Notably, in the 106 group, between 10–15% of the population showed only partial proliferation. This population increased in the LN between day 5 and 7, possibly due to egress of divided cells from the LN to tumor. To determine if this early defect in proliferation at high clonal abundance resulted from incomplete activation of the population, we examined T cell activation markers CD44 and CD62L in tumor draining LNs (Figure 1D,E). In the lower frequency groups, a greater proportion of T cells displayed an activated CD44hiCD62Llo phenotype compared to groups with higher precursor frequencies. However, almost all T cells that infiltrated the tumor had become activated and no difference due to frequency was observed (Figure 1F). This suggests that during the priming phase, self-antigen specific T cells adoptively-transferred in high numbers experience intraclonal competition, which limits their early activation and expansion.
Figure 1. At high precursor frequencies tumor specific CD4+ T cells experience impaired expansion and activation.
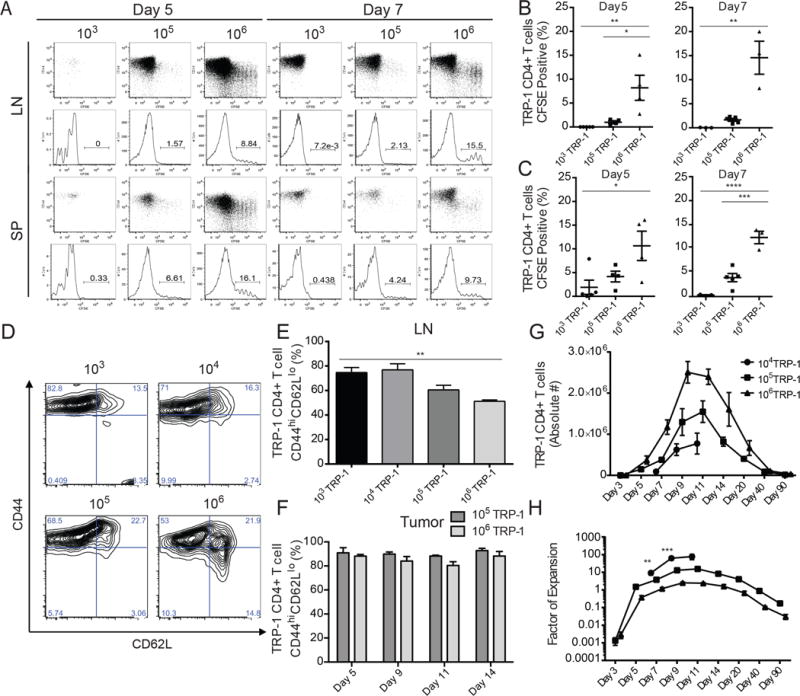
Tumor bearing mice received sublethal irradiation, followed by tail vein injection of 103, 105, or 106 CFSE labeled (where indicated) congenically marked TRP-1 CD4+ T cells co-transferred with naïve splenocytes to assess proliferation and activation. (A) On days 5 and 7, CFSE dilution of the TRP-1 CD4+ T cells was assessed in the lymph node and spleens of host mice. Representative flow plots of CFSE dilution. (B) Percentage of undivided CFSEhi TRP-1 CD4+ T cell population in LN and (C) spleen. (n=3–5 mice/group). Data are represented as mean ± SEM. (D) Representative flow plots of TRP-1 CD4+ T cell activation by CD44 and CD62L expression in LN. (E) Summary of TRP-1 CD4+ T cell activation in LN and (F) tumor (n=3–5 mice/group). (G) Mean absolute number of TRP-1 CD4+ T cells per mouse as determined by combined quantification of congenically marked population in LN, SP, and tumor by flow cytometry. (H) Factor of expansion as determined by comparing absolute number to original transferred precursor frequency (n=5 mice/group). All data representative of three independent experiments. Data are represented as mean ± SEM.
We quantified the absolute number of TRP-1 CD4+ T cells after transfer and measured proliferation over the course of the immune response. The accumulation of total TRP-1 T cells was greatest when cells were initially transferred at larger numbers, with the number of cells peaking at day 9 in the highest frequency group and at day 11 at the lower frequencies (Figure 1G). However, at the highest clonal abundance (106), the transferred population had expanded only 2.5 fold at the height of the immune response, while the population of 105 cells had expanded 15 fold, and 104 TRP-1 CD4+ T cells had expanded over 75 fold (Figure 1H). These data support previous findings demonstrating that precursor frequency is inversely related to the ability of the population to expand.
Intraclonal competition of tumor specific CD4+ T cells does not preclude a successful anti-tumor immune response
We then quantified the anti-tumor response to B16 melanoma, by measuring tumor growth over time in mice reconstituted with variable numbers of TRP-1 T cells. In contrast to other models, in which high clonal abundance impairs the immune response against foreign pathogen and tumors, in this model the opposite is observed (Figure 2A). At the lowest precursor frequency of 103 TRP-1 CD4+ T cells, no tumors undergo complete regression, but if the frequency is increased to 105 T cells, 30–60% of tumors are eradicated. When frequency is further increased to levels where intraclonal competition is most apparent, at a clonal abundance of 106 and 107 TRP-1 CD4+ T cells, the greatest anti-tumor response is observed with 100% tumor rejection and no incidents of recurrence (Figure 2B). The tumor eradication observed in this model is dependent upon engagement of TRP-1 CD4+ T cells with MHC-II expressed by the tumor itself; when MHC-II is blocked via neutralizing antibody in MHC-II deficient hosts receiving the adoptive transfer of TRP-1 CD4+ T cells the anti-tumor response is diminished (Figure S1A,B). In order to determine how the differential accumulation of self-antigen specific T cells might impact tumor response, tumor infiltration of the TRP-1 CD4+ T cells was evaluated. At days 7, 9, and 11 the number of TRP-1 CD4+ T cells infiltrating the tumor is greater in the cohort receiving 106 compared to 105 precursors (Figure S2A,D,E). Additional characterization of the tumor infiltrating TRP-1 CD4+ T cells revealed that at a precursor frequency of 105, a higher percentage of the TRP-1 CD4+ cell population are Foxp3+ regulatory T cells, which resulted in an unfavorable effector to regulatory T cell ratio compared to the higher precursor frequency group (Figure S2B,C).
Figure 2. The intraclonal competition of tumor specific CD4+ T cells does not preclude a successful anti-tumor immune response.

Eighteen days post tumor implantation mice received 600 cGy of irradiation, followed by tail vein injection of 0, 103, 105, 106, or 107 TRP-1 CD4+ T cells co-transferred with naïve splenocytes and tumor regression was monitored. (A) Tumor diameter was measured every 3–5 days by caliper and is represented on the graph by individual lines. (B) The overall survival of each group was plotted. Representative data of over 3 separate experiments (n= 9–10 mice per group). See also Figure S1
At high clonal abundance tumor specific CD4+ T cells differentiate into polyfunctional effector cells
In other models of CD8+ and CD4+ T cell competition, the failure to mount an effective immune response was correlated with impaired development of polyfunctional effector T cells (Foulds and Shen, 2006; Rizzuto et al., 2009). To investigate whether intraclonal competition had affected the differentiation of the tumor specific CD4+ T cells, we focused on day 7 during the initiation of the anti-tumor response. We chose to examine the earliest phase of the response because variability increases at later stages due to the heterogeneous kinetics of regression and levels of tissue necrosis. T cells were harvested from tumors and draining LNs, restimulated ex vivo and assessed for the production of effector cytokines IFNγ and TNFα, as well as the expression of granzyme B, given the cytotoxic nature of TRP-1 CD4+ T cells (Muranski et al., 2008; Quezada et al., 2010; Xie et al., 2010). In lymph nodes, tumor specific CD4+ T cells differentiated into the most potent effector T cells when their clonal abundance was highest (Figure 3A,B), with up to 25% of the population producing both IFNγ and TNFα. The tumor specific T cells at lower clonal abundance produced little IFNγ and TNFα and did not upregulate expression of granzyme B; however, they did produce the greatest amount of the cytokine IL-21, which is one of the phenotypic changes CD4+ T cells undergo during exhaustion (Figure S3A,B) (Crawford et al., 2014). A complementary effector phenotype was observed in the tumor infiltrating TRP-1 CD4+ T cell population (Figure 3C,D; Figure S3C,D). These results demonstrate that high clonal abundance favors generation of polyfunctional effector CD4+ T cells and that the extent of T cell differentiation established during priming in the lymph node is preserved during the effector phase.
