

Abstract
This letter describes a ligand-based virtual screening campaign utilizing SAR data around the M5 NAMs, ML375 and VU6000181. Both QSAR and Shape scores were employed to virtually screen a 98,000-member compound library. Neither approach alone proved productive, but a consensus score of the two models identified a novel scaffold which proved to be a modestly selective, but weak inhibitor (VU0549108) of the M5 mAChR (M5 IC50 = 6.2 μM, M1-4 IC50s >10 μM) based on an unusual 8-((1,3,5-trimethyl-1H-pyrazol-4-yl)sulfonyl)-1-oxa-4-thia-8-azaspiro[4,5]decane scaffold. [3H]-NMS binding studies showed that VU0549108 interacts with the orthosteric site (Ki of 2.7 μM), but it is not clear if this is negative cooperativity or orthosteric binding. Interestingly, analogs synthesized around VU0549108 proved weak, and SAR was very steep. However, this campaign validated the approach and warranted further expansion to identify additional novel chemotypes.
Keywords: M5, Muscarinic acetylcholine receptor, Virtual screen, Structure-Activity Relationship (SAR)
Graphical Abstract
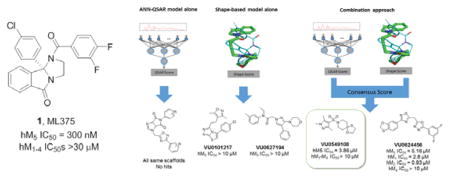
Recently, we reported on the results of a functional high-throughput screen to identify highly selective muscarinic acetylcholine receptor subtype 5 (M5) inhibitors (both negative allosteric modulators (NAMs)1,2 and orthosteric antagonists3). Based on the strong genetic data linking this receptor to addiction,4–6 pharmacological recapitulation with a small molecule is of great interest. Subsequent optimization did lead to the discovery the first highly selective and CNS penetrant M5 NAMs, ML375 (1) and VU6000181 (2); however, SAR was steep. Moreover, we were attracted to the rigid concave/convex topology of the core of 1 and 2 (see X-ray crystal structure 31), and, based on prior machine learning/virtual screening success with mGlu5 NAMs,7 felt this scaffold was a viable lead for a ligand-based virtual screening exercise to identify new M5 chemotypes. In this Letter, we will describe the methodology employed for the discovery of a novel M5 inhibitor chemotype.
The medicinal chemistry effort surrounding the ML375 scaffold resulted in 68 active compounds with varying levels of potency and 145 inactive compounds (M5 IC50s >10 μM). This information made it possible to build artificial neural network (ANN) quantitative structure-activity relationship (QSAR) models to correlate molecular features with biological activity.8 In addition, the rigid structure of the ML375 scaffold (only 3 rotatable bonds) defines a limited conformational space and made shape-based similarity metrics an attractive option as well. 9
Molecular descriptor calculation, ANN training, and model analyses were performed using the BioChemical Library (BCL) developed at Vanderbilt University.8 The dataset was prepared by removing any ions from structures, adding hydrogens, neutralizing charges, and removing duplicate entries. A single three-dimensional conformation was generated for each structure using Corina version 3.60.10 Descriptors which encoded 1D (scalar values), 2D (connectivity), and 3D (shape) information were calculated for each structure. Scalar descriptors included number of hydrogen bond donors and acceptors, calculated LogP, and topological polar surface area. 2- and 3-D information was encoded using autocorrelation functions weighted by properties such as partial charge and polarizability.11 These descriptors resulted in 1315 numerical values for each structure. Calculated descriptor vectors were labeled with the respective human M5 pIC50 value, or 0 if the compound was inactive. A feed-forward neural network with a densely connected 32-node hidden layer and a single-valued output layer was trained using this feature set. For training, error values were calculated by treating pIC50 values as binary values based on whether pIC50 was greater than 5 (active) or less than 5 (inactive). A 5-fold cross validation procedure using monitoring and independent sets and dropout was used to prevent overtraining and to evaluate model performance.11 Receiver-operator characteristic (ROC) curves and figures of merit are shown in Figure 2A and indicate that the models were able to classify active compounds over inactives at a rate substantially higher than random chance.
Figure 2. QSAR and Shape-based Models.
