

Abstract
Since early oligomeric intermediates in amyloid assembly are often transient and difficult to distinguish, characterize and quantify, the mechanistic basis of the initiation of spontaneous amyloid growth is often opaque. We describe here an approach to the analysis of the Aβ aggregation mechanism that uses Aβ-polyglutamine hybrid peptides designed to retard amyloid maturation and an adjusted thioflavin intensity scale that reveals structural features of aggregation intermediates. The results support an aggregation initiation mechanism for Aβ-polyQ hybrids, and by extension for full-length Aβ peptides, in which a modular Aβ C-terminal segment mediates rapid, non-nucleated formation of α-helical oligomers. The resulting high local concentration of tethered amyloidogenic segments within these α-oligomers facilitates transition to a β-oligomer population that, via further remodelling and/or elongation steps, ultimately generates mature amyloid. Consistent with this mechanism, an engineered Aβ C-terminal fragment delays aggregation onset by Aβ-polyglutamine peptides and redirects assembly of Aβ42 fibrils.
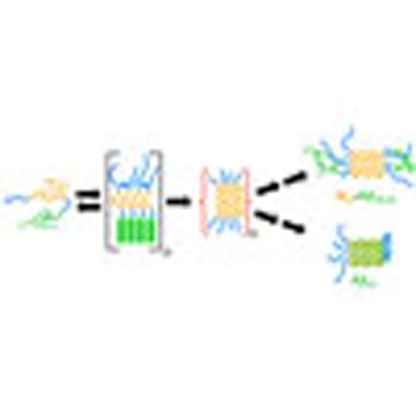 The elucidation of amyloid nucleation mechanisms remains challenging as early oligomeric intermediates are transient and difficult to distinguish. Here the authors use Aβ- polyglutamine hybrid peptides designed to slow and limit amyloid maturation to provide insights into the structures of Aβ self-assembly intermediates.
The elucidation of amyloid nucleation mechanisms remains challenging as early oligomeric intermediates are transient and difficult to distinguish. Here the authors use Aβ- polyglutamine hybrid peptides designed to slow and limit amyloid maturation to provide insights into the structures of Aβ self-assembly intermediates.
In Alzheimer's disease and other amyloid-associated conditions1, it is critically important to understand the mechanisms by which amyloid formation is initiated and the extent to which intermediate oligomeric species contribute to amyloid formation and cytotoxicity. Elucidation of amyloid nucleation mechanisms is especially challenging, however, in systems that feature oligomeric intermediates2,3,4 and secondary nucleation5 pathways. For different proteins, nucleation of amyloid formation might proceed either within an on-pathway oligomeric intermediate6 or via a classical nucleated growth polymerization5 featuring the direct formation of rare amyloid-like conformations in monomers7,8,9,10 or small multimers8.
Most mechanisms proposed to account for Aβ amyloid nucleation invoke an on-pathway role for one or more oligomeric assembly intermediates, but the structural details of these transformations remain mysterious. One early proposal was that amyloid nucleation is mediated by self-association of curvilinear protofibrillar intermediates3. Alternatively, observation of spherical oligomeric intermediates preceding Aβ protofibril and fibril formation2,11 suggested that spontaneous Aβ amyloid formation might proceed via a nucleated conformational conversion mechanism in which oligomer rearrangements serve both as the source of amyloid nucleation and as a means of fibril elongation12,13. Other mechanisms have been elucidated for the role of oligomers in formation of other amyloid fibrils6. Aβ oligomerization begins from intrinsically disordered monomers14, which progress through sub-populations of metastable multimers15 and transient oligomers exhibiting high α-helix contents16 and low ThT responses13,17,18 consistent with low amyloid-like β-structure. Based in part on earlier reports of transient formation of α-oligomers during Aβ fibril growth16, a general mechanism has been proposed for initiation of amyloid assembly (Fig. 1a) in some peptides in which early formation of α-helical oligomers leads to a high local concentration of an adjacent disordered segment, overcoming the concentration barrier to amyloid nucleation19. Once amyloid begins to grow, the α-helical segment appears to quickly unravel to join in the β-sheet network of the mature fibrils20,21 (Fig. 1a). This rapid annealing makes it very challenging to obtain direct structural evidence to support a role for early α-helical intermediates.
Figure 1. Model mechanisms and peptides.

