

Abstract
The recent discovery of significant hydropersulfide (RSSH) levels in mammalian tissues, fluids and cells has led to numerous questions regarding their possible physiological function. Cysteine hydropersulfides have been found in free cysteine, small molecule peptides as well as in proteins. Based on their chemical properties and likely cellular conditions associated with their biosynthesis, it has been proposed that they can serve a protective function. That is, hydropersulfide formation on critical thiols may protect them from irreversible oxidative or electrophilic inactivation. As a prelude to understanding the possible roles and functions of hydropersulfides in biological systems, this study utilizes primarily chemical experiments to delineate the possible mechanistic chemistry associated with cellular protection. Thus, the ability of hydropersulfides to protect against irreversible electrophilic and oxidative modification was examined. The results herein indicate that hydropersulfides are very reactive towards oxidants and electrophiles and are modified readily. However, reduction of these oxidized/modified species is facile generating the corresponding thiol, consistent with the idea that hydropersulfides can serve a protective function for thiol proteins.
Keywords: Hydropersulfides, thiols, thiol protein, oxidative stress, electrophilic stress, cellular protection, hydrogen sulfide
Introduction
Hydrogen sulfide (H2S) has been the topic of significant research interest due to its reported endogenous generation in mammalian systems as a signaling agent [1]. In spite of the numerous proposed signaling and physiological functions of H2S, the chemical/biochemical mechanism(s) associated with its activity remain unknown. Recent work from this lab [2] and others [3] proposes that some, if not most, of the signaling/effects attributed to H2S may instead be due to the presence of hydropersulfides (RSSH), which can act as precursors for H2S. (Note: the term “hydropersulfide” as used herein refers to both the protonated RSSH and deprotonated RSS− forms unless otherwise indicated). Moreover, Toohey has previously discussed the possible importance of sulfane sulfur species, such as hydropersulfides, as physiological signaling agents and effectors [4,5,6]. Due to the biological prevalence of hydropersulfides such as cysteine hydropersulfide (Cys-SSH), glutathione hydropersulfide (GSSH) and protein hydropersulfides (Protein-SSH) as well as polysulfide species (RSnR, n > 2) [2] and their novel chemical properties [7,8], it is proposed that these polysulfur compounds represent an important and active redox state of biological thiols (although they are still at lower concentrations compared to the corresponding thiols). To be sure, biological hydropersulfides and polysuflides have been known for some time. The presence and presumed health benefits of polysulfides in foods such as garlic have been studied extensively [9]. Also, a variety of studies report the presence of hydropersulfide species in a variety of proteins/enzymes [10,11,12,13]. For example, early work by Massey’s group proposed that the molybdenum enzyme xanthine oxidase (XO) contains a persulfide function crucial for catalysis [10]. Although others report that XO contains a molybdenum sulfide, as opposed to a persulfide [14], further work on related molybdenum enzymes proposes the possible presence of a molybdenum persulfide species [15]. Finally, the enzyme rhodanese as well as rhodanese-like proteins have long been known to react with thiosulfate generating an enzyme-bound hydropersulfide, which is capable of transferring the sulfur atom to cyanide ion (CN−) as well as other substrates [16,17]. Although rhodanese is known to be important in the detoxification of CN−, other physiological functions (if any) are not yet established. The recent report of the ubiquitous and prevalent nature of biological hydropersulfide generation seems to indicate that such species may possess a more general function [2]. In spite of their prevalence and unique chemical properties, the exact nature of the role or function these species may play in biological systems is uncertain. Currently there are several ideas regarding the general utility of biological hydropersulfide generation. For example, it is hypothesized that hydropersulfide formation can result in increased nucleophilicity and reducing capability [2,7,8,18] resulting in increased reaction rates with oxidants and electrophiles. As such, hydropersulfides are likely to participate in redox events and electrophilic signaling, possibly to a greater extent than the corresponding thiol.
Based on proposed chemical properties of hydropersulfides, it was recently hypothesized that protein hydropersulfide formation may represent a mechanism by which protein thiols are protected from irreversible oxidative or electrophilic modification [8]. In this situation, cellular oxidizing conditions or the presence of electrophiles could lead to intracellular hydropersulfide generation in both small molecules (that can scavenge oxidants like H2O2 or electrophiles) [2] and in proteins. Hydropersulfide generation in proteins may result in inactive protein but would also provide protection against irreversible oxidative/electrophilic thiol destruction [8,19]. Following the resolution of cellular insult/stress, the re-establishment of cellular reducing conditions could lead to re-formation of the thiol protein and associated activity [2]. While some studies have shown that other oxidized species, such as a sulfenyl amide or a disulfide bond between the catalytic cysteine and an intramolecular cysteine can indeed protect an enzyme from irreversible oxidative inactivation [20,21], the idea that hydropersulfides can accomplish the same function has only indirect, anecdotal support. For example, previous studies have shown that cysteine phosphatases are protected from irreversible oxidation by H2O2 when the protein is pretreated with H2S [19]. One interpretation of this observation is that the protection is due to the generation of the hydropersulfide on the phosphatase [19,22]. Additionally, Ida and coworkers [2] found that overexpression of the Cys-SSH biosynthetic enzyme cystathionine gamma lyase (CSE) in A549 cells results in cellular protection from H2O2 toxicity. Although these previous reports are consistent with the idea that hydropersulfide formation can be protective against oxidative insult, the chemistry responsible for these effects is not established. Thus, the work described herein is focused on the determination of possible mechanisms of hydropersulfide-mediated protection against oxidative and/or electrophilic insult.
Experimental Procedures
Chemicals/Solutions
Dithiothreitol (DTT), N-ethyl maleimide (NEM), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), Activated Thiol-Sepharose® 4B (resin), N-acetyl penicillamine (NAP), penicillamine and 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB) were purchased from Sigma-Aldrich (St. Louis, MO). Sodium hydrogen sulfide (NaHS) was purchased from Strem Chemicals, Inc. (Newburyport, MA). Sodium sulfide (Na2S) and sodium tetrasulfide (Na2S4) were purchased from Alfa Aesar (Ward Hill, MA). Fluorescein diphosphate (FDP) was purchased from AnaSpec (Fremont, CA). Hydrogen peroxide (H2O2, 30%) was purchased from ACROS Organics (Fair Lawn, NJ). Immobilized TCEP disulfide-reducing resin and Ni-NTA resin were purchased from Thermo Scientific (Waltham, MA). All other chemicals and reagents were purchased from commercial suppliers and were of the highest purity available. Angeli’s salt (sodium trioxodinitrate, Na2N2O3) was synthesized as previously described [23].
Recombinant CD148
The cytoplasmic domain of human CD148 (R997 to the C-term) was cloned into pTrcHis-A (Invitrogen; Grand Island, NY) to include a 10xHis N-term tag. The plasmid was transformed into Arctic Express bacteria (Agilent; Santa Clara, CA) and induced with IPTG (500 μM) for 18 hours at 10°C. Cells were lysed in lysis buffer (100 mM phosphate buffer, pH 7.4, 1 mM DTT, and 10 mM imidazole) followed by sonication and centrifugation at 8,000 x g.
Hydropersulfide generation on CD148
Recombinant CD148 from bacterial lysate was bound to Ni-NTA resin and then washed 3x (50 mM HEPES pH 7.2). For each of the subsequent steps, all reactions were performed at 37°C and samples were washed 3x in between treatments. 1) 200 μM DTNB was incubated for 30 min, 2) 100 μM Na2S for 15 min followed by 1 mM NEM for 30 min and 10 mM DTT for 15 min. Resin-bound CD148 was then utilized for phosphatase assays and supernatant collected for GC-MS analysis.
Phosphatase assay
Samples were incubated with 30 μM fluorescein diphosphate (FDP) in 100 μL of 50 mM HEPES, pH 7.2. Following the addition of FDP, phosphatase activity was measured every min for 20 min at 37°C on a Synergy HT Multi-Mode Microplate Reader (Biotek; Winooski, VT) with 485/528 nm filters.