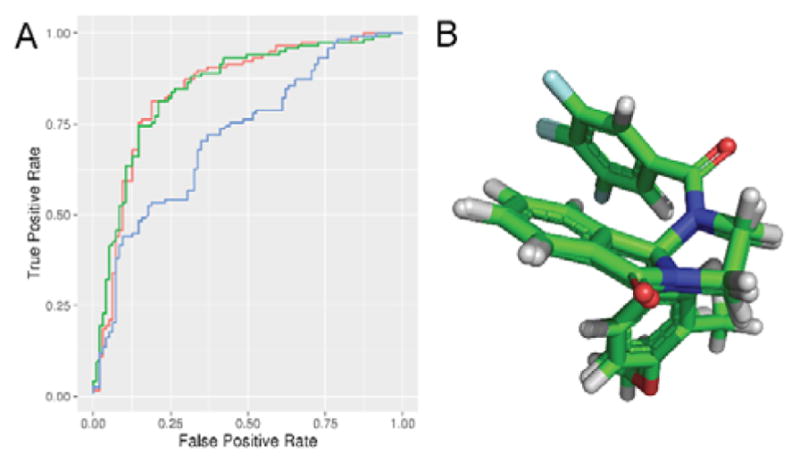
A) Receiver operator characteristic curves for Surflex-Sim shape (blue), QSAR (green), and QSAR+shape consensus (red) models. Area under the curve, QSAR: 0.85, Surflex: 0.72, Consensus: 0.84, random: 0.5. Average enrichment at 10% FPR, QSAR: 1.64, Surflex: 1.48, Consensus: 1.44, random: 1.0. B) Highest-scoring Surflex-Sim hypothesis of VU6000181 and ML375. This hypothesis was used for the shape-based portion of the virtual screening workflow.
In addition, 1 and 2 were selected for the generation of a 3-dimensional binding hypothesis. These two compounds were aligned using the flexible alignment feature of Surflex-Sim from Sybyl 2.1.1.9 Default parameter values for the algorithm were used with the exception that ring flexibility was considered during the alignment. The highest-scoring hypothesis from the alignment was used for virtual screening (Figure 2B). A receiver-operator characteristic (ROC) curve for this hypothesis generated by aligning and scoring the remaining 211 compounds from the M5 NAM dataset with the flexible screening (pscreen) feature of Surflex-Sim is shown in Figure 2A. The shape based model was also able to prioritize active compounds over inactives at a rate higher than random chance. The shape-based model resulted in similar predictive ability to the QSAR model for high-scoring compounds, though the QSAR models appeared to outperform the shape model across the whole dataset.
Each model was used to independently select 30 compounds from an in-house 98,000-compound screening library for experimental testing. The character of the top compounds from each model differed substantially from each other, though there were many substructural commonalities in the compounds of both sets. The QSAR model preferentially chose compounds from the same scaffold pool, whereas the shape-based method contained more diverse chemical structures. From this set of 60 compounds, two compounds from the shape-based set, VU0101217 and VU0627194, showed weak antagonist activity at M5, exhibiting around 40 percent inhibition of M5 response at 30 μM (Figure 3). This indicated that a more diverse selection could be powerful for compound discovery than an exhaustive enumeration of SAR around a high scoring scaffold.
Figure 3.
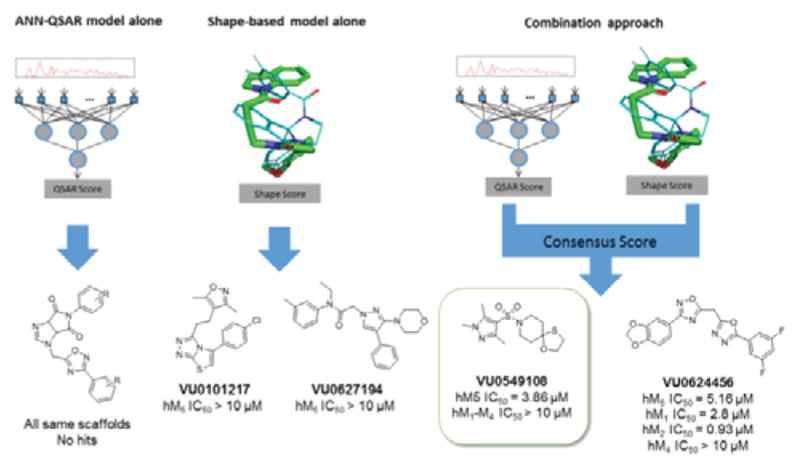
Summary of hit compounds discovered by the three distinct approaches. The consensus approach yielded the best result of the three with VU0549108 demonstrating selectivity for the M5 receptor over other mAChR isoforms, based on assay data employing HTS DMSO stock.