Figure 3. At high precursor frequencies tumor specific CD4+ T cells differentiate into polyfunctional effector cells.
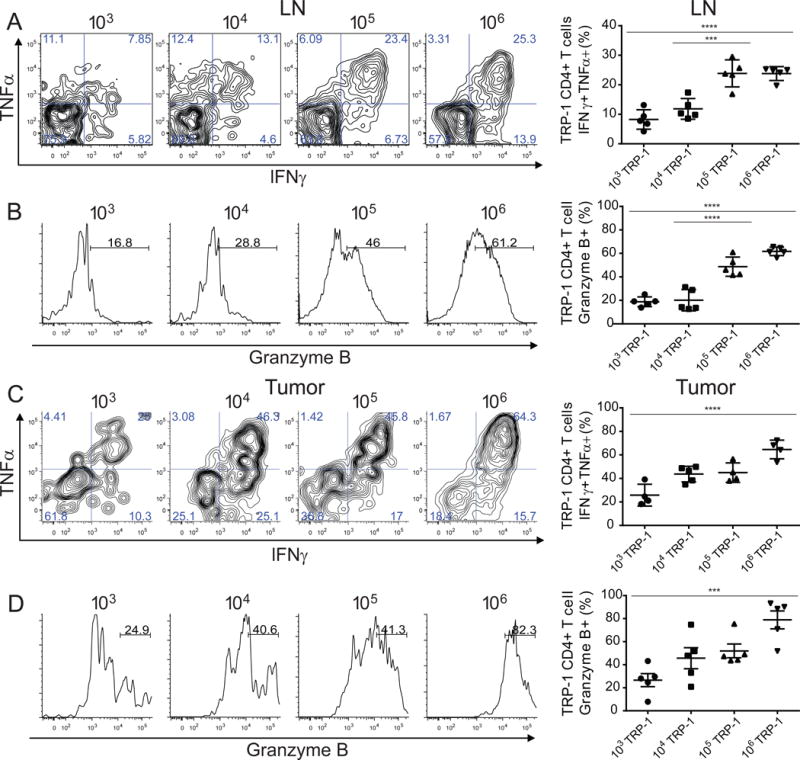
Lymphocytes isolated from tumor bearing mice that had received adoptive transfer of 103, 104, 105, or 106 congenically marked TRP-1 CD4+ T cells were re-stimulated to assess effector function. (A) D7 p.t. lymphocytes restimulated with PMA/ionomycin, representative flow plots of IFNγ and TNFα expression in TRP-1 CD4+ T cell population (left), summary of percentage of IFNγ+ TNFα+ of TRP-1 CD4+ T in LN and (C) tumor. (B) Representative histogram plots of granzyme-B expression in TRP-1 CD4+ T cell population (left), summary of percentage of granzyme B producing population in LN and (D) tumor. Representative data of 3 separate experiments (n=4–5 mice per group). Data are represented as mean ± SEM.
See also Figure S2
Clonal abundance is directly correlated with killing efficiency and Th1 cell differentiation
While production of effector cytokines and expression of granzyme B are useful surrogates for effector function, we wanted to establish if clonal abundance influenced the development of CD4+ T cell cytotoxic function. We performed an in vivo killing assay, in which splenocytes were divided into two populations and labeled CellTrace Violet (CTV)hi or CTVlo. The CTVhi population was loaded with the MHC-II restricted peptide recognized by the TRP-1 CD4+ T cells and the CTVlo cell population was used as control. Both populations were transferred intravenously and killing was evaluated by comparing the ratio of peptide-loaded targets to unloaded controls. Killing of targets in vivo increased in direct proportion to the clonal abundance of the antigen specific T cells (Figure 4A,B). While the in vivo killing assay clearly shows that a higher clonal abundance of tumor-antigen specific CD4+ T cells results in better killing of target cells, it cannot be used to differentiate the individual contributions of T cell quantity versus quality.
Figure 4. Precursor frequency is directly correlated with killing efficiency and Th1 differentiation.
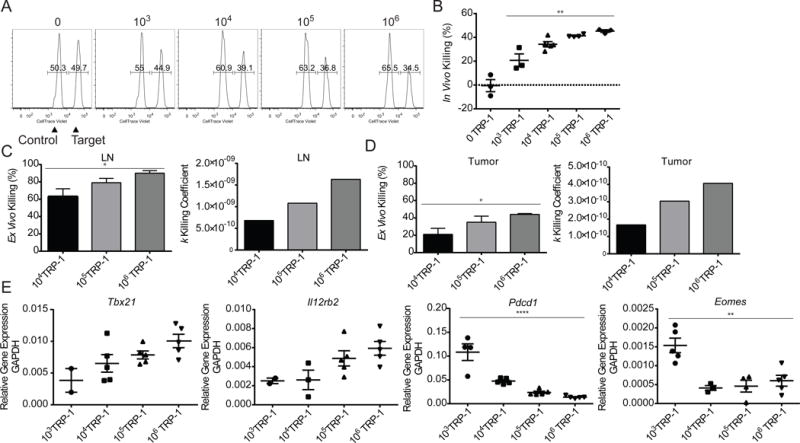
Lymphocytes from tumor bearing mice that had received adoptive transfer of 103, 104, 105, or 106 congenically marked TRP-1 CD4+ T cells were assessed for cytotoxic function and characterized. (A) On day 6, mice were transferred with 500,000 CellTraceViolet (CTV) labeled in vivo killing assay targets (250,000 unpulsed:250,000 antigen pulsed). Representative flow plots of day 7 in vivo killing of target cells in spleen. (B) Summary of in vivo killing as percentage of targets killed. (C) TRP-1 CD4+ T cells were sorted from pooled LN and (D) tumor on d7 p.t. and co-embedded with B16 in an ex vivo killing assay, percentage of B16 killed (left), k coefficient of killing was calculated using empirically determined tumor killing and growth rates (right). Each condition was performed in triplicate. (E) D7 p.t. TRP-1 CD4+ T cells were sorted from individual LN directly into Trizol for RNA extraction and qRT-PCR analysis of targets Tbx21, Il12rb2, Pdcd1, and Eomes. All data representative of 2–3 independent experiments.