GC-MS Analysis of CD148 Activity Assay Supernatant
Supernatant from the CD148 activity assay (see above) was collected after reduction with DTT. Solutions were acidified with 3 M HCl and extracted with approximately 50mL of dichloromethane (DCM). This extract was concentrated under reduced pressure to dryness and the residue redissolved in 1 mL of DCM. Analysis of this solution was carried out using gas chromatography-mass spectrometry (GC-MS) on a Hewlett-Packard 5890 Series II gas chromatograph equipped with a 5972 quadrapole mass spectrometer and a 50 m x 0.2 mm x 0.33 μm film thickness Hewlett Packard Ultra 2 crosslinked 5% PhMe silicone column operating under the following conditions: 40°C for 1 min followed by a 20°C/min temperature increase to a final temperature of 280°C. Under these conditions, a peak at 10.389 min with a parent fragment at 159 m/z was observed (159 amu is the molecular weight of the parent NEM-SH molecule). NEM-SH was synthesized independently by reacting 20 nM NEM with 4 mM NaHS in 10mL pH 7.2 HEPES buffer. After 15 min, this solution was acidified, extracted, concentrated and analysis of the extract using GC-MS performed as described above. Monitoring 159 m/z as before, a peak at 10.389 min was also found in this sample. Further control experiments were performed whereby NEM and NaHS solutions were worked up as described above and analyzed. In these cases, no peak at 10.389 min (159 m/z) was found. The isotopic abundance of 34S (i.e. 161 m/z) was used to confirm the presence of a sulfur species (lit. 4.21% 34S, exper. 4.57%).
Quantitation of Hydropersulfide Generation on Thiol-Sepharose Resin
To begin, 100 mM NaHS was mixed with an equal volume of TCEP resin for 10 min, and then separated from the resin. This treatment removes polysulfide impurities from the NaHS that were noted in previous experiments [24]. Activated thiol Sepharose® 4B resin (11.4 mg) was then treated with 400 μL of NaHS (50 mM) for 20 min. A 200 μL aliquot from the supernatant was diluted in a cuvette of 600 μL DI water and the concentration of 2-thiopyridone (λmax = 343 nm, ε = 8080 M−1 cm−1) determined by UV-Vis spectroscopy. A separate 100 μL aliquot of the supernatant was transferred to a new tube and 100 μL of 100 mM NaCN added and incubated for 10 min. The remaining Thiol Sepharose® 4B resin mixture was washed 3 times with DI water and the resulting solid resin resuspended in 100 μL of 100 mM NaCN for 10 min. Next, 150 μL of a 1 M HNO3 solution was added to both the supernatant and the resin mixtures and incubated for another 10 min. Finally, 100 μL of a 30 mM solution of Fe(NO3)3 was then added to both the supernatant and the resin mixtures and incubated for 10 min. [Fe(SCN)]2+ in both the supernatant and the resin mixtures was quantified by measuring the absorbance at 460 nm (ε = 5000 M−1 cm−1) and comparison to a standard curve. Absorbance measurements were made using a Shimadzu 2501 UV-Vis spectrophotometer. The amount of 2-thiopyridine released into the supernatant via treatment with H2S represents the total thiol equivalent of the resin. The amount of Resin-bound persulfide, as measured via SCN− formation, can be divided by this value to give the approximate percentage of Resin-bound persulfide generated. This procedure was performed twice and both times revealed an approximate 20–30% yield of resin-bound hydropersulfide.
As a negative control, an aliquot of the NaHS solution used to treat the thiol-sepharose resin was examined for hydropersulfide (i.e. to test whether the treatment of the NaHS solution with the immobilized TCEP resin indeed removed any hydropersulfide impurities). Thus, 200 μL of the NaHS treatment solution also underwent this thiocyanate assay. Briefly, 100 μL of a 100 mM NaCN solution was added to 200 μL of the 50 mM NaHS treatment solution and incubated for 10 min. Then, 600 μL of 1M HNO3 was added and incubated for 10 min followed by addition of 300 μL of a 30 mM Fe(NO3)3 solution and a subsequent 10 min incubation. The resulting solution showed no absorbance at 460 nm.
Hydropersulfide Formation on Thiol-Sepharose 4B Resin: Protection from Irreversible Electrophilic Modification by HNO or Oxidation by H2O2
Activated Thiol-Sepharose® 4B (5 mg, 0.02 μmol equivalent of glutathione mixed disulfide with 2-thiopyridine) was suspended and then washed 4x with 200 μL of DI H2O and then a fifth time with 200 μL HEPES buffer (pH 7.2, 200 mM). To the hydrated resin was then added NaHS in HEPES buffer (200 mM pH 7.2 for HNO experiments or 100 mM, pH 10 for H2O2 experiments) to a final concentration of 100 mM, DTT at a final concentration of 100 mM in HEPES buffer or HEPES buffer only and allowed to react for 20–30 min. To monitor the reaction, samples were centrifuged and 100 μL of supernatant from each resin reaction was analyzed for the displaced 2-thiopyridine (λmax = 343 nm, ε = 8080 cm−1 M−1) by UV-Visible spectroscopy. The reacted resin samples were then washed 5x as described immediately above. For HNO modification, Angeli’s Salt was added to a final concentration of 20 mM and allowed to react for 2 hrs at room temperature. For H2O2 oxidation, H2O2 in HEPES buffer to a final concentration of 10 mM was added and reacted 12 hours at 28°C on a rotating incubator. The reacted resin samples were then washed 5x as before. To reduce the resin, DTT in HEPES buffer was added to a final concentration of 100 mM and allowed to react for 30 min. The reacted resin samples were then washed 5x as before. For HNO treated samples, DTNB was added to a final concentration of 1 mM for 20 min, washed, and then 100 mM DTT was added for another 20 min. For H2O2 treated samples, 100 mM DTNB in HEPES buffer was added for 20 min. Supernatants were finally analyzed for the TNB anion (TNB-S−) (λmax = 412 nm, ε = 14150 cm−1 M−1) [25] by UV-Visible spectroscopy on a Shimadzu 2501 UV-Vis spectrophotometer.
NMR Analysis of Thiol versus Hydropersulfide Reactivity
NMR studies were performed using a hydropersulfide donor species, S-methoxycarbonyl penicillamine disulfide (MCPD) which will spontaneously rearrange via an S-to-N methoxycarbonyl transfer to give the corresponding hydropersulfide, N-methoxycarbonyl penicillamine persulfide (MCPP). MCPD was synthesized according to a previous literature procedure [26]. Stock solutions (10 mM) of both NAP and MCPD were made in D2O. Stock solutions of iodoacetamide (IAA) (100 mM) and DTT (200 mM) were prepared in 0.1 M phosphate buffer (pH 7.4). For any given reaction, iodoacetamide was diluted in an NMR tube containing 0.1 M phosphate buffer (pH 7.4) to a final concentration of 10 mM. Next, either NAP or MCPD was added to the NMR tube to a final concentration of 1 mM and the reactions monitored by (H)PRESAT NMR using an Agilent 400 mHz NMR spectrometer. Upon completion of the reaction of either the thiol or the persulfide with iodoacetamide, DTT was added to the NMR tube to a final concentration of 20 mM. Reduction of the trapped thiol or persulfide species was monitored by (H)PRESAT NMR.
Analysis of Hydropersulfide Reactivity via Mass Spectrometry
A stock solution of MCPD (10 mM) was made in deionized (DI) water. Stock solutions of both IAA (100 mM) and DTT (200 mM) were prepared in 0.1 M ammonium bicarbonate buffer (pH 7.4). For the initial reaction of IAA and MCPD, IAA was diluted into a test tube containing 0.1 M ammonium bicarbonate buffer (pH 7.4) to a final concentration of 10 mM. MCPD was then added to the test tube to a final concentration of 1 mM and the mixture stood at room temperature for thirty minutes. An aliquot of the reaction mixture was then diluted 5x with methanol and injected directly into a Thermo TSQ Quantum Triple Quadrupole Mass Spectrometer controlled with XCalibur 2.1 via syringe pump. Spectra were acquired in positive ion mode with a spray voltage of 3500 V, a capillary temperature of 250 °C and, a flow rate of 1 μL/min. Ion optics were optimized for all ions of interest.
After verifying alkylation of MCPP with IAA, DTT was then added to the test tube to a final concentration of 20 mM and the mixture stood at room temperature for ninety minutes. An aliquot of the reaction mixture was then diluted 5x with methanol and analyzed via MS.
Results and Discussion
Effect of Hydropersulfide Generation in CD148 on the Susceptibility to Irreversible Electrophilic Modification
As discussed above, it is hypothesized that hydropersulfide formation on thiol proteins represents a protected state of the protein since oxidative or electrophilic modification of a hydropersulfide could be reductively reversed to the thiol. That is, the chemical modification should occur on the sulfane sulfur atom (the terminal sulfur atom), which can be reductively removed afterwards. On the other hand, thiols will not exhibit the same behavior after similar modifications and will remain irreversibly modified. This concept is schematically depicted in Figure 1.
