

Abstract
Congenital nephrotic syndrome in the newborn is most frequently related to mutations in genes specific for structural integrity of the glomerular basement membrane and associated filtration structures within the kidney, resulting in massive leakage of plasma proteins into the urine. Occurrence of congenital nephrotic syndrome in a multi-system syndrome is less common. We describe an infant with deteriorating neurological status, seizures, edema, and proteinuria who was found to have a mutation in ALG1 and a renal biopsy consistent congenital nephrotic syndrome. Furthermore, we briefly review rare existing case reports documenting congenital nephrotic syndrome in patients with ALG1 and treatment strategies, including novel use of peritoneal dialysis.
Keywords: edema, hypoalbuminemia, fluid overload, peritoneal dialysis, microcephaly, pontocerebellar atrophy
Introduction
Congenital nephrotic syndrome describes a clinical entity of edema, hypoalbuminemia, hyperlipidemia, and proteinuria within the first three months of life. The majority of congenital nephrotic syndrome cases are primary and due to mutations in NPHS1 (nephrin), WT1 (Wilms tumor suppressor), or NPHS2 (podocin) genes leading to derangement of the glomerular basement membrane and filtration apparatus with resultant proteinuria. In the perinatal period, this disorder can present as a component of a multi-system, generalized syndrome. In particular, congenital nephrotic syndrome has been reported to occur concurrently with failure to thrive, microcephaly, deteriorating neurological status, and sepsis in the setting of congenital disorders of glycosylation due to mutations in the ALG1 gene [1–6, 10].
Congenital disorders of glycosylation (CDG) are rare multisystem diseases due to impaired synthesis, transfer, and processing of sugars resulting in hypoglycosylation of ubiquitously expressed proteins throughout the body [7]. The congenital disorder of glycosylation associated with ALG1 (ALG1-CDG) gene mutations (OMIM #608540) is described as an inherited error of metabolism resulting in deficiency of β-1,4 mannosyltransferase (OMIM #605907). The ALG1-CDG phenotype is characterized by microcephaly, developmental delay, abnormal fat distribution, strabismus, and coagulation abnormalities [8]. ALG1-CDG has also been associated with recurrent infantile seizures, cerebellar hypoplasia, hypotonia, and failure to thrive. There are 58 described cases of ALG1-CDG in the existing literature with twelve of those cases also reporting the presence of renal failure and congenital nephrotic syndrome [2, 8, 9, 10]. We report details of one case of congenital nephrotic syndrome associated with ALG1-CDG, formerly known as CDG type 1k, and discuss the use of peritoneal dialysis in management of this rare patient population.
Case Presentation
The infant (patient # 18, reference [10]) was a full term male, born at a community hospital. Due to recurrent apnea, feeding difficulties, and Enterobacter cloacae sepsis, he was transferred to neonatal intensive care unit at one month of age. He was the first child of healthy, non-consanguineous parents with unremarkable family history. Examination at admission was notable for microcephaly, hypertelorism, weak gag reflex, hypotonia, clenched hands, shortened limbs, bilateral cryptorchidism, and sacral dimple with a tuft of hair. Medical history from was notable for hypertension, mild generalized edema, and poor nutritional status. He was hypoalbuminemic (1.1 g/dL) with proteinuria (urine protein to creatinine ratio of 14.0) and microscopic hematuria (30 red blood cells per high power field).
The infant became progressively edematous and oliguric with persistent hypoalbuminemia requiring albumin replacement. Due to progressive renal dysfunction, a renal biopsy was performed. The biopsy revealed complete podocyte effacement without mesangial sclerosis and unremarkable immunofluorescence, suggestive of congenital nephrotic syndrome (Figures 1 and 2). He developed worsening fluid overload that became refractory to albumin and furosemide, and a peritoneal dialysis catheter was placed to provide mechanism for ultrafiltration and liberalization of total fluids due to the infant’s poor nutritional state.
Figure 1.