(a) Generic mechanistic model for α-helical oligomer mediated amyloid nucleation (reprinted with permission from Oxford University Press; ref. 19. (b) Proposed mechanism for α-helical oligomer mediated amyloid nucleation of HTT exon1-like fragments (reprinted from ref. 24), with green=HTTNT; orange=polyQ; black=proline rich domain. (c) Model for an HTT exon1 homo-oligomer (reprinted with permission from Elsevier Ltd; ref. 2828). (d) Model for hetero-oligomer generated from a mixture of HTT exon1 and HTTNT (blue) (reprinted with permission from Elsevier Ltd; ref. 2828). (e) Structural model for Aβ in inclusion bodies from bacterial expression based on FTIR, solid-state NMR and hydrogen/deuterium exchange solution NMR (reprinted with permission from WILEY-VCH Verlag GmbH & Co.; ref. 4343). (f) Model peptides used in this study. Note that a–d are purely schematic and are not intended to imply particular knowledge of either the multimer sizes or the parallel or anti-parallel arrangement of the helices.
Intriguingly, an almost identical mechanism was deduced for the nucleation of polyglutamine (polyQ) amyloid formation in the Huntingtin (HTT) exon1-like fragments implicated in Huntington's disease22. In this mechanism (Fig. 1b), the 17 amino acid HTTNT segment of HTT exon1 readily undergoes a polyQ repeat length-dependent transition from disordered monomer to α-helix rich tetramer and higher oligomers22,23,24. In these non-β aggregates, the HTTNT segments act as quasi-independent, modular units to form α-helical bundles while the tethered, largely disordered polyQs are brought together within the oligomers at a high local concentration that greatly facilitates polyQ amyloid nucleation. Evidence in support of this mechanism includes (a) a dramatic rate increase on polyQ amyloid formation by covalent attachment of HTTNT, (b) early formation of ThT-negative oligomeric intermediates and (c) a unique, very low concentration dependence of initial aggregation rates that is inconsistent with a classical nucleated growth polymerization mechanism22. The rate enhancement by HTTNT has a modular aspect in that can be observed whether it is attached to the N terminus or C terminus of a polyQ track, and whether or not there is an insertion of Lys residues between the HTTNT and the polyQ22. With or without attached polyQ, HTTNT readily forms tetramers and metastable, α-helix rich oligomers23,24. Amyloid fibrils of HTT exon1-like peptides have a polyQ core but retain some HTTNT-associated α-helical elements25,26 whose interactions provide additional stability to the fibrils27. Finally, introduction of isolated HTTNT peptides in trans delays onset of aggregation, presumably via formation of mixed oligomers28 (Fig. 1d). In contrast to the experimental challenges to demonstrating the Aβ amyloid nucleation mechanism (Fig. 1a), elucidation of the analogous mechanism for HTT exon1 (Fig. 1b) was greatly aided by several features, including a low tendency of HTTNT itself to make amyloid29 and therefore to remain in stable α-helical oligomers23, the relatively slow aggregation kinetics of isolated polyQ sequences, and the strong tendency of polyQ amyloid to exclude flanking sequences from the developing amyloid core25,26.
In this paper, we take advantage of these features of polyQ to test the Fig. 1a hypothesis that a segment of Aβ might act similarly to HTTNT as a modular unit to mediate formation of non-β oligomeric intermediates required for efficient amyloid nucleation by an adjacent sequence (Fig. 1a). Our results provide broad support for this hypothesis while suggesting a mechanistic rationale for the dramatic difference in amyloidogenicity between Aβ42 and Aβ40. In addition, an appreciation of the strong dependence of ThT intensity on amyloid-like β-structure content reveals some surprising features of the assembly mechanism.
Results
Assigning pro-α and pro-β segments within the Aβ molecule
We first had to define the most likely segments of Aβ to serve the various roles suggested by the generic mechanism in Fig. 1a, including (a) the pro-α segment responsible for rapid, initial assembly into a helical, oligomeric intermediate and (b) the pro-β segment responsible for rapid nucleation of amyloid structure within that intermediate. Solution NMR of the monomer in aqueous solution provides no clues, revealing a completely disordered peptide14. The relatively hydrophilic nature and experimental accessibility of the Aβ N terminus (Fig. 1f) in various assembled states30 suggests that this segment is unlikely to play a central role in the assembly mechanism. In contrast, NMR studies of monomeric Aβ in the presence of SDS31 and lyophilized Aβ oligomers32 show that both the 10–24 and 28–42 regions of Aβ are capable of forming stable α-helix, thus suggesting that either might be capable of serving as the pro-α segment. At the same time, a variety of studies suggest that the most amyloid-prone portion of Aβ is the 16–20 Leu-Val-Phe-Phe (LVFF) segment33,34,35. It therefore seemed most likely that the central portion of Aβ including LVFF is the segment that initiates amyloid structure formation within oligomers, while the C-terminal Aβ segment mediates initial α-helical oligomer formation (Fig. 1f).