To leverage the predictive ability of both models, a larger screen using a consensus approach was performed. To address the issue of low compound diversity when prioritizing by QSAR score alone, the workflow involved first scoring the full 98,000-compound database with the ANN-QSAR models and clustering the highest-scoring 10 percent of the data to select a diverse set of high-scoring chemical structures. To enable clustering, a random subset was sampled from the prioritized compounds, Murcko scaffolds12 were generated for each selected compound, and the maximum common substructure between each pair of Murcko scaffolds was calculated. Ring and chain fragments from each scaffold were also added to supplement the database. Fingerprint vectors were computed by searching for the presence of each substructure in the prioritized compounds. The distance metric between pairs of compounds was calculated as the Tanimoto13 coefficient between fingerprint vectors.
A consensus score was calculated by scoring the top two highest QSAR-scoring compounds from each cluster (or a single compound if the cluster size was 1) with the shape-based method and combining the individual model scores. 986 compounds were aligned to the ML375 binding hypothesis in this manner using the Surflex-Sim “pscreen” algorithm. The QSAR and Surflex scores were normalized such that their ranges fell between 0 and 1, and both normalized scores were added together to provide the final consensus score for each molecule. The 320 highest scoring compounds according to consensus score were selected and submitted for pharmacological screening against mAChR M5.
Using the two-model consensus approach, two compounds VU0549108 and VU0624456 demonstrated significant M5 antagonist activity (maximum M5 inhibition of 50 and 70 percent, respectively in a single point M5 inhibition assay at 10 μM), and were confirmed with 10-point concentration response curves (M5 IC50s of 3.86 μM and 5.16 μM, respectively employing the HTS DMSO stock). By QSAR score, VU0549108 was rank 1284 and VU0624456 was rank 3073 (with rank 1 being the best score). By shape score VU108 was rank 3520 and VU0624456 was rank 2446. In order to rapidly explore SAR surrounding the initially more attractive VU0624456 scaffold, SAR-by-catalog was performed by ordering a set of analogs from commercial sources. In addition, compounds that had a high similarity to the two hit compounds (2D fingerprint similarity with Tanimoto > 0.5), and a second set of virtually screened compounds using updated models were chosen from the in-house screening library to follow up these results (Table 1). Single point screening revealed several possible antagonists which were subsequently confirmed using 10-point CRCs against M5. However, only analogs of the original hits showed significant M5 activity, and none proved more potent than the original compounds.
Table 1.
Sources and counts of compounds used to explore SAR around VU0549108 and VU0624456.
| Selection approach | Source | Number of compounds |
|---|---|---|
| 2D similarity, VU0549108 | In-house library | 46 |
| 2D similarity, VU0549456 | In-house library | 37 |
| VU0549456 analogs, commercial | Commercial | 67 |
| Virtual screening (consensus) | In-house library | 237 |
These data prompted the resynthesis of VU0549108 (4) to reconfirm mAChR activity from fresh powder, as the lack of mAChR selectivity deprioritized VU0624456.14 VU0549108 (4) was readily prepared in two steps (Scheme 1). Starting from commercial piperidine hydrate 5, treatment with sulfonyl chloride 6 provides 7 in 76% yield. Condensation with 2-mercaptoehthanol under Lewis acid catalysis affords 4 in 79% yield. This expedited route was also employed for analog synthesis.
Scheme 1.
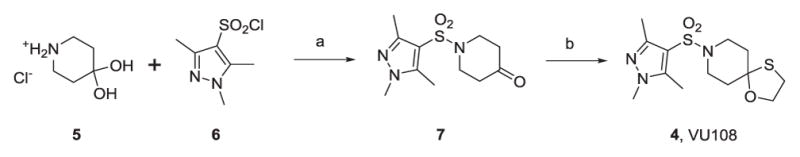
Synthesis of VU0549108 (4). Reagents and Conditions: (a) Triethylamine, DCM, rt 12 hrs., 76%; (b) 2-mercaptoethanol, BF3·OEt2, DCM, 12 hrs. rt 79%.