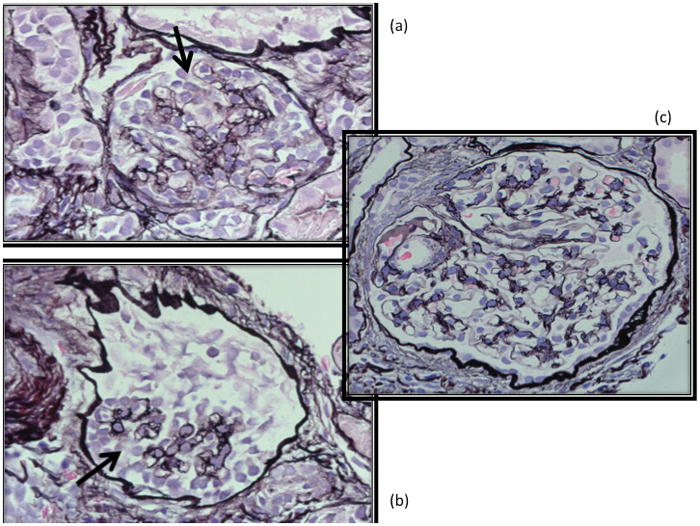
Two glomeruli (insets A, B) with features of collapsing focal segmental glomerulosclerosis on silver stain. There is epithelial (podocyte) hyperplasia (at arrows) with collapsed capillary loops. Compare with a normal glomerulus (inset C) in photo on the right.
Figure 2.
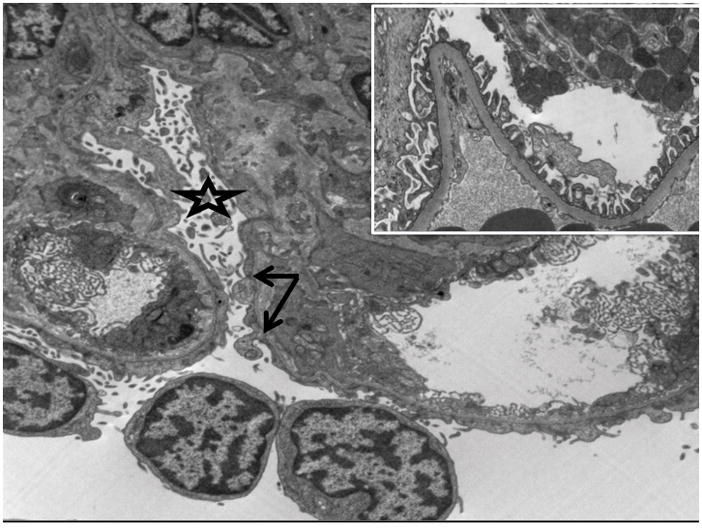
Electron microscopy demonstrating epithelial (podocyte) foot process effacement with arrow denoting rare intact podocytes and star demonstrating podocyte effacement and microvillous transformation. Comparison electron microscopy depicting a normal glomerulus with intact epithelial (podocyte) foot processes shown at inset.
His neonatal course was further complicated by neurological, growth, and infectious complications. He had progressive neurological deterioration with seizures refractory to multiple anti-epileptic medications and required continuous ventilatory support due to recurrent seizure-related apnea. Magnetic resonance imaging revealed pontocerebellar atrophy with an absent cerebellar vermis and small posterior fossa. The infant experienced failure to thrive despite optimized enteral and parenteral nutrition. Enteral feeding was complicated by recurrent aspiration, requiring Nissen fundoplication. In addition to Enterobacter cloacae sepsis at the time of transfer, the infant had Stenotrophomonas maltophilia sepsis during his hospital course. In the setting of hypogammaglobulinemia (IgG 77 mg/dL) and congenital nephrotic syndrome, intravenous immunoglobulin was provided in addition to antibiotic therapy for sepsis therapy.
At three months of age, the decision was made to transition to palliative care. The infant demonstrated rapidly worsening respiratory and renal failure, and life-sustaining measures were withdrawn. The infant died at three months of age.