Based on these assignments we designed three hybrid peptides (Fig. 1f) to test the Fig. 1a mechanism. To replace the presumptive Aβ pro-β segment, we chose a polyQ repeat length of Q23, since a simple K2Q23K2 peptide aggregates very slowly8. We maintained the Aβ C-terminal segments at the C termini of the designed hybrid peptides, to give the critical difference between Aβ40 and Aβ42 the greatest opportunity to express itself at the peptide C termini, in analogy to the Aβ peptides. A Lys-Lys pair was inserted N-terminal to the polyQ, to replace the hydrophilic Aβ N-terminal domain. In addition, to mimic the hydrophilic Aβ21–28 segment and thereby further improve peptide kinetic solubility we inserted a Lys–Lys sequence between the polyQ and Aβ portions in two of the peptides. We justify the latter insertion based on our model (Fig. 1b) that pro-α segments act in a modular way in the nucleation mechanism, and on previous data that helped generate that model which show that a Lys–Lys insert between HTTNT and polyQ has little effect on the ability of HTTNT to enhance aggregation kinetics22. Nonetheless, to ensure that the Lys–Lys insert is not playing an outsized role in the hybrid Aβ-polyQ peptides, we also designed a third peptide in which we left out the Lys–Lys insert and replaced it with the wild-type Aβ GSNK segment. This design had the added benefit of testing a possible mechanistic role of GSNK, which has been suggested to influence Aβ monomer conformation36 and amyloid assembly kinetics and structure20,35,37.
This led to a total of three designed peptides, each harbouring presumptive pro-α segments derived from Aβ and an installed polyQ pro-β segment meant to replace the presumptive pro-β LVFF segment of Aβ (Fig. 1).
Behaviour of K2Q23K2Aβ30–42
Addition of the Aβ30–42 fragment to K2Q23K2 markedly increases its spontaneous aggregation, in a manner similar to the effect of HTTNT on polyQ sequences22. Thus, while 103 μM K2Q23K2 exhibits a long lag time before onset of aggregation with a t½ of aggregation of 207 h (Fig. 2a, black square), a similar concentration of K2Q23K2Aβ30–42 aggregates with a t½ of 1.9 h and no lag time (Fig. 2a, red circle). The concentration dependence of initial aggregation rates (Supplementary Fig. 1) in a log–log plot (a way to dissect some nucleation mechanisms5) gave a slope of 1.1 (Fig. 2b; Table 1) for this peptide. This is in the same range as the signature shallow slope exhibited by HTTNT-polyQ peptides22 and contrasts with the slope of 5.9 (Table 1) for K2Q23K2 itself that translates to a critical nucleus (n*) of 4 (ref. 8). In further contrast with K2Q23K2, which exhibits no detectible aggregates by DLS or EM during the lag phase8, K2Q23K2Aβ30–42 at very early time points generates aggregates with the size, shape and staining characteristics of spherical oligomers and protofibrils as described for Aβ42 (ref. 18). Thus, in a time frame where about 15% of the reaction mixture can be sedimented by centrifugation (Fig. 2a, red circle), we observed relatively small particles by EM (spherical oligomers of 10–20 nm; Fig. 2d) and DLS (∼60 nm; Fig. 2c and Supplementary Fig. 2).
Figure 2. Aggregation of K2Q23K2Aβ30–42.