Figure 1.

Possible mechanism by which hydropersulfide formation can protect thiol proteins from irreversible damage by either oxidation or electrophilic modification.
In order to test this idea, the cysteine-phosphatase CD148 was used as a model thiol protein. This tyrosine phosphatase is well-characterized and a member of the type III family of protein tyrosine phosphatases (PTPs) which have critical active-site cysteine residues [27]. We have shown previously that protein hydropersulfides can be made by treating thiol proteins with DTNB to make the mixed disulfide (Reaction 1), followed by treatment with H2S to generate the protein hydropersulfide (Reaction 2) [28].
| (1) |
| (2) |
Importantly, this method for making protein/peptide hydropersulfides has also been used by others and confirmed [18]. Thus, using the DTNB/H2S method outlined above, the His-tagged cytoplasmic domain of human recombinant CD148 (containing the phosphatase activity) was bound to an affinity resin (Ni) and treated with DTNB to generate the resin-bound protein mixed disulfide (CD148-S-S-TNB) (Reaction 1). Allowing the His-tagged protein to remain bound to the Ni resin allows facile manipulation of the protein, which remains active when bound [data not shown] and rapid removal of reagents via centrifugation and washing. After washing away the excess DTNB (and released TNB-S−) the resin-bound CD148-S-S-TNB was treated with H2S to generate the hydropersulfide CD148-S-S-H (Reaction 2). The CD148-SSH bound to the resin was then washed to remove excess H2S. It needs to be noted that quantitative conversion of Protein-SH to Protein-SSH via this method is difficult and unlikely due to further reactions associated with the Protein-SSH species. Previous work by us has shown that Protein-SSH species can further react with H2S to give the corresponding thiol (Protein-SH) and, presumably, dihydrogen disulfide (H2S2) (Reaction 3) [28].
| (3) |
Thus, under the conditions of these experiments it is likely that a mixture of CD148-SH and CD148-SSH are generated. More recent work has shown that Reaction 3 represents an equilibrium and that subsequent reactions of H2S2 can lead to further loss of the Protein-SSH species [29,30]. Regardless, this remains a useful way of generating Protein-SSH species, albeit not quantitatively.
It is worth noting that conversion of thiols on CD148 to hydropersulfides results in inhibition of enzyme activity [24]. Resin-bound CD148-SSH (made via the procedure outlined above and therefore mixed with some resin-bound CD148-SH) was then treated with the electrophile N-ethyl maleimide (NEM) (1 mM, 30 min at 37°C). The Protein-SSH species will react with NEM to give the Protein-NEM mixed disulfide (Protein-SS-NEM) (Reaction 4) while Protein-SH will react to give the dialkyl sulfide or thioether (Protein-S-NEM) (Reaction 5).
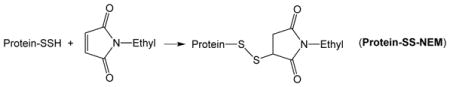 |
(4) |
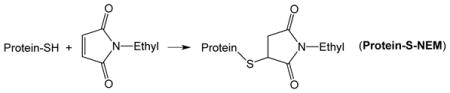 |
(5) |
After incubation with NEM was complete, the resin mixture was then washed 3 times to eliminate excess NEM followed by treatment with 10 mM of the potent thiol reductant DTT. A Protein-SS-NEM adduct should be reduced to the active Protein-SH species (Reaction 6) while the Protein-S-NEM species will not be reduced by DTT.
| (6) |
Recoverable enzyme activity was then determined using the artificial substrate fluorescein diphosphate (FDP). The results of this experiment are shown in Figure 2. As shown in Figure 2, there was approximately 30% recovery of enzyme activity in the case where CD148-SSH was formed compared to the case where there was no electrophilic modification of the protein. As a negative control, the thiol protein (where no hydropersulfide is present) shows no activity after treatment with NEM/DTT (Reaction 5). The presence of a Protein-SSH species in this experiment can be inferred from the generation of NEM-SH via Reactions 4 and 6. Thus, analysis of the solution resulting from the series of Reactions 1, 2, 4 and 6 reveals the presence of NEM-SH, consistent with the intermediacy of a Protein-SSH species (Figure 3). Unfortunately, reliable quantitation of the amount of released NEM-SH was found to be difficult due to the complex workup procedure. However, this finding qualitatively demonstrates that Protein-SSH was indeed formed and that the electrophilic modification by NEM could be removed by DTT treatment. These results indicate that hydropersulfide formation in CD148 leads to significant protection against electrophile-mediated irreversible loss of activity (Figure 1, RSSH + E+, [H] = DTT). The lack of quantitative recovery (approximately 30% DTT-mediated recovery is observed) is likely due to an incomplete conversion of CD148 to the hydropersulfide species (vide infra).
Figure 2.
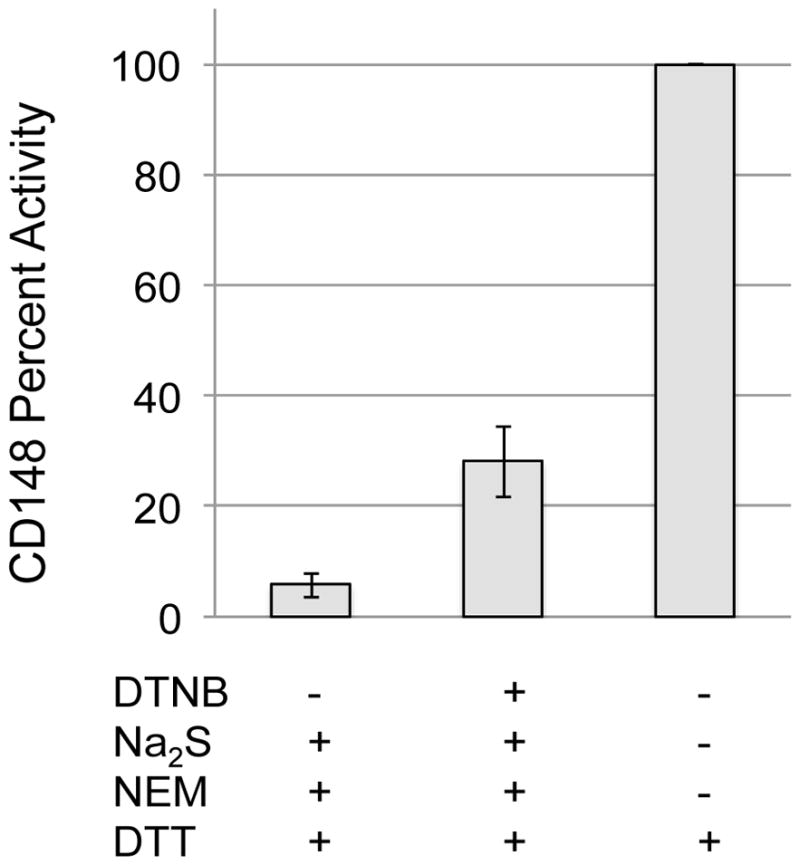
The effect of hydropersulfide formation on recoverable activity of NEM treated enzyme followed by DTT reduction. Recombinant CD148 was treated with or without DTNB followed by Na2S to generate the persulfide or the reduced thiol respectively. Proteins were then treated with NEM followed by DTT. Phosphatase activity was then measured using FDP and normalized at 100% to the sample where only DTT was used. These results are the normalized averages from 4 separate experiments. Error bars represent standard deviation.
Figure 3.