Genetic and metabolic disorders were investigated as causative etiologies for the infant’s clinical presentation of pontocerebellar atrophy, seizures, and renal failure encompassing a broad disease differential, including congenital disorders of glycosylation. Chromosome microarray demonstrated a copy number variant (167 kb duplication on 9q32) not currently associated with a human constitutional disorder. Carbohydrate-deficient transferrin analysis was performed by electrospray ionization mass spectrometry. This showed elevation in the CDG mono/di ratio (mono/di = 2.151) and CDG A/DI oligosaccharide ratio (A/DI = 1.772), suggestive of a CDG type I pattern. Molecular testing for CDG-Ia (PMM2) and CDG-Ib (MPI) was negative for mutations in genes known to cause disease; however, post-mortem, research-based whole exome and Sanger sequencing confirmed patient homozygosity for a known CDG causing mutation in ALG1, specifically a c.773C>T substitution that causes a missense mutation p.S258L (dbSNP ID - rs28939378), causative for ALG1-CDG. This was confirmed by a CLIA certified lab (GeneDx).
Discussion
ALG1-CDG is a rare but recognized cause of congenital nephrotic syndrome. There is considerable overlap in phenotype due to isolated congenital nephrotic syndrome and congenital nephrotic syndrome in the setting of ALG1-CDG. Specifically, both diseases are notable for significant failure to thrive, coagulopathy and increased risk for severe bacterial infections [8, 11]. ALG1-CDG should be considered in the differential diagnosis of congenital nephrotic syndrome, particularly if patient presentation involves neurological deterioration.
Recent genetic analysis of six individuals with ALG1 mutations and renal failure demonstrated homozygosity for S258L [10]. Furthermore, there are six additional cases of congenital nephrotic syndrome associated with ALG1-CDG (Table 1). Range of patient age at onset of nephrotic syndrome was 3 weeks to 15 months. Some reports describe lifespan of patients with ALG1-CDG extending into adolescence or early adulthood; however, all reported cases of ALG1-CDG associated with nephrotic syndrome experienced death before 2 years of age [1, 3, 5, 6, 8]. Pathology from renal biopsies described in the literature report diffuse mesangial sclerosis as a prominent biopsy feature in two of the cases [1, 3], in contrast to our patient who had only podocyte effacement.
Table 1.
Summary of congenital nephrotic syndrome cases reported to date within pediatric literature. There are seven currently characterized cases of congenital nephrotic syndrome, including the current case.
| Author | Age of Onset Nephrotic Syndrome | Renal Pathology | Genetics | Age at Death |
|---|---|---|---|---|
| Hutchesson (1995) | 3 week | Not performed | CDG-I | 3 months |
| Van der Knapp (1996) | < 2 months | Diffuse mesangial sclerosis | CDG-I, phosphomannomutase-2 | 2 months |
| De Vries (2000) | < 1 month | Fusion of podocyte foot processes | CDG-I, unknown type | 2 months |
| Kranz (2004) | Not described | Not described | CDG-Ik (ALG1) | 11 weeks |
| Morava (2008) | Not described | Not described | CDG-Ik (ALG1) | 5 months |
| Sinha (2009) | 15 months | Diffuse mesangial sclerosis | CDG-Ix | 17 months |
Hypoalbuminemia, typically without generalized edema, has been described in congenital disorders of glycosylation but may more often be attributed to malnutrition and/or gastrointestinal protein losses [12]. Given the relatively high occurrence of congenital nephrotic syndrome (N = 12/58 or 21% of reported cases of ALG1, including our patient), the question could be raised whether surveillance for proteinuria is warranted to ascertain whether hypoalbuminemia is not from urinary losses.
Management options in this rare population are limited. Existing reports describe use of albumin and loop diuretic, fluid restriction, and/or no specific therapy described at all. One case report describes use of angiotensin converting enzyme inhibitor therapy in combination with diuretics for a patient [1]. This is the first report to document the use of peritoneal dialysis in an infant affected by ALG1-CDG. This modality allowed for improved fluid management and provision of nutrition in the setting of nephrotic syndrome. Ultimately, management of this unique population may prove difficult even with maximal medical renal therapy as described here given the neurological manifestations of disease, risk for coagulopathy, sepsis, and ultimately high mortality rate in infancy.
Acknowledgments
Exome sequencing was provided by the University of Washington Center for Mendelian Genomics (UW CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 1U54HG006493 to Drs. Debbie Nickerson, Jay Shendure and Michael Bamshad. H.H.F. is supported by the Rocket Fund and the National Institutes of Health (NIH) (R01DK099551).