The resynthesized 4 proved to be a functional inhibitor of M5 (Figure 4A), with greater activity in the 10 μM single point assay (88%), an IC50 of 6.2 μM (pIC50 = 6.18±0.09, ACh min 12.6±2.5), and modest selectivity versus M1-4 (IC50s >10 μM). The divergence from classical orthosteric antagonist chemotype, coupled with the observed selectivity, led us to perform radioligand binding assays to assess if 4 was an allosteric ligand (NAM) or an atypical orthosteric ligand. Here, employing the standard [3H]-NMS ligand (Figure 4B),1–3 and compared to atropine, 4 proved to interact with or modulate with orthosteric site with a Ki of 2.7 μM (atropine control, Ki = 2.7 nM).1–3,15–17 However, the effect on NMS binding could be due to cooperativity with the orthosteric site by binding of 4 to an allosteric site, or it could reflect direct, competitive interaction at the orthosteric site. Based on 4 being a small, non-basic chemotype, distinct from prototypical mAChR antagonists, and some measure of mAChR selectivity, more potent analogs are required to definitively address the mode of inhibition of M5. Moreover, shape-based alignment of 1 and 4 (Figure 5), while showing reasonable overlap for achiral 4, significant lipophilic regions are not occupied. This hypothesis shows an alignment of one sulfonyl oxygen and the free pyrazole nitrogen in 4 align with the carbonyl groups in 1, and the 4 oxathiolane oriented along the 9b-4-chlorophenyl group of 1. Since the 4-chlorophenyl group confers stereochemistry to ML375, this three-point pharmacophore could hold the structure in a position that allows 4 to mimic the stereochemistry of 1 without itself being chiral, and suggests why the virtual screen identified 4.
Figure 4.
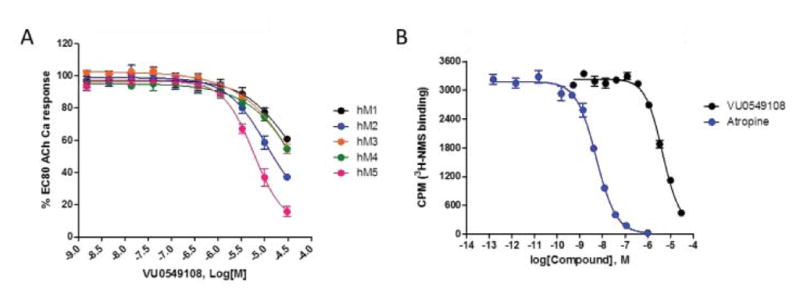
Molecular pharmacology profile of VU0549108 (4). A) Concentration–response curves of 4 for human hM5, as well as hM1–4 (IC50 >10 μM), n=3). At 30 μM, % Ach Min (Ave±SEM): hM1, 61±2%; hM2, 37±2%; hM3, 55±3%; hM4, 55±1%; hM5, 16±2% B) [3H]-N-methylscopolamine (NMS) competition binding (n=3) in membranes prepared from human M5-expressing cells, showing interaction (either allosteric cooperativity or orthosteric displacement; Ki=2.7 μM.
Figure 5.
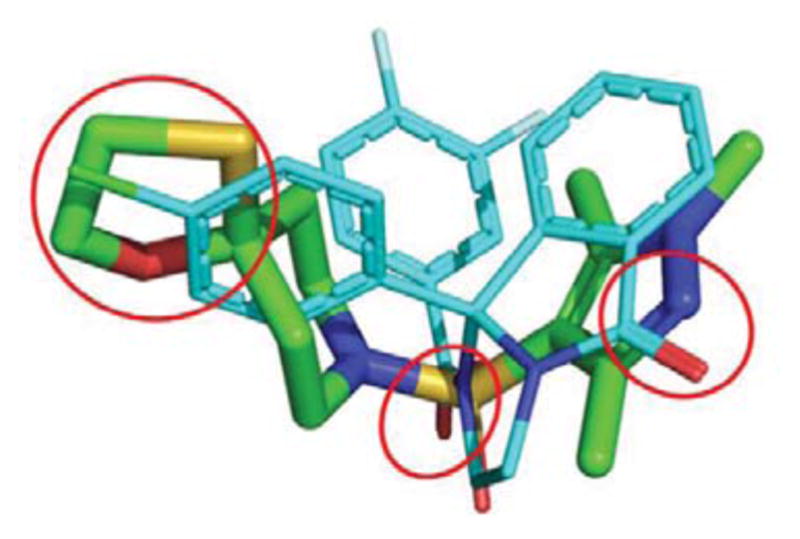
Overlay of 4 (green) on 1 (cyan). Hydrogen bond acceptors at the pyrazole and sulfonyl groups of 4 align with corresponding hydrogen bond acceptors in 1, and the oxathiolane of 4 overlaps with the 4-chlorophenyl moiety of 1. The overlap of these features could explain why the virtual screen selected 4.