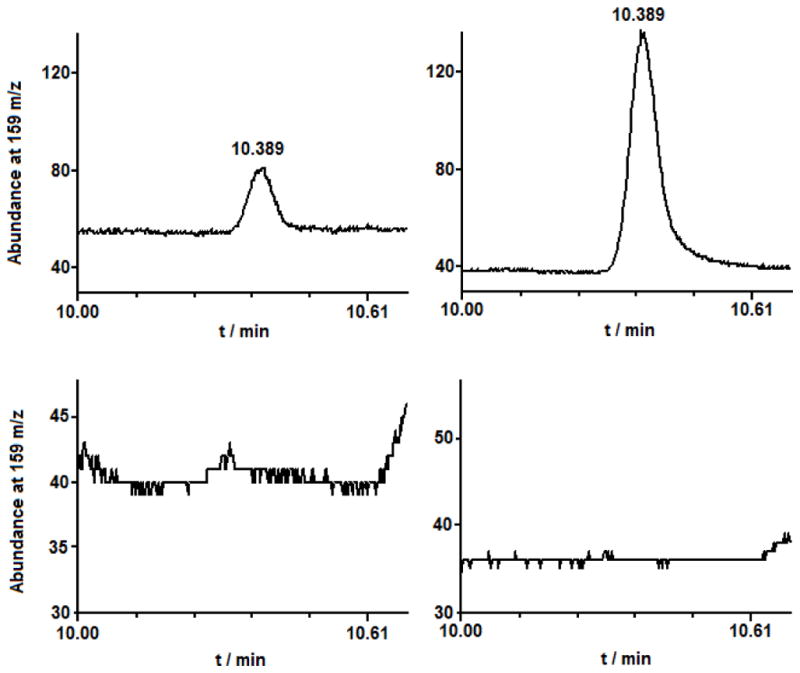
Detection of NEM-SH (M.W. = 159, retention time 10.389 min.) via select ion monitoring at m/z of 159 in (upper left) Protein-SSH sample treated with NEM and then DTT, (upper right) standard sample made via reaction of H2S with NEM, (lower left) Protein-SH treated with NEM and DTT, (lower right) Protein-SH solution treated with DTT.
CD148 contains 5 cysteines in the phosphatase domain, one active-site residue and 4 others. It is not known whether the non-catalytic cysteines serve any regulatory role. To be sure, persulfide modification could have conceivably occurred on numerous residues leading to numerous sites of NEM modification. Although it seems reasonable that the active-site residue would be quickly modified via the electrophilic chemistry used herein (provided access of the electrophile is not hindered), this does not preclude reaction at the other thiol sites and possible persulfide-mediated effects associated with these sites.
In order to further examine the idea that 1) hydropersulfide generation can protect against irreversible electrophile-mediated thiol modification and 2) generation of hydropersulfides using the above methodology is not quantitative due to the instability of the hydropersulfide in the presence of excess H2S, this chemistry was further examined using purely chemical (non-enzymatic) systems.
Hydropersulfide Generation on Thiol Sepharose 4B Resin
As described above, hydropersulfides can be generated via the simple reaction of a disulfide with H2S (Reaction 1) [31]. Indeed, this reaction has been used as a source of hydropersulfide in previous studies [28]. Experimentally, this method for in situ hydropersulfide generation can be problematic in some cases since the hydropersulfide solution will also contain other potentially reactive species such as H2S and RSH (as well as higher order polysulfides). Thus, interpretation of experiments examining hydropersulfide chemistry can be confounded by the presence of these other species. Ideally, it would be best to separate hydropersulfides from these other compounds prior to examining their chemistry. However, considering the inherent instability of hydropersulfides [8], many separation methods (i.e. column chromatography, crystallization, etc.) are not viable options. The generation of a resin-bound hydropersulfide species can be accomplished by reacting H2S with an “activated” thiol Sepharose® 4B resin (a resin that contains a glutathione disulfide with an easily displaced thiopyridine). Therefore, in order to simplify the separation of thiols from reactants, an activated Sepharose® resin with bound glutathione (initially present as the 2-thiopyridinyl disulfide) was used as the model thiol species for examining hydropersulfide chemistry. By using this resin, a hydropersulfide can be synthesized and then easily removed from other chemical reagents/contaminants via centrifugation/washing. Thus, the activated Sepharose-glutathione 2-pyridinyl disulfide resin (Resin-GS-S-Pyr) was treated with H2S to make the corresponding Sepharose-glutathione hydropersulfide (Reaction 7).
| (7) |
The resin was then washed several times to remove excess H2S and other products leaving, presumably, Resin-GSSH. Evidence for the above reaction was obtained by simple monitoring of the generation of released HS-Pyr, which absorbs at 343 nm.
Previous use of thiol Sepharose 4B resin by others has relied on selective reaction of a thiol with the sulfur atom associated with glutathione (as shown above in Reaction 7 where the thiol is H2S) giving a resin bound thiol (as a disulfide) and 2-thiopyridine release. This is a reasonable expectation since the thiopyridine (HS-Py/−S-Py) is a good leaving group making the internal sulfur (attached to the resin-bound GSH) the electrophilic site. However, as described previously, a lack of quantitative generation of Resin-GSSH from the reaction of H2S with Resin-GSS-Py may be difficult since the initially formed Resin-GSSH species (from Reaction 7) may react further with excess H2S to give Resin-GSH and dihydrogen disulfide (H2S2) (Reaction 8, which is analogous to Reaction 3).
| (8) |
The existence/likelihood of Reaction 8 simply indicates that quantitative conversion of Resin-GSS-Pyr to Resin-GSSH via treatment with H2S is unlikely or, at the very least, difficult to attain. However, if generation of resin-bound hydropersulfide can be confirmed and the amount quantitated, then this system remains useful for synthesizing and examining hydropersulfide chemistry. Thus, in order to determine the percentage of Resin-GSSH formed under the conditions of our experiment, activated resin was reacted with H2S (Reaction 7) and then washed to eliminate excess reactant, H2S, as well as the possible products HS-Py and H2S2.
The resin-bound products, now free of excess reactant and other species, were then treated with CN−. It is established that CN− will react with the sulfane sulfur atom of hydropersulfides to give thiocyanate (−SCN) (Reaction 9), which can be characterized and quantified via formation of a colored ferric complex [28,32].
| (9) |
Thus, in an experiment to quantitate the amount of Resin-bound persulfide made via this method, approximately 11 mg of the activated” thiol Sepharose® 4B was reacted with 50 mM NaHS yielding 56 M HS-Pyr released into the supernatant. Analysis for polysulfides in the supernatant (e.g. H2S2) via CN− addition and quantitation of SCN− formation gave only 4 M sulfane sulfur content released into the supernatant. Treatment of the isolated Sepharose® 4B resin (after exposure to NaHS) with CN− and quantitation of SCN− via [Fe(SCN)]2+ generation gave SCN− at approximately 27% of the amount of HS-Pyr released. These results indicate that approximately 27% of the resin-bound GSH exists as the GSSH species. As discussed above, the lack of quantitative conversion to the Resin-GSSH can be due to the equilibrium represented by Reaction 8. Interestingly, attempted quantitation of putative H2S2 (presumably formed via Reaction 8), via treatment of the supernatant with CN− to give SCN− resulted in only 7% of the total HS-Pyr released, indicating a lack of mass balance (i.e. HS-Pyr released versus hydropersulfide species). Although the reasons for this lack of mass balance are currently not understood, it is noteworthy that H2S2 is especially unstable with respect to decomposition to H2S and elemental sulfur (S8) [33,34] and S8 is poorly reactive with CN− [35]. Regardless, it is clear that approximately 27% hydropersulfide can be generated on the Sepharose resin under these conditions.
It is clear that H2S/HS− is capable of reacting with the mixed disulfide of the resin and under the conditions of the experiments described herein, generates approximately 30% Resin-GSSH. Interestingly, the results with CD148 described above also allude to an approximate 30% CD148-SSH generation using DTNB and H2S. Although there is no reason to think that these two values should be similar, both results indicate a non-quantitative conversion of thiol to hydropersulfide using these methods (likely due to the existence of Reactions 3 and 8).
It is worth noting that use of Resin-bound species is likely to greatly decrease the prevalence of sulfur-based catenation reactions (i.e. polysulfur species generation), since these reactions would rely on second-order processes whereby hydropersulfides react further to give, for example, trisulfides. Since Resin-binding greatly limits diffusion and interaction of these reactive species, it is unlikely that polysulfur species are participating significantly in the processes described herein.