To determine the contribution of T cell quantity on the anti-tumor response we utilized a clonogenic ex vivo killing assay (Budhu et al., 2010). TRP-1 CD4+ T cells isolated from naïve donor mice were directly embedded at varied T cell concentrations with a fixed quantity of B16 tumor cells in collagen-fibrin gels. At 24, 48, and 72 hours the gels were lysed and plated to form colony-forming units, which were counted 7 days later. We observed the greatest level of killing in the condition where the T cell concentration was the highest (Figure S4A). In the groups with high concentrations of TRP-1 CD4+ T cells, 106 and 5 × 106, tumor killing was sustained over time and was associated with an increase in cytokine accumulation (Figure S4C). However, at the lower concentrations, tumor growth began to outpace killing, and lower responses were seen at 72 hours compared to the peak killing at 48 hours (Figure S4B). These findings implicate T cell concentration within the tumor as one of the factors that shifts that balance between regression and progressive tumor growth.
Conversely, to directly evaluate the ability of clonal abundance to enhance the development of cytotoxic function on a per cell basis, titrated quantities of 104, 105 or 106 TRP-1 CD4+ T cells with normal repertoire diversity naïve splenocytes were co-transferred into irradiated recipients bearing established B16 melanoma. On day 7, the TRP-1 CD4+ T cells were sorted directly from tumor draining LNs and tumor, and then co-embedded at identical frequencies with B16 melanoma cells in the collagen-fibrin gel based killing assay. Killing percent and killing coefficient k were determined by comparing the killing rate of each group to the empirically determined tumor growth rate (Figure 4C,D). We found that the per cell killing capability of the effector cells increased with increasing clonal abundance of the tumor specific CD4+ T cells.
To better understand the underlying factors driving this increase in effector function, we investigated candidate transcriptional regulators of cytotoxicity by RT-PCR. The expression of the master regulator of the Th1 cell lineage, T-bet or Tbx21, directly correlated with the increase in killing function (Figure 4E); Similarly, expression of the T-box transcription factor Eomesodermin (Eomes) was increased at low clonal abundances –a hallmark of terminal differentiation and exhaustion (Paley et al., 2012). Additionally, we found that IL-12RB2 expression correlated with T-bet expression and the initial precursor frequency of the tumor specific CD4+ T cells. These findings suggest that as clonal abundance of antigen specific CD4+ T cells increases, the population more efficiently drives its own lineage commitment. We also found that PD-1 (Pdcd1) expression was inversely correlated with T-bet expression in the TRP-1 specific CD4+ T cells, which supports that T-bet may be suppressing PD-1 expression (Kao et al., 2011). The high Eomes and PD-1 expression and reduced T-bet expression occurring in T cells derived from low clonal abundances strongly suggested that the impaired development of effector function could be the consequence of T cell exhaustion.
Poorly differentiated tumor specific CD4+ T cells express high levels of T cell exhaustion markers independent of tumor burden
Characterization of T cell exhaustion in chronic viral infection and tumor models has primarily focused on the CD8+ T cell compartment. However, recent studies have identified markers of T cell exhaustion shared with or unique to CD4+ T cells. To determine if the impaired effector phenotype of tumor specific CD4+ T cells at low clonal abundance was linked to T cell exhaustion we investigated expression of the inhibitory and costimulatory molecules PD-1, CTLA-4, LAG-3, ICOS, BTLA, and CD27, which are components of the CD4+ T cell exhaustion profile (Crawford et al., 2014). We found that in the LN, exhaustion markers were upregulated on tumor specific T cells derived from low clonal abundance, compared to T cells from high clonal abundance (Figure 5A). The expression pattern of T cell exhaustion markers was similar, although attenuated, in the tumor, with the exception of LAG-3 (Figure 5A). We also observed IL-21 production in the low precursor frequency TRP-1 specific CD4+ T cells, which further implicates a state of exhaustion (Figure S3A–D).
Figure 5. At low precursor frequencies tumor specific CD4+ T cells express high levels of T cell exhaustion markers independent of tumor burden.
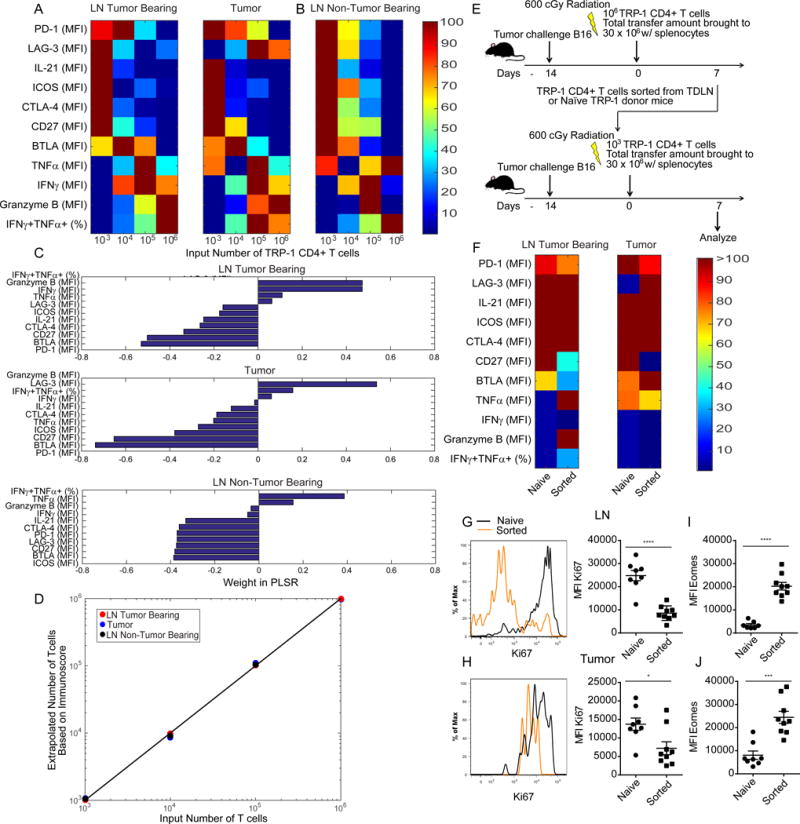
Lymphocytes from tumor bearing mice that had received adoptive transfer of 103, 104, 105, or 106 TRP-1 CD4+ T cells were assessed on d7 p.t. for markers of exhaustion. (A) Heat maps depicting measurements of activation and exhaustion markers on TRP-1 CD4+ T cell d7 p.t. in LN and tumor of tumor bearing hosts. (B) As described in (A) for day 7 p.t. LN of naïve tumor free hosts. Mean MFI or percentage (n=5 mice/group). (C) PLSR weights for activation/exhaustion measures across tissues in tumor bearing and non-tumor bearing mice. (D) PLSR of the precursor frequencies of TRP-1 T cells as a function of activation/exhaustion measures leads a PLSR score that captures from 99, 95 and 92% of the variability in precursor frequency and 70, 75 and 76% of the variability of activation/exhaustion measures. (E) TRP-1 CD4+ T cells derived directly from Tgn donors or AT recipients of 106 TRP-1 CD4+ T cells on d7 p.t. were serially transferred into new tumor bearing adoptive hosts. (F) Heat maps for measures of activation and exhaustion on TRP-1 CD4+ T cell d7 in LN and tumor.
(G) Representative histogram plots and summaries of Ki67 expression on TRP-1 CD4+ T cell d7 p.t. in LN and (H) tumor. (I) Summary of Eomes expression of TRP-1 CD4+ T cell d7 p.t. in LN and (I) tumor (n= 9–10 mice/group). All data representative of 2–3 independent experiments.
See also Figure S3.