We would like to acknowledge Drs. Richard Smith and M. Adela Mansilla for the confirmatory testing performed at the Iowa Institute of Human Genetics.
The preparation of this case report did not require any outside funding or grant support.
Footnotes
Transparency Declarations
The authors have no conflicts of interest to disclose. The genetic sequencing information presented in this paper has been described in reference 10.
Institutional Review Board at our institution deemed that informed consent was not required for the purposes of this case report.
References
- 1.Sinha MD, Horsfield C, Komaromy D, Booth CJ, Champion MP. Congenital disorders of glycosylation: a rare cause of nephrotic syndrome. Nephrol Dial Transplant. 2009;24(8):2591–4. doi: 10.1093/ndt/gfp226. Epub 2009/05/29. [DOI] [PubMed] [Google Scholar]
- 2.Kranz C, Denecke J, Lehle L, Sohlbach K, Jeske S, Meinhardt F, et al. Congenital disorder of glycosylation type Ik (CDG-Ik): a defect of mannosyltransferase I. American journal of human genetics. 2004;74(3):545–51. doi: 10.1086/382493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Knaap MS, Wevers RA, Monnens L, Jakobs C, Jaeken J, van Wijk JA. Congenital nephrotic syndrome: a novel phenotype of type I carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis. 1996;19(6):787–91. doi: 10.1007/BF01799174. Epub 1996/01/01. [DOI] [PubMed] [Google Scholar]
- 4.Morava E, Wosik H, Karteszi J, Guillard M, Adamowicz M, Sykut-Cegielska J, et al. Congenital disorder of glycosylation type Ix: review of clinical spectrum and diagnostic steps. J Inherit Metab Dis. 2008;31(3):450–6. doi: 10.1007/s10545-008-0822-0. [DOI] [PubMed] [Google Scholar]
- 5.Hutchesson AC, Gray RG, Spencer DA, Keir G. Carbohydrate deficient glycoprotein syndrome; multiple abnormalities and diagnostic delay. Arch Dis Child. 1995;72(5):445–6. doi: 10.1136/adc.72.5.445. Epub 1995/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vries BB, van’tHoff WG, Surtees RA, Winter RM. Diagnostic dilemmas in four infants with nephrotic syndrome, microcephaly and severe developmental delay. Clin Dysmorphol. 2001;10(2):115–21. doi: 10.1097/00019605-200104000-00008. Epub 2001/04/20. [DOI] [PubMed] [Google Scholar]
- 7.Grunewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation: a review. Pediatr Res. 2002;52(5):618–24. doi: 10.1203/00006450-200211000-00003. Epub 2002/11/01. [DOI] [PubMed] [Google Scholar]
- 8.Morava E, Vodopiutz J, Lefeber DJ, Janecke AR, Schmidt WM, Lechner S, et al. Defining the phenotype in congenital disorder of glycosylation due to ALG1 mutations. Pediatrics. 2012;130(4):e1034–9. doi: 10.1542/peds.2011-2711. [DOI] [PubMed] [Google Scholar]
- 9.de Koning TJ, Toet M, Dorland L, de Vries LS, van den Berg IE, Duran M, et al. Recurrent nonimmune hydrops fetalis associated with carbohydrate-deficient glycoprotein syndrome. Journal of inherited metabolic disease. 1998;21(6):681–2. doi: 10.1023/a:1005496920435. [DOI] [PubMed] [Google Scholar]
- 10.Ng BG, Shiryaev SA, Rymen D, Eklund EA, Raymond K, Kircher M, et al. ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients. Hum Mutat. 2016 doi: 10.1002/humu.22983. Epub 2016/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jalanko H. Congenital nephrotic syndrome. Pediatric nephrology. 2009;24(11):2121–8. doi: 10.1007/s00467-007-0633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohlfing AK, Rust S, Reunert J, Tirre M, Du Chesne I, Wemhoff S, et al. ALG1-CDG: a new case with early fatal outcome. Gene. 2014;534(2):345–51. doi: 10.1016/j.gene.2013.10.013. [DOI] [PubMed] [Google Scholar]