Electrophilic Modification of a Hydropersulfide and Reductive Conversion to a Thiol
As described above, Resin-bound glutathione hydropersulfides can be generated at a level of approximately 20–30% when activated thiol Sepharose 4B resin is reacted with H2S (vide supra) under the conditions described herein. In order to test the idea that hydropersulfides can be oxidized or electrophilically modified and then reductively converted to the corresponding thiol (Figure 1), resin-bound GSSH (Resin-GSSH) was made as described above (treating Resin-GSS-Pyr with H2S, Reaction 7). This reaction can be easily followed by simply monitoring the absorbance at 343 nm (for released −S-Pyr) (see Experimental Procedures section). The resin was then treated with nitroxyl (HNO) since it has been demonstrated to be an extremely thiophilic species and leads to sulfinamide formation when excess thiol is not present (Reaction 10) [36,37]. Since HNO is self-reactive, it is generated in situ using the HNO-donor species Angeli’s salt (Na2N2O3), a well-characterized and often-utilized donor [38]. Thus, the resin was reacted with the HNO-donor Angeli’s salt for 2 hours at room temperature (approximately 7 half-lives of the donor). Following treatment with Angeli’s salt the resin was washed to eliminate excess Angeli’s salt (as well as other decomposition products) and then treated with DTT (Reactions 7,10,11).
| (10) |
| (11) |
As a positive control, the Resin-GSS-Pyr was reduced with DTT, to generate the Resin-GSH species (Reaction 12), and then reacted with HNO (Reaction 13). The resin-sulfinamide (Resin-G-S(O)-NH2) species formed from this sequence should not be reducible by DTT under the conditions of this procedure and therefore represents what will occur in this experiment when a thiol rather than a hydropersulfide is made.
| (12) |
| (13) |
As a negative control, the Resin-GSS-Pyr was simply reduced by DTT (Reaction 12) and not treated with HNO. This experimental procedure is depicted schematically (Scheme 1).
Scheme 1.

Experimental protocol for examining the reversibility of thiol and hydropersulfide modification.
Based on the proposed chemistry, a Resin-GSH species should be modified by HNO to give the sulfinamide (Resin-GS(O)-NH2), a species which is not readily reduced to the thiol state (Resin-GSH) by DTT (Reaction 13). On the other hand, exposure of a resin-hydropersulfide (Resin-GSSH) to HNO will generate a different product (presumably Resin-GSS(O)-NH2) whereby reductive cleavage of the S-S bond by DTT can form the thiol Resin-GSH (Reactions 10 and 11). Therefore, a comparison of the thiol content from these resin treatments can serve as a measure of the ability for oxidized/modified hydropersulfides to be reduced to the thiol species (compared to oxidized/modified thiols). The thiol content of the resins from these treatments was therefore determined using DTNB (also referred to as Ellman’s reagent, which is used routinely for quantitation of thiols) [39] via monitoring the absorbance of TNB-S− at 412 nm (Reaction 1). As shown in Table 1, HNO-mediated modification of a thiol (Resin-GSH) gives products whereby approximately 18% can be converted back to the thiol with DTT treatment (the likely reason for this level of DTT-mediated reversibility is discussed below). However, treatment of the resin containing approximately 20–30% Resin-GSSH with HNO followed by reduction by DTT gives about 37% thiol produced. The approximate increase in DTT-dependent thiol formation for HNO-treated Resin-GSSH versus HNO-treated Resin-GSH can be attributed to the presence of the hydropersulfide.
Table 1.
Effect of Hydropersulfide Formation on Electrophilic and Oxidative Modification: Reduction of Modified Species to the Thiol via DTT.
| Sample (Oxidant) | Treatment Conditions | Thiol Production1,2 |
|---|---|---|
| Control |
|
100% |
| Thiol (HNO) |
|
18% |
| Hydropersulfide (HNO) |
|
37% |
|
| ||
| Control |
|
100% |
| Thiol (H2O2)3 |
|
39% |
| Hydropersulfide (H2O2)3 |
|
68% |
all values normalized to the control.
all experiments were performed at least twice and the results of representative experiments are presented.
H2O2 treatment at 10 mM for 12 hrs.
It is important to note at this time that work from the Toscano lab reported that sulfinamides can be reduced by DTT to the corresponding thiol [40]. However, this reaction is very slow and requires elevated temperature (i.e., 37°C) and long reaction times (typically 5 hours to achieve an approximate 30% yield of product) and should not be a significant factor in the studies herein where analogous reactions were run at room temperature for < 30 minutes.
The fact that 18% of the HNO-treated Resin-GSH can be reduced to the thiol by DTT is not necessarily surprising since modification of thiols by HNO can follow two distinct and competing reaction pathways. One pathway, discussed above, leads to the sulfinamide product and the other pathway leads to a disulfide and hydroxylamine (Figure 4) [36]. Since the disulfide can be easily reduced to the corresponding thiol by DTT (while the sulfinamide cannot), the results herein simply indicate that treatment of Resin-GSH with HNO gives a significant percent of reducible resin-disulfide.
Figure 4.
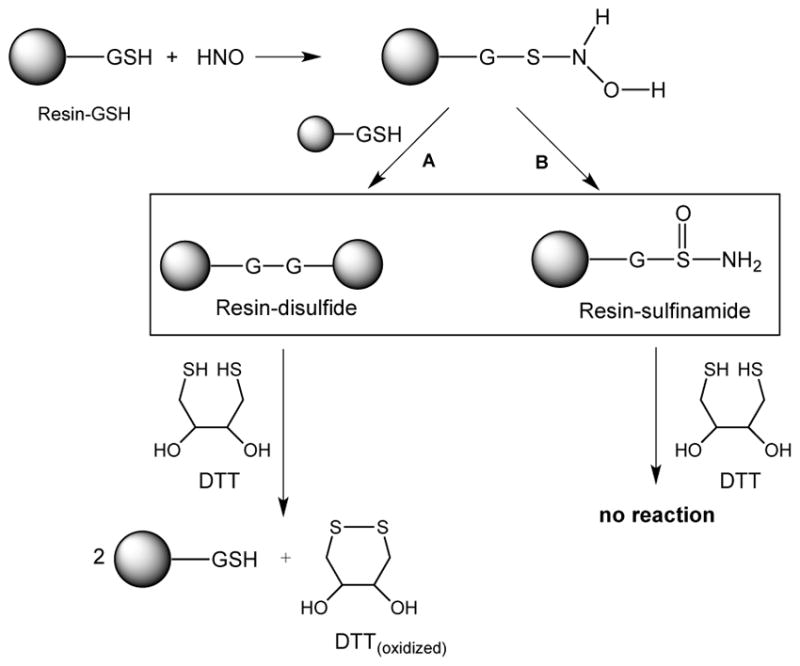
Reactions of HNO with Resin-GSH.
Overall, these results are consistent with the idea that HNO-mediated electrophilic modification of hydropersulfides can be reduced off to generate the corresponding thiol using, in this case, DTT as the reductant (Figure 1, E+ = HNO, [H] = DTT).
Oxidative Modification of a Hydropersulfide and Reductive Conversion to a Thiol
Using a nearly identical procedure outlined above for HNO, the interaction of Resin-GSSH/Resin-GSH with the oxidant H2O2 was examined. Thus, Resin-GSH and Resin-GSSH (synthesized as described above) were oxidized using 10 mM H2O2 for 12 hours at 28°C. Reduction of the oxidized resins using DTT followed by thiol quantitation using DTNB (vide supra) gives the results shown in Table 1. As with HNO-mediated modification, generation of Resin-GSSH prior to treatment with H2O2 allowed a greater percentage of DTT-mediated reduction to the corresponding thiol. In the case of Resin-GSH oxidation by H2O2, approximately 39% of the oxidized resin species can be reduced back to the thiol by DTT. However, generation of the Resin-GSSH prior to H2O2 exposure resulted in 68% of the oxidized resin species reduced to the corresponding thiol. The fact that approximately 39% of the Resin-GSH treated with H2O2 could be reduced back to the thiol by DTT is likely due to numerous effects. First, H2O2- mediated oxidation of thiols gives a multitude of products such as the sulfenic (RSOH), sulfinic (RS(O)OH) and sulfonic (RS(O)2OH acids (see Figure 1). Of these three species, the sulfenic acid species can be reduced back to the thiol. Also, generation of the sulfenic acid and subsequent reaction with another thiol will give a disulfide (Reaction 14), which, as described above, is resistant to further oxidation (compared to a thiol).
| (14) |
Since both sulfenic acids and disulfides are susceptible to reduction to the corresponding thiol by DTT, their presence in this system allows some of the oxidized resin to be reduced. Regardless, it is clear that formation of the hydropersulfide species, Resin-GSSH, prior to oxidation by H2O2 results in a greater percentage of DTT-mediated reduction back to the corresponding thiol.
NMR Analysis of the Electrophilic Modification and Subsequent Reduction of Hydropersulfides
The above studies rely primarily on the generation of a hydropersulfide via the reaction of H2S with a disulfide species (e.g. Reactions 2 or 7). Although this method of hydropersulfide formation is established, for some applications it is not ideal since hydropersulfides are generated in the presence of H2S, possibly leading to further reaction (e.g. Reaction 8), among other possible problems. Thus, in order to confirm the hydropersulfide chemistry proposed above, another method of hydropersulfide generation would be useful. Importantly, a facile and H2S-independent method for in situ generation of hydropersulfides has been developed in other labs involving disulfide precursors that spontaneously rearrange in neutral pH buffer to give hydropersulfides [26,41] (Reaction 15).