To understand if the T cell exhaustion was dependent on tumor burden or potentiated by the higher proliferative potential of T cells at low initial abundance, we examined development of exhaustion in tumor free mice. Once again, expression of T cell exhaustion markers was higher when tumor specific CD4+ T cells were present at the low (physiologic) frequency (Figure 5B). The ability of the T cells to produce effector cytokines had a similar trend to that in tumor bearing mice (Figure 5B). CD4+ T cells in low abundance favor IL-21 production but reduce TNFα production. However, the impairment in effector phenotype is not as marked in naïve animals, likely due to the lack of tumor-derived antigen in the lymph nodes driving the extreme polarization.
To quantitatively estimate the multifactorial aspects of T cell exhaustion, we created an exhaustion score compiling the overall quality of T cell responses, and demonstrated its dependence on precursor frequency. To do so, we normalized the variable abundance of markers (Mi) on the surface of T cells harvested from the LN of tumor bearing and tumor free mice, as well as tumor, derived from different clonal abundances. We then applied a partial-least square regression (PLSR) against the logarithm of the number of input T cells in order to generate an exhaustion immunoscore YT cells, as a weighted sum of the abundance of markers. PLSR is a supervised statistical method that best identifies the latent variables necessary to predict the multivariate immune response in our system, while avoiding the pitfall of overfitting (Wold et al., 2001). The PLSR weights correlated positively for effector functions and negatively for exhaustion markers in all tissues examined independent of tumor burden (data not shown); Granzyme B and PD-1 expression were the greatest phenotypic contributors to exhaustion scoring in tumor bearing mice (Figure 5C). Our exhaustion immunoscore captured 95, 92, and 99% of the variance in the number of cells and 75, 76, and 70% of the variance in activation and exhaustion marker expression respective to the tissue examined. The linear correlation between exhaustion score YT cells and initial precursor frequency was excellent (R> 0.999) (Figure 5D).
To further characterize the impact of initial precursor frequency on development of exhaustion, we examined whether exhaustion could be induced in the effector CD4+ T cells generated at high clonal abundance by performing a serial adoptive transfer experiment. Tumor bearing recipients received adoptive transfer of 106 TRP-1 CD4+ T cells and on day 7 the TRP-1 CD4+ T cells were sorted directly from the tumor draining LNs of these mice. The sorted cells were then transferred at a precursor frequency of 103 T cells, which has been established to potentiate T cell exhaustion, into new hosts bearing established B16 melanoma (Figure 5E). As a control, TRP-1 CD4+ T cells were also sorted from naive donor mice and transferred at the same 103 precursor frequency into a separate cohort of tumor bearing hosts. Upon adoptive transfer, the T cells that had previously differentiated into effector T cells show a strong proliferative defect characteristic of terminal differentiation (Figure 5G,H). Compared to naïve TRP-1 cells transferred at a clonal abundance of 103, the sorted cells from the high clonal abundance condition display comparable or increased expression of exhaustion markers, but retain some effector function characteristic of high clonal abundance and consistent with the hierarchical loss of function during T cell exhaustion; the onset of T cell exhaustion is accompanied by increased Eomes expression (Figure 5E,I–J). These results demonstrate that exhaustion is influenced both by initial precursor frequency and previous states of differentiation.
Lower clonal abundance promotes an irreversible exhausted phenotype
Considering the recent successes of checkpoint blockade for the treatment of advanced malignancies, we tested whether this approach could reverse the exhaustion observed in our model (Barber et al., 2006; Hirano et al., 2005). We chose to target PD-1 due to its high PLSR weight and the established clinical efficacy of PD-1 blockade (Topalian et al., 2015). Tumor bearing mice with a clonal abundance of 103 TRP-1 CD4+ T cells received PD-1 blockade every 3 days beginning on the day of adoptive transfer and were compared to mice with a clonal abundance of 106 due to their ability to mount a successful anti-tumor response. In the LNs and tumors of mice treated with PD-1 blockade, the tumor antigen specific CD4+ T cells failed to show any increase in effector function (Figure 6A–D). Additionally, the TRP-1 CD4+ T cells receiving PD-1 blockade showed upregulation of Eomes expression, which increased in proportion to T-bet (Figure 6G,H). When the TRP-1 CD4+ T cells were sorted from the LNs and tumor and then embedded at identical numbers in a clonogenic ex vivo killing assay, there was a reduction in cytotoxicity on a per cell basis in the group that received PD-1 blockade (Figure 6E, F). To assess if PD-1 blockade could be improved by combining it with another form of checkpoint blockade, we combined PD-1 blockade with anti-CTLA-4 therapy. This target was chosen after a pilot experiment comparing LAG-3 blockade to CTLA-4 blockade had demonstrated similar efficacy (data not shown). Yet, no improvement in effector function was observed in any treatment condition (Figure S5A,B).
Figure 6. Lower precursor frequencies promote an irreversible exhausted phenotype.
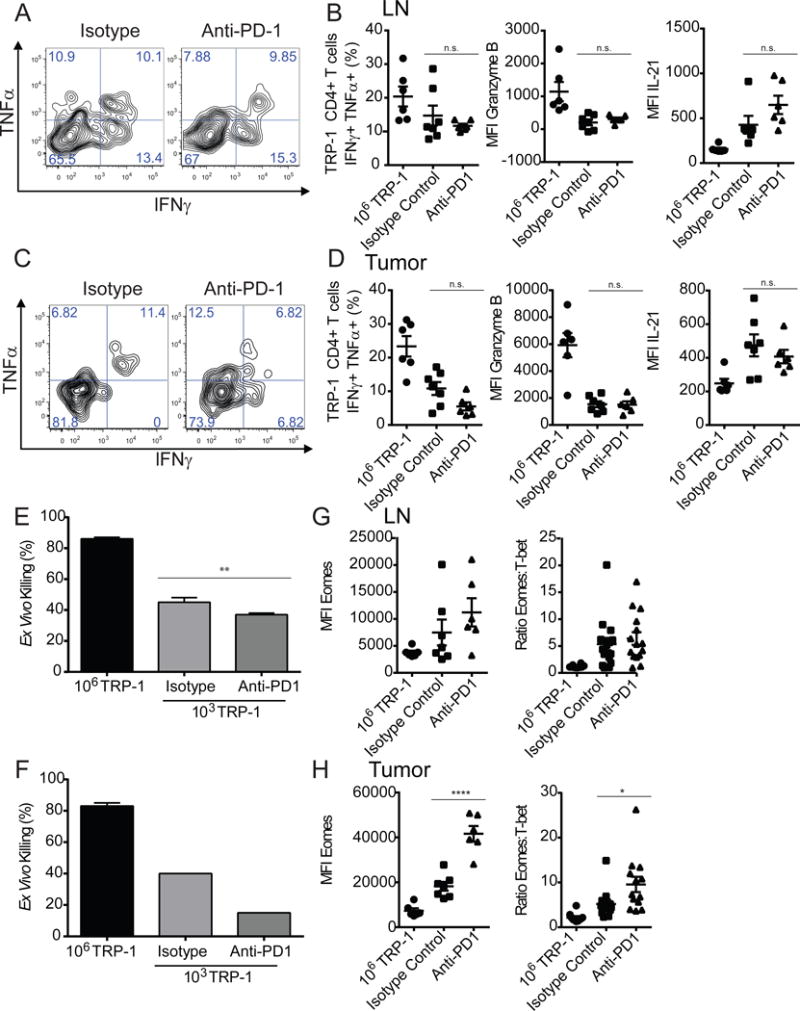
Tumor bearing mice receiving adoptive transfer of 103 TRP-1 CD4+ T cells were treated with anti-PD-1 antibody or an isotype control every 3 days beginning on the day of adoptive transfer. On d7 p.t. lymphocytes were isolated from LN and tumor of treated tumor bearing mice and restimulated with PMA/ionomycin. (A) Representative plots of IFNγ+ TNFα+ TRP-1 CD4+ T cells in lymph node and (C) tumor. (B) Summary of percentage of IFNγ+ TNFα+ TRP-1 CD4+ T cells, MFI of granzyme-B and IL-21 respectively in LN and (D) tumor (n=5–7 mice/group). (E) TRP-1 CD4+ T cells were sorted from LN or tumor (F) of treated mice on day 7 and co-embedded with B16 in an ex vivo killing assay, percentage of B16 killed. (G) MFI of Eomes of TRP-1 CD4+ T cells in day 7 LN and tumor (H) and as a ratio in comparison of MFI of T-bet within population from pooled duplicate experiments. Data are representative of three independent experiments. Data are represented as mean ± SEM.