(a) Aggregation assessed by disappearance of monomer for 37 °C incubation in PBS of 87 μM K2Q23K2Aβ30–42 (red circle) compared with 103 μM K2Q23K2 (black square). (b) Log–log plot of initial aggregation rates of K2Q23K2Aβ30–42 at various starting concentrations (Table 1 and Supplementary Fig. 1a). (c) Particle histograms from DLS analysis at different incubation times of K2Q23K2Aβ30–42 reaction described in a (Supplementary Fig. 2a). (d–i) TEM images (scale bar, 50 nm) of incubation of K2Q23K2Aβ30–42 at 37 °C in PBS at either 87 μM (d, 30 min; e, 90 min; f, 180 min; g, 2,880 min) or 6 μM (h, 2 h; i, 5 h). (j) Time-dependent changes in secondary structure content from CD analysis (Supplementary Fig. 3a) of reaction mixtures of the incubation of 6 μM K2Q23K2Aβ30–42 at 37 °C in PBS (120 h, anti-parallel β-sheet=41%, parallel β-sheet=0%); (k) aggregation kinetics of the reaction described in j as assessed by changes in monomer concentration (black square) and weight-normalized ThT fluorescence (AWN-ThT) (red circle). The grey-shaded bar spanning j and k highlights the time domain in which the α-helix content increases while ThT fluorescence is nil.
Table 1. Amyloid nucleation and stabilization data.
To more closely dissect the reaction time course of K2Q23K2Aβ30–42, we slowed the kinetics using a 6 μM starting concentration. In this and other assembly reactions reported here, we exclusively used our adjusted measure of ThT fluorescence, an ‘aggregate-weight-normalized (AWN)' ThT intensity, which is the raw ThT fluorescence units divided by the mole fraction of starting monomer that is in pelletable aggregates at each time point18,38 (Methods). We found that K2Q23K2Aβ30–42 undergoes a rapid drop in monomer concentration within the first 2 h of incubation, after which there is little further change in monomer concentration up to 10 h (Fig. 2k, black square). Interestingly, aggregates formed up to this point possess essentially no amyloid-like β-structure according to their AWN-ThT values (Fig. 2k, red square). Within this initial 10 h period EM images show a transition from initial spherical oligomers (2 h, Fig. 2h) to filaments (5 h, Fig. 2i). At around 10 h monomer concentrations once again begin to decrease, and this is accompanied by a gradual increase in the AWN-ThT fluorescence (Fig. 2k). At about 90 h there is a plateau in both the monomer loss curve and the AWN-ThT curve (Fig. 2k), suggesting that the assembly reaction has come to a final equilibrium position. It is important to note that the very gradual rise in AWN-ThT is not necessarily expected. If the onset of further aggregation at 10 h involved formation of a homogeneous suspension of mature amyloid as the only aggregate present, the AWN-ThT should have immediately climbed to a value at or near that of mature fibrils as seen at 120 h. That this did not happen suggests a very gradual transition within a heterogeneous suspension of aggregates through a series of species with ever increasing levels of amyloid-like β-structure (Discussion). CD spectroscopy of reaction mixtures cannot distinguish between monomers and aggregates in suspension, but does confirm a gradual increase in β-structure in the entire reaction mixture from 10 h to the end of the reaction (Fig. 2j; Supplementary Fig. 3). Importantly, total α-helix content of the aggregation reaction increases to a maximum value in the first 10 h of the reaction (shaded grey bar), then decays sharply over the next 10–20 h (Fig. 2j). This rise and fall of α-helix content, which perfectly coincides with the ThT-negative phase of aggregation (Fig. 2k), is similar to what was previously observed for spontaneous assembly of Aβ itself16.