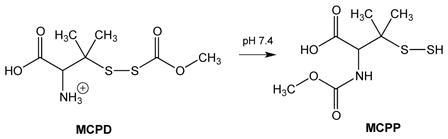 |
(15) |
Herein, MCPD was synthesized according to the method of Artaud and Galardon [26] and used to generate the hydropersulfide MCPP in an H2S-independent fashion. Initial experiments show that both NAP and MCPP can be alkylated by the electrophile IAA present in approximately 10-fold excess, Reactions 16 and 17 (Figure 5).
Figure 5.

1H-NMR Spectra of the reaction of IAA with (A) NAP and (B) MCPP. IAA was present at 10-fold excess. Methylene protons on IAA resonate at approx. 3.62 ppm (a). Methylene protons of the IAA adducts (shown in bold) resonate upfield at approx. 3.2 ppm for the NAP adduct (b) and 3.2 ppm for the MCPP (b’) adduct. Note: for the NAP adduct (A) the methine (CH) proton resonates at 4.1 ppm (c), the acetyl methyl protons (CH3) resonates at 1.9 ppm (d) and the diastereotopic gem-dimethyl protons (CH3,CH3) resonate at 1.2 – 1.3 ppm (e); for the MCPP adduct (B) the methine proton (CH) resonates at 4 ppm (c’), the carbonyl O-methyl protons (OCH3) resonate at approx. 3.5 ppm (d’) and the gem-dimethyl protons (CH3,CH3) resonate at 1.2–1.3 ppm (e’).
| (16) |
| (17) |
In Figure 5A adduct formation between NAP and IAA is clearly evidenced by the upfield shift in the methylene protons of the acetamide from 3.62 ppm to 3.21 ppm. A similar upfield shift of the acetamide methylene protons can also be seen when MCPP (generated from MCPD rearrangement) is allowed to react with IAA, Reaction 17 (Figure 5B).
Subsequent 1H-NMR experiments examining the reactivity of the thiol versus hydropersulfide adducts show that the NAP-IAA adduct cannot be reduced by the reductant DTT, whereas the MCPP-IAA adduct is readily reduced by DTT (Figure 6). That is, examining only the gem-dimethyl protons (for the sake of simplicity), it is clear that the 1H chemical shifts of the gem-dimethyl groups of NAP (Figure 6A) shift upfield when NAP is reacted with IAA (Figure 6B).
Figure 6.

1H-NMR resonances associated with the gem-dimethyl groups of (A) 1 mM NAP, (B) product resulting from the reaction of 1 mM NAP with 10 mM IAA and, (C) product remaining after treatment of B with 20 mM DTT, (D) 1 mM MCPD in D2O, (E) product resulting from the reaction of 1 mM MCPD and 10 mM iodoacetamide in pH 7.4 buffer and, (F) product resulting from the reduction of E with 20 mM DTT. Except for (D) spectra were measured in 0.1 M phosphate buffer (pH 7.4) with 10 % D2O.
Expectedly, when the adduct of NAP and IAA is treated with DTT, no change in the NMR spectrum is seen (Figure 6C) indicating that the adduct cannot be reduced to back to NAP. The reactions of MCPP with IAA can also be examined by following the changes in the 1H-NMR chemical shifts associated with the diastereotopic gem-dimethyl groups. Figure 6D shows the NMR spectrum of the gem-dimethyl groups of MCPD in pure D2O (no buffer). Under these conditions, no S-to-N methoxy carbonyl transfer occurs. When an aliquot of the MCPD-D2O solution is added to a solution of IAA in 0.1 M phosphate buffer (conditions whereby MCPD will rearrange to form the hydropersulfide MCPP [26]), a trapped species is formed (Figure 6E) (Reaction 16) as indicated by the upfield shift in the gem-dimethyl protons (and shown previously in Figure 5). Subsequent treatment of the MCPP-IAA adduct with DTT then leads to reduction of the adduct, consistent with the idea that the fully reduced thiol methoxycarbonyl penicillamine (MCP) was formed (Figure 6F) (Reaction 18).
| (18) |
These results are consistent with the idea that hydropersulfides can be trapped by an electrophile and the adduct easily reduced to the corresponding thiol by the thiol reductant DTT. On the other hand, similar thiol-electrophile adducts (i.e. thiol ethers) are not reversed by DTT.
Analysis of Hydropersulfide Reactivity via Mass Spectrometry
The NMR-based experiments discussed above are consistent with the idea that electrophilic modification of the thiol NAP with IAA is irreversible whereas analogous modification of the hydropersulfide MCPP is reversible with use of the reductant DTT. To further validate this proposal, characterization of both the trapped MCPP-IAA adduct and the resulting species generated from the reduction of this trapped adduct with DTT was carried out using electrospray ionization (ESI) MS. The mass spectrum shown in Figure 7A displays the products resulting from the initial reaction of MCPP with a 10-fold excess of IAA. The trapped MCPP-IAA adduct (m/z = 297.05) (Reaction 17) and the ammonium salt of the trapped MCPP-IAA adduct (m/z = 314.06) are identified as the major products resulting from this reaction.
Figure 7.

Identification by ESI-MS of (A) the trapped MCPP-IAA adduct and the ammonium ion of this adduct formed by the reaction of 1mM MCPP with 10 mM IAA and, (B) the ammonium ion of the free thiol MCP formed after treatment of A with 20 mM DTT. All reactions were performed in 0.1 M ammonium bicarbonate buffer (pH 7.4). Aliquots of the reaction mixture were diluted 5x with methanol prior to ESI-MS analysis.
Treatment of the MCPP-IAA species with DTT results in the complete reduction of both the trapped adduct and the ammonium ion of the trapped adduct to the ammonium ion of the free thiol methoxycarbonyl penicillamine MCP (m/z = 225.10) (Figure 7B). These results are in good agreement with the NMR data and further support the notion that although hydropersulfides can be electrophilically modified, such modified species are easily reduced to the free thiol with use of the reductant DTT.
Summary and Possible Implications
Much of the work presented herein utilizes solid-phase systems for binding the chemical species of interest. This approach allows facile and rapid separation of resin-bound reactive species from other soluble reactants and products, which may otherwise interfere with subsequent reactions and confound experimental interpretation. It should be noted, however, that relying on these solid-phase systems is not without its limitations and problems. First and foremost, generation of the reactive species of interest (i.e. hydropersulfides, thiols and derived species) on the insoluble resins precludes rigorous and direct characterization of the bound species (i.e. via NMR, UV-vis, etc.). Thus, the identity of the bound species needs to be inferred from established chemistry. Also, since the resin-bound species cannot be directly detected using traditional methods, precise quantitation becomes difficult and, again, levels of resin-bound species need to be determined indirectly via established chemistry (e.g. cyanolysis of resin-bound hydropersulfides followed by −SCN analysis). In spite of these drawbacks and limitations, solid-phase systems used in this way provide a viable experimental approach (albeit somewhat qualitative compared to more traditional homogenous systems) to address the questions presented in this work. Importantly, studies utilizing the H2S-independent generation and characterizable hydropersulfide, MCPP, support the idea that electrophilically modified hydropersulfides are easily reduced to the corresponding thiol whereas similarly modified thiols are not reversed.
The results presented herein indicate that electrophilic or oxidative modification of a hydropersulfide can be readily reduced to the corresponding thiol. However, as expected, analogous modification of a thiol is not amenable to reductive conversion back to the thiol. In the case of NEM-mediated modification of CD148, the modified enzyme hydropersulfide can be reduced back to the catalytically active thiol species, whereas the modified thiol cannot. In the case of simple HNO-mediated modification of a resin-GSH, a significant portion of the Resin-GSH-HNO adduct (likely the sulfinamide) cannot be reduced to the corresponding thiol using DTT under the conditions of these experiments. However, partial generation of resin-GSSH in this system leads to an increase in the degree of DTT-mediated reduction to the thiol species. Similar results were observed with H2O2-mediated modification of resin-GSH versus resin- GSSH. Lastly, for the case of IAA-mediated modification of MCPP, reduction to the corresponding thiol is achieved with the use of DTT, however reduction of the NAP modified species is not. All of these results are consistent with the idea that hydropersulfides can protect thiols from irreversible electrophilic or oxidative modification via formation of a hydropersulfide prior to electrophile/oxidant exposure. Taken altogether, it is clear that hydropersulfides can react with electrophiles/oxidants to give species that can be reduced to the corresponding thiol using reductants such as DTT. Figure 1 summarizes this chemistry. Importantly, the idea of reductive conversion of modified hydropersulfides to thiols (and the lack of reduction of modified thiols) is not new since several assays for hydropersulfides rely on this phenomenon. For example, the Xian and Nagy labs have developed hydropersulfide specific assays based on this chemistry [30,42].