See also Figure S4
Generation of a polyfunctional effector phenotype is associated with helper functions of tumor specific CD4+ T cells
Traditionally, CD4+ T cells are known to orchestrate an immune response through the maturation of antigen presenting cells and the secretion of cytokines (Bevan, 2004). We first looked for evidence of dendritic cell maturation in the lymph nodes of animals with TRP-1 specific CD4+ T cells of different precursor frequencies. In CD11b+CD11c+ dendritic cells, we observed increased expression of both CD80 and MHC-II as the frequency of tumor specific CD4+ T cells increased (Figure 7A), while CD86 expression was unchanged (data not shown). The increase in MHC-II expression correlated with an increased concentration of IFNγ found in lymph node extracts above the threshold of functional significance (Figure 7C). The cytokine IFNγ has been described to induce MHC-II expression on cells that are devoid of MHC-II at homeostasis, including B16 melanoma. This tumor specific expression has been shown to be vital to the cytotoxic function of TRP-1 CD4+ T cells (Quezada et al., 2010). We investigated if IFNγ could be enforcing a positive feedback loop within the tumor. We found that MHC-II expression on both CD45− tumor cells, as well as the CD45+ lymphocytic infiltrate is highest when the tumor specific T cells are at the greatest clonal abundance (Figure 7B). This increase in MHC-II expression correlated with accumulation of higher levels of IFNγ as measured in tumor extract (Figure 7D).
Figure 7. Generation of polyfunctional effector phenotype is associated with helper function of tumor specific CD4+ T cells.
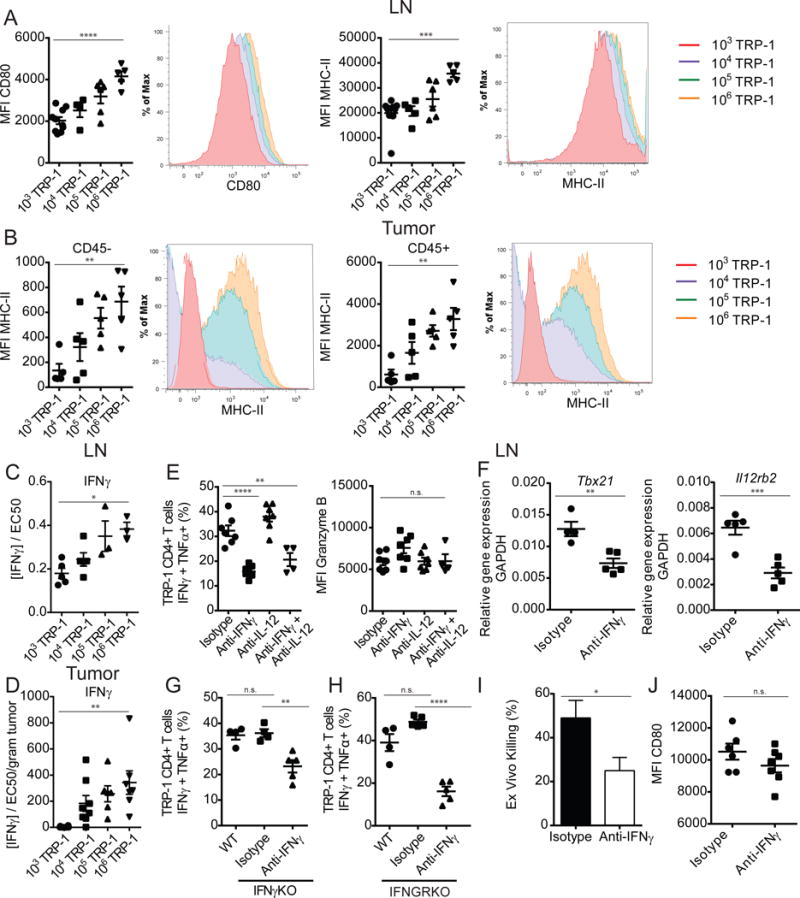
Tumor bearing adoptive hosts that had received adoptive transfer of 103, 104, 105, or 106 TRP-1 CD4+ T cells were characterized. (A) MFI of CD80 and MHC-II expression on d7 p.t. Cd11b+Cd11c+ LN dendritic cells, summary (right) and representative histogram plots (left). (B) MFI of MHC-II expression on CD45− and CD45+ tumor populations, summary (right) and representative histogram plots (left). (C) Quantification of IFNγ concentration in LN and (D) tumor homogenates, calculated to demonstrate activity as a function of half-maximal response of mIFNγ (n=3–5 mice/group). (E) D7 p.t. lymphocytes were isolated from LN of treated WT tumor bearing mice and restimulated with PMA/ionomycin, summary of percentage of IFNγ+ TNFα+ TRP-1 CD4+ T cells in LN, MFI IFNγ, granzyme-B respectively. (F) D7 p.t. TRP-1 CD4+ T cells were sorted from LN of treated mice directly into Trizol for RNA extraction and qRT-PCR analysis of targets Tbx21, Il12rb2. (G) As described in (E) for IFNγ knockout mice and (H) IFNGR knockout mice. (I) TRP-1 CD4+ T cells were sorted from LN of treated mice on d7 p.t. and co-embedded with B16 in an ex vivo killing assay, percentage of B16 killed. (J) CD80 expression on LN Cd11b+Cd11c+ dendritic cells of mice treated with IFNγ neutralization. Data are representative of three independent experiments. Data are represented as mean ± SEM.
See also Figure S5
IFNγ is also the canonical effector cytokine of the Th1 lineage; lineage commitment begins during T cell priming when IFNγ signaling initiates early expression of T-bet. T-bet induction promotes expression of IL-12 receptor, which then shifts Th1 differentiation into a primarily IL-12 driven process (Lazarevic et al., 2013). To address the role of Th1 polarization in the development of effector function we sought to neutralize the cytokines IFNγ and IL-12 in vivo, alone and in combination. In the two groups receiving IFNγ neutralizing antibodies, the generation of effectors capable of producing both IFNγ+ and TNFα+ was impaired, but granzyme B expression was not significantly altered (Figure 7E). IL-12 did not contribute to the effector differentiation of the tumor specific T cells, so was not investigated further. We sorted and embedded the T cells that had differentiated in vivo in the presence and absence of IFNγ neutralization in an ex vivo kill assay. These T cells possessed decreased killing ability when differentiation had occurred in the absence of IFNγ (Figure 7I). The decrease in effector function was associated with decreased Th1 polarization, as measured by expression of Tbx21 and IL-12rb2 (Figure 7F). When IFNγ was neutralized during the in vitro differentiation of TRP-1 CD4+ T cells an even more dramatic reduction in effector function and Th1 polarization was observed (Figure S6A–F).