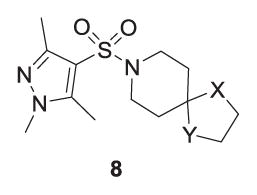
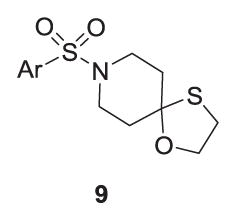
A small library was synthesized to further explore SAR surrounding the 4 scaffold in hopes of increasing M5 inhibitory potency. Two points were considered for modification, namely the oxathiolane spirocycle, analogs 8 (Table 2) and the heterocyclic sulfonamide congeners, 9 (Table 3). SAR was steep,15–17 with all analogs displaying IC50s >10 μM; however, the EC80 was diminished. The 1,3-oxathiolane (4) was critical for activity, as the parent piperidinone, 1,3-dithiolane (8a), 1,3-dioxolane (8b) and spiro furan (8c) analogs all showed weak inhibition of M5 (IC50s >10 μM, single point % inhibition at 10 mM form 37–86%). Similarly, only the 1,3,5-pyrazole sulfonamide of 4 maintained M5 inhibition.
Table 2.
Structure and activities of analogs 8a.
| |||
|---|---|---|---|
| Compound | X | Y | hM5 Percent inhibition |
| 4 (VU0549108) | S | O | 84% (hM5 IC50 = 5.23 μM) |
| 8a | S | S | 86% |
| 8b | O | O | 45% |
| 8c | O | CH2 | 37% |
Average of three determinations with hM5 cells with an EC80 of ACh as a single point % inhibition at 10 μM. If not IC50 reported, the IC50 was >10 μM.
Table 3.
Structure and activities of analogs 9 a.
| ||
|---|---|---|
| Compound | Ar | hM5 Percent inhibition |
| 4 (VU0549108) | 1,3,5-TriMe-1H-4-pyrazole | 88% (hM5 IC50 = 6.18 μM) |
| 9a | 1-Me-1H-4-pyrazole | 27% |
| 9b | 1,3-DiMe-1H-4-pyrazole | Inactive |
| 9c | 1,5-DiMe-1H-4-pyrazole | 83% |
| 9d | 3-Cl-4-F-phenyl | 83% |
| 9e | 3,5-DiF-phenyl | 62% |
| 9f | 3,4,5-TriF-phenyl | 55% |
| 9g | 3,5-DiMe-4-isoxazole | 40% |
| 9h | 1-Me-4-imidazole | Inactive |
Average of three determinations with hM5 cells with an EC80 of ACh as a single point % inhibition at 10 μM. If not IC50 reported, the IC50 was >10 μM.
In summary, a virtual screening campaign was conducted which employed a combination of an ANN-QSAR modeling approach with a shape-based modeling approach based on a rigid M5 NAM chemotype. This exercise identified a novel M5 ligand VU0549108 (4) derived from an unusual 8-((1,3,5-trimethyl-1H-pyrazol-4-yl)sulfonyl)-1-oxa-4-thia-8-azaspiro[4,5]decane scaffold, characterized by steep SAR, modest selectivity versus M1-4 and an a noted interaction with the orthosteric site in [3H]-NMS binding studies (studies to determine if allosteric cooperativity or orthosteric binding are in progress). Interestingly, neither model alone produced a hit from the virtual screen, but the combination of the two proved successful to identify a new M5 inhibitor chemotype. These initial data argue well for validation of these approaches and warrant further expansion to identify additional novel M5 chemotypes. Further work is in progress and will be reported in due course.
Figure 1.
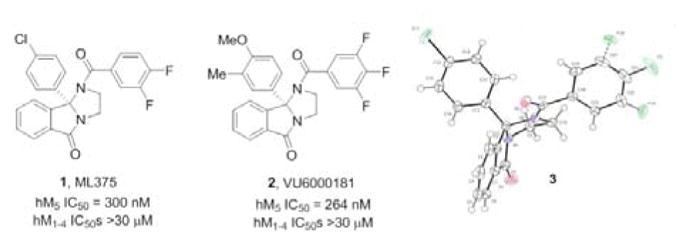
Structures of M5 NAM ML375 (1), VU6000181 (2) and the x-ray crystal structure 3 of 1, highlighting the rigid structure of the core.