To be sure, the idea of protection of thiol proteins via generation of an oxidized and readily reversible adduct is not new. For example, the formation of a thiol protein-glutathione mixed disulfide (Protein-SS-G) is thought to provide some protection of the protein thiol since this oxidized species lacks the inherent reactivity of a thiol and is readily reversed back to a thiol under reducing conditions [43]. The proposed mechanism of protection herein differs somewhat in that hydropersulfides are hypothesized to react readily with the chemical modifiers (e.g. electrophiles or oxidants) and then under reducing conditions are reversed back to the original thiol species.
It is important to note that previous work has shown that hydropersulfide formation in cells can occur via the actions of the enzymes cystathionine gamma lyase (CSE) and/or cystathionine beta synthase (CBS), both of which will convert cystine (Cys-SS-Cys) to cysteine hydropersulfide (Cys-SSH) (Reaction 19) [2].
| (19) |
Moreover, cellular Cys-SS-Cys uptake into cells is mediated by a cystine-glutamate antiporter xc−, which is composed of two proteins, xCT, which mediates transport and 4F2hc, which localizes the protein complex to the membrane [44]. Importantly, both CSE and xCT are inducible under conditions of oxidative/electrophilic stress. For example, xCT can be induced under electrophilic/oxidative stress via activation of Nrf2, a primary electrophile/oxidant response transcription factor [45] and CSE expression can be induced by H2O2 [46] and/or pro- inflammatory cytokines via NF-κB activation [47]. Since CSE and xCT expression can lead to increased levels of Cys-SSH, which can serve as a precursor to the generation of other biological hydropersulfides [2], it appears that hydropersulfide generation can be a cellular response to oxidative/electrophilic stress. Importantly, recent work from the Banerjee lab indicates that increased levels of intracellular Cys-SS-Cys may stimulate Cys-SSH formation via CSE, although under normal conditions other, non-hydropersulfide forming processes predominate [48]. Moreover, hydropersulfides can be fleeting species, likely present predominately under oxidizing conditions. For example, a hydropersulfide generated in a biological system can also be reduced back to the thiol species via reaction with other thiols, generating H2S and the corresponding disulfide (Reaction 20).
| (20) |
Since the disulfide can be further reduced to the thiol species, hydropersulfides may be unstable species in a normally reducing biological environment (but prevalent in a system under oxidative stress). Also, the Nagy lab has recently reported that the enzyme thioredoxin is capable of reducing hydropersulfides, indicating that there is an enzymatic pathway for removing hydropersulfides as well [30]. From a chemical perspective, the results shown herein support this idea. That is, oxidative/electrophilic cellular stress leads to the generation of highly nucleophilic and reducing hydropersulfides, which can either react directly and intercept oxidants and electrophiles or protect thiol proteins from irreversible modification via generation of a protein-hydropersulfide. Then, after the oxidative/electrophilic crisis and when the cell regains its reductive status, the protected proteins can be regenerated to active forms via simple reduction. Scheme 2 outlines these ideas. It needs to be emphasized that these ideas are highly speculative at this time and other reasons for CSE/xCT expression during cellular stress conditions have been proposed. For example, CSE/xCT expression during cellular stress has been reported to be for the purposes of increasing GSH levels [49, 50]. Regardless, it is intriguing to speculate that hydropersulfides may also be a part of cellular protection.
Scheme 2.
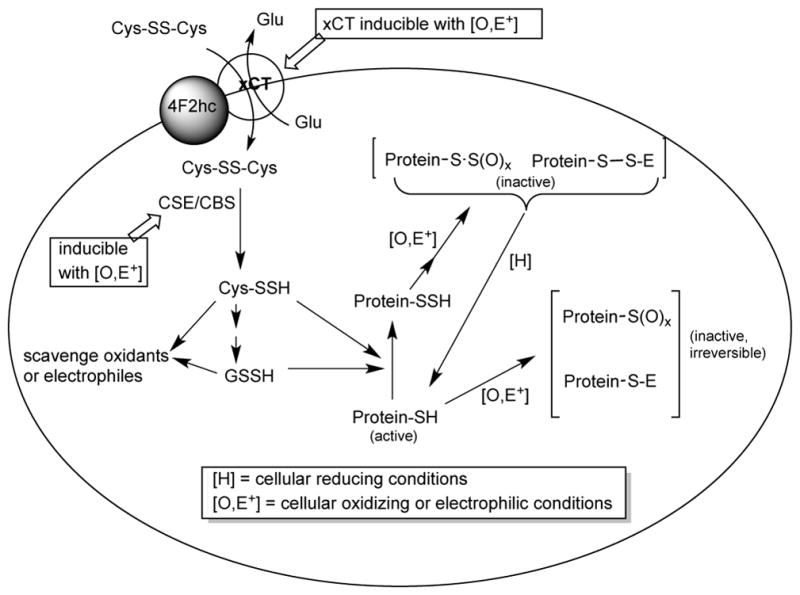
Highlights.
Hydropersulfides can be easily modified by oxidants and electrophiles
Modified hydropersulfides are easily reduced to the corresponding thiols
Hydropersulfide formation in proteins may serve a protective role against irreversible oxidative/electrophilic modification
Acknowledgments
Funding Sources
This work was supported by grants from the National Institutes of Health (HL106598) to JMF and the National Science Foundation (CHE-1148641) to JMF, (CHE-1213438) to JPT and an MRI grant from the National Science Foundation (CHE-1126482) to the Sonoma State Chemistry Department.
The authors are indebted to the lab of Prof. Arthur Weiss (UCSF) for the generous donation of genetic materials. The authors are grateful to Dr. Yitmin Liang for providing important analytical resources for this work and for his enthusiastic and unselfish support of this lab for the past 25 years.