To address if paracrine IFNγ produced by the TRP-1 CD4+ T cell population was driving their own differentiation, we performed the same adoptive transfer of 106 TRP-1 specific CD4+ T cells, using splenocytes and hosts deficient in IFNγ production. In this model, IFNγ could only be derived from the transferred TRP-1 transgenic CD4+ T cells or the B16 tumor itself, which has not been described to produce IFNγ. Beginning the day before adoptive transfer, mice received an isotype control or an IFNγ neutralizing antibody, with the exception of the control group. Formation of a polyfunctional population of tumor specific CD4+ T cells was the same in wild-type and IFNγ deficient hosts (Figure 7G), demonstrating that IFNγ produced by the TRP-1 specific CD4+ T cell population alone is sufficient to drive their own differentiation. When IFNγ is blocked in this experiment, the development of effector function is decreased, but not completely extinguished. While IFNγ is an important clonally derived mediator of the population’s differentiation, it is most likely not the only necessary factor. To elucidate how IFNγ might be regulating cell extrinsic factors important to the differentiation of the CD4+ T cells, we performed a similar experiment utilizing IFNγ receptor (Ifngr) knockout tumor bearing hosts. In this experiment, the only cells expressing the IFNGR, and thus directly responsive to IFNγ, were the TRP-1 CD4+ T cells and the implanted tumor. When IFNγ was neutralized in these mice, differentiation is still not completely abolished and is maintained at concentrations similar to previous experiments asserting that the other factors contributing to the differentiation of the T cells are not directly regulated by IFNγ (Figure 7H). In support of this, we observe that IFNγ neutralization has no effect on the expression of CD80 (Figure 7J) in the lymph node, implicating other mechanisms of T cell help, likely through costimulation.
Although neutralization of IFNγ resulted in a decrease in T cell differentiation in the lymph nodes, the effector phase of the immune response in the tumor was also altered. The neutralization of IFNγ resulted in a paradoxical increase in the population of T cells producing effector cytokines upon restimulation of the cells isolated from the tumors of wild type, IFNγ knockout, and IFNGR knockout hosts (Figure S7A,B). This increase in cytokine production only marginally altered the overall ex vivo cytotoxicity of the T cells isolated from wild-type hosts, possibly due to the converse decrease in granzyme B production (Figure S7C). Neutralization of IFNγ affects regulation of sources of both positive and negative feedback in the tumor. Blocking IFNγ results in the downregulation of MHC-II on both CD45− and CD45+ tumor populations, but also causes a decrease in the expression of the inhibitory ligand PD-L1 on the CD45− tumor, which could simultaneously release negative feedback on the T cells and may result in restored effector function (Figure S7D).
Despite the complexity of the pleiotropic effects mediated by IFNγ, this cytokine appears to be an integral component of a successful CD4+ T cell mediated anti-tumor response. When IFNγ is neutralized, tumor regression is dramatically impacted. Overall survival of the population drops from 100% to less than 50% and many of the tumors fail to regress (Figure S7E,F). Whether this loss of tumor regression is due to IFNγ as an effector cytokine itself, driver of T cell differentiation, regulator of tumor recognition, or more likely a combination of these factors, it is clear that it is a fundamental mediator of the intraclonal cooperation within the tumor-responding CD4+ T cell population.
DISCUSSION
The constrained activation and proliferation we observe within populations of high clonal abundance is consistent with observations made in other systems and our previous findings with elevated frequencies of antigen specific CD8+ T cells. However, here the impaired proliferative capacity was uncoupled from impaired T cell differentiation. Interestingly, it is more common for the strict linkage between division and differentiation in the CD4+ subset to break down when precursor frequency exceeds physiological levels (Laouar and Crispe, 2000). Although extensive division may be unnecessary, we observe that the vast majority of tumor specific CD4+ T cells that have acquired effector function have also undergone proliferation (data not shown). It has been described that large antigen doses can overcome barriers to initial activation at high precursor frequencies and it is possible that this is occurring in our model due to the ubiquitous expression of the cognate antigen (Catron et al., 2006). Even though a smaller percentage of the total T cell population is activated and recruited into the response at any single time point at high precursor frequencies, when the greater cumulative size of this population is taken into account, a larger absolute number of cells are engaged. It is likely the total accumulation of activation signals that antigen presenting cells are integrating from the tumor specific T cell population, through costimulatory and cytokine signaling, that contributes to their increased maturation and antigen presentation. Consequently, this lowers the threshold of activation for the tumor specific CD4+ T cell population as a whole.
When tumor specific T cells are present at low precursor frequencies they become functionally exhausted. It may seem counter-intuitive that T cells would be susceptible to exhaustion by day 7 of the anti-tumor immune response; however, early T cell dysfunction appears to be a characteristic of CD4+ T cell exhaustion and has been previously observed during chronic LCMV infection (Brooks et al., 2005; Crawford et al., 2014). One could argue that the T cells at low precursor frequencies are displaying an activated phenotype, as all exhaustion markers are also markers initially upregulated upon T cell activation. However, the T cells derived from low clonal abundance also show the impaired production of IFNγ and TNFα and increased IL-21 production that are characteristic of exhausted CD4+ T cells. It is tempting to speculate that induction of exhaustion in self-antigen specific CD4+ T cells at low precursor frequencies could act as a failsafe mechanism to enforce peripheral tolerance for clones that have escaped negative selection. Further studies will be required to determine if pools of exhausted self-antigen specific clones exist in the periphery and if their IL-21 production contributes to the development of the aberrant autoantibody production occurring in early stage autoimmune disease.
The presence of the tumor was shown to be unnecessary for the generation of the exhausted T cell phenotype. It is possible that the TRP-1 CD4+ T cells are experiencing chronic antigen exposure in the absence of tumor, due to the fact that TRP-1 is a self-antigen ubiquitously expressed in mouse melanocytes. However, another possibility is that placing these T cells into the lymphopenic environment induced by sub-lethal irradiation is exacerbating the TCR stimulation received from self peptide-MHC-II during homeostatic proliferation. Interestingly, in many of the model systems commonly used for examining T cell exhaustion such as HIV and LCMV infection, some degree of lymphopenia accompanies the chronic infection (Corbeau and Reynes, 2011; Walsh et al., 2010). Regardless of the source of the chronic stimulation, it seems that while the intraclonal competition of T cells may impair proliferation it can also protect against exhaustion by limiting repeated antigen exposure.