Acknowledgments
We thank the NIH for funding via the National Institute of Drug Abuse (1R01DA037207) and the Chemistry-Biology Interface Research Training Program. Work in the Meiler laboratory is supported through NIH (R01 GM080403, R01 GM099842, R01 DK097376) and NSF (CHE 1305874). We also thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gentry PR, Kokubo M, Bridges TM, Byun N, Cho HP, Smith E, Hodder PS, Niswender CM, Daniels JS, Conn PJ, Lindsley CW, Wood MR. J Med Chem. 2014;57:7804–7810. doi: 10.1021/jm500995y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurata H, Gentry PR, Kokubo M, Cho HP, Bridges TM, Niswender CM, Byers FW, Wood MR, Daniels JS, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2015;25:690–694. doi: 10.1016/j.bmcl.2014.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gentry PR, Kokubo M, Bridges TM, Cho HP, Smith E, Chase P, Hodder PS, Utley TJ, Rajapakse A, Byers F, Niswender CM, Morrison RD, Daniels JS, Wood MR, Conn PJ, Lindsley CW. ChemMedChem. 2014;9:1677–1682. doi: 10.1002/cmdc.201402051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, Yamada M, Wess J. Proc Natl Acad Sci USA. 2002;99:11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fink-Jensen A, Fedorova I, Wörtwein G, Woldbye DP, Rasmussen T, Thomsen M, Bolwig TG, Knitowski KM, McKinzie DL, Yamada M, Wess J, Basile A. J Neurosci Res. 2003;74:91–96. doi: 10.1002/jnr.10728. [DOI] [PubMed] [Google Scholar]
- 6.Thomsen M, Woldbye DP, Wortwein G, Fink-Jensen A, Wess J, Caine SB. J Neurosci. 2005;25:8141–8149. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mueller R, Dawson ES, Meiler J, Chauder BA, Bates BS, Felts AS, Lamb JP, Menon UN, Jadhav SB, Kane AS, Jones CK, Rodriguez AL, Conn PJ, Olsen CM, Winder DG, Emmitte KA, Lindsley CW. ChemMedChem. 2012;7:406–414. doi: 10.1002/cmdc.201100510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butkiewicz M, Lowe EW, Jr, Mueller R, Mendenhall JL, Teixeira PL, Weaver CD, Meiler J. Molecules. 2013;18:735–756. doi: 10.3390/molecules18010735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain AN. J Med Chem. 2004;47:947–961. doi: 10.1021/jm030520f. [DOI] [PubMed] [Google Scholar]
- 10.Sadowski J. J Comput Aid Mol Des. 1997;11:53–60. doi: 10.1023/a:1008023427310. [DOI] [PubMed] [Google Scholar]
- 11.Mendenhall J, Meiler J. J Comput Aided Mol Des. 2016;30:177–189. doi: 10.1007/s10822-016-9895-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bemis GW, Murcko MA. J Med Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 13.Willett P. J Chem Inf Comput Sci. 1998;38:983–996. doi: 10.1021/ci970431+. [DOI] [PubMed] [Google Scholar]
-
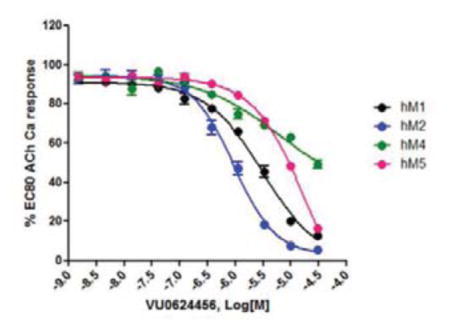
14.Concentration response curves for VU0624456 against hM1-5:
- 15.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conn PJ, Lindsley CW, Meiler J, Niswender CM. Nat Rev Drug Discov. 2014;13:692–708. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindsley CW, Emmitte KA, Hopkins CR, Bridges TM, Gregory KA, Niswender CM, Conn PJ. Chem Rev. 2016;116:6707–6741. doi: 10.1021/acs.chemrev.5b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]