Abbreviations
- RSSH
generic hydropersulfide
- Cys-SSH
cysteine hydropersulfide
- GSSH
glutathione hydropersulfide
- DTT
dithiothreitol
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- NEM
N-ethyl maleimide
- DTNB
5,5'-dithiobis-(2-nitrobenzoic acid)
- FDP
fluorescein diphosphate
- MCPD
S-methoxycarbonyl penicillamine disulfide
- MCPP
N-methoxycarbonyl penicillamine persulfide
- MCP
methoxy carbonyl penicillamine
- IAA
iodoacetamide
- DCM
dichloromethane
- NAP
N-acetyl penicillamine
- MCP
methoxycarbonyl penicillamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 2.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Reactive cysteine hydropersulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl, Acad, Sci USA. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mustafa AK, Gadalla MM, Snyder SH. Signaling by gasotransmitters. Science Signal. 2009;2:1–8. doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toohey JI. Persulfide sulfur is a growth factor for cells defective in sulfur metabolism. Biochem Cell Biol. 1986;64:758–765. doi: 10.1139/o86-103. [DOI] [PubMed] [Google Scholar]
- 5.Toohey JI. Sulphane sulphur in biological systems: a possible regulatory role. Biochem J. 1989;264:625–632. doi: 10.1042/bj2640625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toohey JI. Sulfur signaling: Is the agent sulfide or sulfane. Analyt Biochem. 2011;413:1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 7.Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA, Chen A, Wink DA. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide and their derived species. Chem Res Toxicol. 2012;25:769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J, Fukuto JM. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications of their possible biological activity and utility. Free Radic Biol Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Münchberg U, Anwar A, Mecklenburg S, Jacob C. Polysulfides as biologically active ingredients of garlic. Org Biomol Chem. 2007;5:1505–18. doi: 10.1039/b703832a. [DOI] [PubMed] [Google Scholar]
- 10.Massey V, Edmondson D. On the mechanism of inactivation of xanthine oxidase by cyanide. J Biol Chem. 1970;245:6595–6598. [PubMed] [Google Scholar]
- 11.Branzoli U, Massey V. Evidence for an active site hydropersulfide residue in rabbit liver aldehyse oxidase. J Biol Chem. 1974;249:4346–4349. [PubMed] [Google Scholar]
- 12.Kim EJ, Feng J, Bramlett MR, Lindahl PA. Evidence for a proton transfer network and a required hydropersulfide-bond-forming cysteine residue in Ni-containing carbon monoxide dehydrogenase. Biochemistry. 2004;43:5728–5734. doi: 10.1021/bi036062u. [DOI] [PubMed] [Google Scholar]
- 13.Wright CM, Christman GD, Snellinger AM, Johnston MV, Mueller EG. Direct evidence for enzyme hydropersulfide and disulfide intermediates during 4-thiouridine biosynthesis. Chem Commun. 2006:3104–3106. doi: 10.1039/b604040c. [DOI] [PubMed] [Google Scholar]
- 14.Newton WE, McDonald JW, Yamanouchi K, Enemark JH. Synthesis and molecular structure of molybdenum(V) dimers with mixed oxo and sulfido terminal ligands: removal of the terminal sulfide by triphenylphosphine and cyanide. Inorg Chem. 1979;18:1621–1626. [Google Scholar]
- 15.Hofmann M. Density functional theory study of model complese for the revised nitrate reductase active site Desulfovibrio desulfuricans NapA. J Biol Inorg Chem. 2009;14:1023–1035. doi: 10.1007/s00775-009-0545-1. [DOI] [PubMed] [Google Scholar]
- 16.Kessler D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol Rev. 2006;30:825–840. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 17.Cipollone R, Ascenzi P, Visca P. Common themes and variations in the Rhodanese superfamily. IUBMB Life. 2007;59:51–59. doi: 10.1080/15216540701206859. [DOI] [PubMed] [Google Scholar]
- 18.Pan J, Carroll KS. Hydropersulfide reactivity in the detection of protein-S- sulfhydration. ACS Chem Biol. 2013;8:1110–1116. doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:1–12. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivaramakrishnan S, Cummings AH, Gates KS. Protection of a single-cysteine redox switch from oxidative destruction: On the functional role of sulfenyl amide formation in the redox-regulated enzyme PTP1B. Bioorg Med Chem Lett. 2010;20:444–7. doi: 10.1016/j.bmcl.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CY, Willard D, Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48:1399–409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 22.Greiner R, Palinkas Z, Basell K, Becher D, Antelmann H, Nagy P, Dick TP. Polysulfides link H2S to protein thiol oxidation. Antiox Redox Signal. 2013;19:1749–1765. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith PAS, Hein GE. The alleged role of nitroxyl in certain reactions of aldehydes and alkyl halides. J Am Chem Soc. 1960;82:5731–5740. [Google Scholar]
- 24.Saund SS, Sosa V, Henriquez S, Nguyen QNN, Bianco CL, Soeda S, Millikin R, White C, Le H, Ono K, Tantillo DJ, Kumagai Y, Akaike T, Lin J, Fukuto JM. The chemical biology of hydropersulfides (RSSH): Chemical stability, reactivity and redox roles. Arch Biochem Biophys. 2015;588:15–24. doi: 10.1016/j.abb.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collier HB. A note of the molar absorptivity of reduced Ellman’s reagent 3- carboxylato-4-nitrothiophenolate. Analyt Biochem. 1973;56:310–311. doi: 10.1016/0003-2697(73)90196-6. [DOI] [PubMed] [Google Scholar]
- 26.Artaud I, Galardon E. A persulfide analogue of the nitrosothiol SNAP: Formation, characterization and reactivity. ChemBioChem. 2014;15:2361–2364. doi: 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]
- 27.Hermiston ML, Zikherman J, Zhu JW. CD45, CD148, and Lyp/Pep: Critical phosphatases regulating Src family kinase signaling networks in immune cells. Immunol Rev. 2009;228:288–311. doi: 10.1111/j.1600-065X.2008.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francoleon NE, Carrington SJ, Fukuto JM. The reaction of H2S with oxidized thiols: Generation of hydropersulfides and implications to H2S biology. Arch Biochem Biophys. 2011;516:146–154. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Vasas A, Doka E, Fabian I, Nagy P. Kinetic and thermodynamic studies on the disulfide-bond reducing potential of hydrogen sulfide. Nitric Oxide. 2015;46:93–101. doi: 10.1016/j.niox.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Doka E, Padar I, Biro A, Johansson K, Cheng Q, Ballago K, Prigge JR, Pastorflores D, Dick TP, Schmidt EE, Arner ESJ, Nagy P. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci Adv. 2016;2:e1500968. doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavallini D, Federici G, Barboni E. Interaction of proteins with sulfide. Eur J Biochem. 1970;14:169–174. doi: 10.1111/j.1432-1033.1970.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 32.Flavin M. Microbial transusulfurization: The mechanism of an enzymatic disulfide elimination reaction. J Biol Chem. 1962;237:768–777. [PubMed] [Google Scholar]
- 33.Minkwitz R, Kornath A, Sawodny W, Hahn J. Synthesis and characterization of the mercaptosulfonium cation, HSSH2+ Inorg Chem. 1996;35:3622–3625. [Google Scholar]
- 34.Butler KH, Maass O. Hydrogen sulfide. J Am Chem Soc. 1930;52:2184–2198. [Google Scholar]
- 35.Wood JL. Sulfane sulfur. Meth Enzymol. 1987;143:25–29. doi: 10.1016/0076-6879(87)43009-7. [DOI] [PubMed] [Google Scholar]
- 36.Wong PS-Y, Hyun J, Fukuto JM, Shiroda FN, DeMaster EG, Nagasawa HT. The Reaction between nitrosothiols and thiols: Generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 37.Fukuto JM, Bartberger MD, Dutton AS, Paolocci N, Wink DA, Houk KN. The physiological chemistry and biological activity of nitroxyl (HNO): The neglected, misunderstood and enigmatic nitrogen oxide. Chem Res Toxicol. 2005;18:790–801. doi: 10.1021/tx0496800. [DOI] [PubMed] [Google Scholar]
- 38.Fukuto JM, Jackson MI, Kaludercic N, Paolocci N. Examining nitroxyl in biological systems. Meth Enzymol. 2008;440:411–431. doi: 10.1016/S0076-6879(07)00826-9. [DOI] [PubMed] [Google Scholar]
- 39.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 40.Keceli G, Toscano JP. Reactivity of nitroxyl-derived sulfinamides. Biochemistry. 2012;51:4206–4216. doi: 10.1021/bi300015u. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Bhushan S, Yang C, Otsuka H, Stein JD, Pacheco A, Peng B, Devarie-Barz NO, Aguilar HC, Lefer DJ, Xian M. Controllable hydrogen sulfide donors and their activity against myocardial ischemia-reperfusion injury. ACS Chem Biol. 2013;8:1283–1290. doi: 10.1021/cb400090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang D, Macinkovic I, devarie-Baez NO, Pan J, Park CM, Carroll KS, Filipovic MR, Xian M. Detection of protein S-sulfhydration by a tag-switch technique. Angew Chem Int Ed. 2014;53:575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dalle-Donne I, Milzani A, Gagliano N, Colombo R, Giustarini D, Rossi R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antiox Redox Signal. 2008;10:445–473. doi: 10.1089/ars.2007.1716. [DOI] [PubMed] [Google Scholar]
- 44.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system Xc−: Cystine supplier and beyond. Amino Acids. 2012;42:231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 45.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antiox Redox Signal. 2013;18:522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang M, Guo Z, Wang S. Cystathionine gamma-lyase expression is regulated by exogenous hydrogen peroxide in mammalian cells. Gene Express. 2013;15:235–241. doi: 10.3727/105221613x13571653093286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fox B, Schantz J-T, Haigh R, Wood E, Moore PK, Viner N, Spencer JPE, Winyard PG, Whiteman M. Inducible hydrogen sulfide synthesis in chondrocytes and mesenchymal progenitor cells: Is H2S a novel cytoprotective mediator in the inflamed joint? J Cell Mol Med. 2012;16:896–910. doi: 10.1111/j.1582-4934.2011.01357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yadav P, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR, Banerjee R. Biosynthesis and reactivity of cysteine persulfides in signaling. J Am Chem Soc. 2016;138:289–299. doi: 10.1021/jacs.5b10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee ZW, Low YL, Huang S, Wang T, Deng LW. The cystathionine - lyase/hydrogen sulfide system maintains cellular glutathione status. Biochem J. 2014;460:425–435. doi: 10.1042/BJ20131434. [DOI] [PubMed] [Google Scholar]
- 50.Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/Glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]