Heterogeneity within the overarching phenotype of T cell exhaustion exists not only between CD4+ and CD8+ T cell lineages, but also within each lineage. One way in which exhaustion is characterized is by examining the ratio of the T-box transcription factors T-bet and Eomes (Buggert et al., 2014; Paley et al., 2012). It is largely believed that within the exhausted CD8+ T cell subset EomeshiTbetloPD-1hi T cells have less proliferative potential and are more refractory to checkpoint blockade than exhausted populations expressing higher levels of T-bet. Within this context, it is more understandable why checkpoint blockade has failed in our model. The tumor antigen specific CD4+ T cells at low precursor frequencies express low levels of T-bet with Eomes levels comparable to or greater than T cells at high precursor frequencies. This combination suggests they would be resistant to checkpoint blockade. Indeed, we observe that blocking PD-1 results in an increase in the expression of Eomes, supporting that checkpoint blockade is pushing the population further towards terminal differentiation. The occurrence of exhaustion and the failure of checkpoint blockade to reverse it have profound implications for the use of CD4+ T cells in adoptive cell therapy (ACT) for cancer patients. While it would be tempting to use small numbers of tumor specific T cells for adoptive cell therapy, due to the speed at which they can be derived, and compensate by combining ACT with immune modulation, our findings would caution against this approach.
We believe the improved effector function observed at high clonal abundance is dependent upon a combination of CD4+ T cell help and paracrine IFNγ signaling. Historically, CD4+ T cell help has been described in the context of the CD8+ T cell response. T cell help is indispensible to the success of the cytotoxic T cell response to infection and has also been implicated in the prevention of CD8+ T cell exhaustion during chronic infection (Bevan, 2004; Zajac et al., 1998). While not as much is known in regards to help provided to CD4+ T cells themselves, there is evidence for cooperation between CD4+ T cell clones of different specificities (Creusot et al., 2003). The upregulation of CD80 expression on lymph node dendritic cells observed in our model is likely due to increased engagement with CD40 or MHC-II by the tumor specific T cells at high clonal abundance (Nabavi et al., 1992; Ranheim, 1993). This increased CD80 expression may be contributing to the residual effector differentiation occurring during IFNγ neutralization. During normal conditions, increased antigen presentation and co-stimulation would be acting in synergy with the accumulation of paracrine cytokines, such as IFNγ and possibly IL-2. This population-mediated cooperation is likely deterring the development of exhaustion, through the expression of protectively high levels of T-bet.
Despite the importance of IFNγ production during priming and differentiation of tumor specific CD4+ T cells in the lymph node, we paradoxically find that IFNγ neutralization increases the population of T cells secreting effector cytokines in the tumor. It is known that IFNγ itself can regulate the expression of its own receptor. In our model, we have found that as a result of IFNγ blockade, the expression of IFNGR is increased (data not shown). It is possible that within the tumor, this increase in IFNGR expression raises the threshold of sensitivity of the TRP-1 specific CD4+ T cells to autocrine IFNγ or IFNγ that has not been neutralized due to high intra-tumor concentrations. Alternatively, it is also known that IFNγ is a major driver of negative feedback loops that dampen immune activation. It is possible that by removing IFNγ from the tumor microenvironment, we are decreasing IFNγ mediated immunosuppression. This could occur in a T cell intrinsic manner through the decreased production of IL-10, but could also involve a secondary mediator produced by tumor cells themselves. Possibilities include the decreased binding of inhibitory receptors PD-1 or LAG-3 on the T cell to their tumor expressed ligands PDL1 and MHC-II, as well as a reduction in other immunosuppressive mediators such as IDO and arginine. While it is not clear why neutralization of IFNγ is having such disparate effects in the lymph node and tumor environment, our findings necessitate an in depth characterization of the dynamics of CD4+ T cell immune modulation of the tumor microenvironment.
EXPERIMENTAL PROCEDURES
Mice and tumors
All mouse procedures were performed in accordance with institutional protocol guidelines at Memorial Sloan-Kettering Cancer Center under an approved protocol. C57BL/6J, Ifng−/−, Ifngr1−/− were obtained from The Jackson Laboratory. TRP-1 CD4+ TCR transgenic mice were obtained from the N. Restifo laboratory (National Institutes of Health, Bethesda, MD). TRP-1 CD4+ TCR-Luciferase reporter mice were obtained from the J. Allison laboratory (MD Anderson Cancer Center, Houston, TX). TRP-1 CD4+ TCR transgenic were crossed to a Rag1−/− Tyrp1−/− CD45.1 background. The B16-F10 melanoma line was originally obtained from I. Fidler (M.D. Anderson Cancer Center, Houston, TX). Tumor implantation was via intradermal injection containing 2.5 × 105 B16–F10 and PBS in the flank of shaved recipients. Tumor growth was tracked every 3–5 days via caliper measurement.
Irradiation and adoptive transfer
Recipient mice received 600 cGy total body irradiation several hours prior to adoptive transfer. Donor cells were isolated from LN and spleen of male TRP-1 TCR transgenic mice or TRP-1 TCR-Luciferase reporter mice when indicated. Purification was by positive selection magnetic cell sorting using CD4 beads (L3T4) (Miltenyi Biotech) according to the manufacturer’s instructions and injected intravenously. Normal repertoire diversity splenocytes were derived from the ACK lysis buffer (Lonzo) incubated spleens of naïve donors. When indicated, CD4+ T cells were labeled with 5 μM CFSE (Life technologies) prior to transfer according to manufacturer’s instructions.
Flow cytometry
Cells from tumor draining lymph nodes, spleens, and tumors were prepared by mechanical dissociation in 40 μM filters and red blood cells were removed. When tumor mass exceeded. 1 grams lymphocytes were isolated using Percoll (GE Healthcare) gradient centrifugation. TRP-1 CD4+ T cells were detected by staining for congenic CD45.1 marker. All intracellular staining was conducted using the Foxp3 fixation/permeabilization buffer (eBioscience) according to the manufacturer’s instructions. Flow cytometry was performed on an LSRII (BD Biosciences). FlowJo software (version 9.4.10; Tree Star) was used for all flow cytometry analysis. FACS sorting was conducted on a FACSAria II cell sorter (BD Biosciences).
In vivo neutralization via monoclonal antibody
Anti-PD-1(RMP1–14), anti-MHC-II (M5/114), anti–CTLA-4 (9D9,) anti–IFNγ (XMG1.2), anti-IL-12p40 (C17.8) and respective isotype controls (2A3, LTF-2, HRPN, MPC-11) were purchased from BioXCell. For cytokine neutralization experiments mice received i.p. injection of 200 μg/dose of isotype control, anti-IFNγ, or anti-IL-12p40 antibody with the exception of the wildtype control group. Treatment was initiated the day before adoptive transfer and was administered every other day. For checkpoint blockade experiments mice received intra-peritoneal injection of 250 μg/dose of anti-PD-1, 100 μg/dose of anti-CTLA-4, or their respective isotype controls, with the exception of the 106 adoptive transfer control group. Treatment was initiated the day of adoptive transfer and was administered every 3 days. See Supplemental Experimental Procedures for more details.
In Vivo killing assays
For in vivo killing assays splenocytes from C57BL/6J mice were labeled with 5 or 0.5 μM Cell Trace Violet. CTVhigh splenocytes were loaded with 20 μM of TRP-1 peptide (Genemed Synthesis, Inc) for 2 hours at 37°C. On day 6 after adoptive transfer 5 × 105 cells of a 50:50 mixture of TRP-1 peptide pulsed and unpulsed splenocytes were transferred via tail vein injection in a 200 μl bolus. The following day, mice were sacrificed and spleens were removed, and the percentage of loaded and unloaded splenocytes was analyzed by flow cytometry. Cytotoxicity was calculated using the following equation: In vivo killing percentage = 100% (1−((unloaded/loaded)control/(unloaded/loaded)experimental)).
Supplementary Material
Acknowledgments
This study was partially supported by NIH grants R01CA56821, the NIH/NCI Cancer Center Support Grant P30 CA008748; Ludwig Collaborative Laboratory; Swim Across America, and the Lita Annenberg Hazen Foundation. NM was supported by a Cancer Research Institute Pre-Doctoral Fellowship. We would like to thank Karen Tkach, Jen Oyler-Yaniv, and Alon Oyler-Yaniv for excellent discussion and insights.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization NM, GAB, TM and JW; Methodology NM, GR, TM, and JW; Investigation NM, SB NK, CL, JM, and CC; Resources HZ and XY; Writing – Original Draft NM; Funding Acquisition JW; Supervision GAB, TM, and JW
References
- Badovinac VP, Haring JS, Harty JT. Initial T Cell Receptor Transgenic Cell Precursor Frequency Dictates Critical Aspects of the CD8+ T Cell Response to Infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Bevan MJ. Helping the CD8(+) T-cell response. Nature reviews. Immunology. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- Blair DA, Lefrançois L. Increased competition for antigen during priming negatively impacts the generation of memory CD4 T cells. Proceedings of the National Academy of Sciences. 2007;104:15045–15050. doi: 10.1073/pnas.0703767104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattman JN, Antia R, Sourdive DJD, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the Precursor Frequency of Naive Antigen-specific CD8 T Cells. The Journal of experimental medicine. 2002;195:657–664. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. Journal of virology. 2005;79:10514–10527. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhu S, Loike JD, Pandolfi A, Han S, Catalano G, Constantinescu A, Clynes R, Silverstein SC. CD8+ T cell concentration determines their efficiency in killing cognate antigen-expressing syngeneic mammalian cells in vitro and in mouse tissues. The Journal of experimental medicine. 2010;207:223–235. doi: 10.1084/jem.20091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaelsson J, Lund O, Hejdeman B, Jansson M, Sonnerborg A, et al. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS pathogens. 2014;10:e1004251. doi: 10.1371/journal.ppat.1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnet FM. A modification of Jerne’s theory of antibody production using the concept of clonal selection. Aust J Sci. 1957;20:67–77. doi: 10.3322/canjclin.26.2.119. [DOI] [PubMed] [Google Scholar]
- Burnet FM. The Clonal Selection Theory of Acquired Immunity. Cambridge University Press; Cambridge: 1959. [Google Scholar]
- Catron DM, Rusch LK, Hataye J, Itano AA, Jenkins MK. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central– memory cells. The Journal of experimental medicine. 2006;203:1045–1054. doi: 10.1084/jem.20051954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbeau P, Reynes J. Immune reconstitution under antiretroviral therapy: the new challenge in HIV-1 infection. 2011;117 doi: 10.1182/blood-2010-12-322453. [DOI] [PubMed] [Google Scholar]
- Crawford A, Angelosanto Jill M, Kao C, Doering Travis A, Odorizzi Pamela M, Barnett Burton E, Wherry EJ. Molecular and Transcriptional Basis of CD4+ T Cell Dysfunction during Chronic Infection. Immunity. 2014;40:289–302. doi: 10.1016/j.immuni.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creusot RJ, Thomsen LL, Tite JP, Chain BM. Local Cooperation Dominates Over Competition Between CD4+ T Cells of Different Antigen/MHC Specificity. The Journal of Immunology. 2003;171:240–246. doi: 10.4049/jimmunol.171.1.240. [DOI] [PubMed] [Google Scholar]
- Foulds KE, Shen H. Clonal Competition Inhibits the Proliferation and Differentiation of Adoptively Transferred TCR Transgenic CD4 T Cells in Response to Infection. The Journal of Immunology. 2006;176:3037–3043. doi: 10.4049/jimmunol.176.5.3037. [DOI] [PubMed] [Google Scholar]
- Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 2006;312:114–116. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer research. 2005;65:1089–1096. [PubMed] [Google Scholar]
- Houghton AN. Cancer antigens: immune recognition of self and altered self. The Journal of experimental medicine. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MK, Moon JJ. The Role of Naive T Cell Precursor Frequency and Recruitment in Dictating Immune Response Magnitude. The Journal of Immunology. 2012;188:4135–4140. doi: 10.4049/jimmunol.1102661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, Wherry EJ. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nature immunology. 2011;12:663–671. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. T Cells Compete for Access to Antigen-Bearing Antigen-Presenting Cells. The Journal of experimental medicine. 2000;192:1105–1114. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laouar Y, Crispe IN. Functional Flexibility in T Cells: Independent Regulation of CD4+ T Cell Proliferation and Effector Function In Vivo. Immunity. 2000;13:291–301. doi: 10.1016/s1074-7613(00)00029-7. [DOI] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nature reviews. Immunology. 2013;13:777–789. doi: 10.1038/nri3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo AL, Klonowski KD, Bon AL, Borrow P, Tough DF, Lefrancois L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl Ross M, Jenkins MK. Naive CD4+ T Cell Frequency Varies for Different Epitopes and Predicts Repertoire Diversity and Response Magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi N, Freeman GJ, Gault A, Godfrey D, Nadler LM, Glimcher LH. Signalling through the MHC class II cytoplasmic domain is required for antigen presentation and induces B7 expression. Nature. 1992;360:266–268. doi: 10.1038/360266a0. [DOI] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science. 2012;338:1220–1225. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. The Journal of experimental medicine. 2010;207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiel J, Caucheteux S, Laurence A, Singh NJ, Bocharov G, Ben-Sasson SZ, Grossman Z, Paul WE. Antigen-stimulated CD4 T-cell expansion is inversely and log-linearly related to precursor number. Proceedings of the National Academy of Sciences. 2011;108:3312–3317. doi: 10.1073/pnas.1018525108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranheim E, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. The Journal of experimental medicine. 1993;177:925–935. doi: 10.1084/jem.177.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto GA, Merghoub T, Hirschhorn-Cymerman D, Liu C, Lesokhin AM, Sahawneh D, Zhong H, Panageas KS, Perales MA, Altan-Bonnet G, et al. Self-antigen-specific CD8+ T cell precursor frequency determines the quality of the antitumor immune response. The Journal of experimental medicine. 2009;206:849–866. doi: 10.1084/jem.20081382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Wikstrom ME, Fazekas de St Groth B. Visualizing T Cell Competition for Peptide/MHC Complexes. Immunity. 2000;13:783–794. doi: 10.1016/s1074-7613(00)00076-5. [DOI] [PubMed] [Google Scholar]
- Topalian Suzanne L, Drake Charles G, Pardoll Drew M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KB, Marsolais D, Welch MJ, Rosen H, Oldstone MBA. Treatment with a sphingosine analog does not alter the outcome of a persistent virus infection. Virology. 2010;397:260. doi: 10.1016/j.virol.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire JK, Benning N, Whitton JL. Precursor Frequency, Nonlinear Proliferation, and Functional Maturation of Virus-Specific CD4+ T Cells. The Journal of Immunology. 2006;176:3028–3036. doi: 10.4049/jimmunol.176.5.3028. [DOI] [PubMed] [Google Scholar]
- Willis RA, Kappler JW, Marrack PC. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proceedings of the National Academy of Sciences. 2006;103:12063–12068. doi: 10.1073/pnas.0605130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold S, Sjöström M, Eriksson L. PLS-regression: a basic tool of chemometrics. Chemometrics and Intelligent Laboratory Systems. 2001;58:109–130. [Google Scholar]
- Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. The Journal of experimental medicine. 2010;207:651–667. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, Ahmed R. Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. The Journal of experimental medicine. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–270. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.