Behaviour of K2Q23K2Aβ30–40
It is a common observation that Aβ40 is much less amyloidogenic than Aβ42 (refs 15, 39). To test if our model system might replicate this trend, we studied the K2Q23K2Aβ30–40 peptide. For peptides incubated at ∼100 μM, we found a t½ for spontaneous aggregation of K2Q23K2Aβ30–40 of 94 h (Fig. 3a, red circle), in contrast to 207 h for K2Q23K2 (Fig. 3a, black square). This rate increase of only about twofold is markedly smaller than the 100-fold increase conferred by the Aβ30–42 segment (Fig. 2a). In addition, the slope of the log–log plot for K2Q23K2Aβ30–40 is 2.9 (Fig. 3b, Table 1), inconsistent with an oligomer-mediated amyloid assembly reaction22 but consistent with a classical nucleated growth polymerization reaction with n*=1 (ref. 8). (This nucleus size contrasts with that of K2Q23K2 itself, which gives n*=4 (Table 1)8. However, small structural changes in this transitional repeat length range8 have been shown to greatly affect the size of the critical nucleus40.) Like K2Q23K2 (ref. 8), and unlike HTT exon1-like fragments22 and K2Q23K2Aβ30–42 (Fig. 2), K2Q23K2Aβ30–40 incubated at 112 μM exhibits no evidence by EM (Fig. 3d,e) or DLS (Fig. 3c) of any particles prior to the onset of formation of sedimentable, fibril-like particles (Fig. 3f) at ∼1 day incubation time. These trends are mirrored in the AWN-ThT values that rapidly increase around 10 h, when sedimentable aggregates just begin to appear (Fig. 3i, dashed line). In contrast to K2Q23K2Aβ30–42, K2Q23K2Aβ30–40 exhibits no evidence of α-helix in this time frame, but rather undergoes small increases in β-sheet and β-turns (Fig. 3h). At 5 days, when the aggregation reaction is 70% complete by the sedimentation assay (Fig. 3i), the β-sheet plus β-turn content of the reaction mixture reaches about 40%. This implies that the mature amyloid fibrils that have formed by five days (as evidenced by the nearly maximized AWN-ThT value) should possess about 60% of the maximum possible β-structure, consistent with a peptide whose 23 Gln residues account for 61% of the total residues. Together these data paint a picture of a polyQ-containing peptide that aggregates very much like simple polyQ peptides. Thus, the two C-terminal residues of Aβ30–42 are responsible for a qualitative difference in aggregation rates and aggregation mechanisms in these polyQ fusion peptides.
Figure 3. Aggregation of K2Q23K2Aβ30–40.

(a) Aggregation assessed by disappearance of monomer for 37 °C incubation in PBS of 108 μM K2Q23K2Aβ30–40 (red circle) compared with 103 μM K2Q23K2 (black square). (b) Log–log plot of initial aggregation rates of K2Q23K2Aβ30–40 at various starting concentrations (Table 1 and Supplementary Fig. 1b). (c) Raw DLS data at different incubation times in the incubation of K2Q23K2Aβ30–40 at 112 μM in PBS at 37 °C (data indicated no oligomeric particles at 1–2 h and no interpretation at 24 h due to large highly scattering particles). (d–g) TEM images (scale bar, 50 nm) from the reaction described for c (d, 1 h; e, 2 h; f, 23 h; g, 432 h). (h) Time-dependent changes in secondary structure content from CD analysis (Supplementary Fig. 3b) of the reaction described in c (360 h, anti-parallel β-sheet=31%, parallel β-sheet=0%). (i) Aggregation kinetics of the reaction described in c as assessed by changes in monomer concentration (black square) and aggregate weight-normalized ThT fluorescence (AWN-ThT) (red square). The grey dashed line spanning h and i highlights the time just before substantial sedimentable aggregate begins to form; at this time, weight-normalized ThT for existing aggregates is already high, and total α-helix content is negligible.
Behaviour of K2Q23Aβ25–42
We found that K2Q23Aβ25–42 behaves similarly to K2Q23K2Aβ30–42, while exhibiting somewhat faster amyloid growth rates. Thus, even while incubated at less than half the starting concentration, 42 μM K2Q23Aβ25–42 exhibits a t½ for aggregation that is ∼200 times lower than that for 103 μM K2Q23K2 (Fig. 4a). Like K2Q23K2Aβ30–42, it appears to form amyloid via a pathway featuring oligomeric intermediates, as indicated by its log–log slope of 1.9 (apparent n*=−0.1) (Fig. 4b, Table 1), and by the appearance of light scattering (Fig. 4c), oligomeric (Fig. 4d,e,g–i) species at early time points. Similarly to K2Q23K2Aβ30–42, even at a very low concentration of 3.4 μM it exhibits formation within the first 5 h of sedimentable (Fig. 4k, black square), oligomeric (Fig. 4g–i) aggregates that are ThT-negative (Fig. 4k, red square) and rich in α-helix (Fig. 4j). After a rapid drop in α-helix and gain in β-structure that is complete at ∼7 h (Fig. 4j,k; grey bar), a second phase of the aggregation reactions begins featuring steadily increasing AWN-ThT (Fig. 4k) and β-sheet (Fig. 4j). Secondary structure compositions, sedimentable monomer, and AWN-ThT all reach plateaus at ∼45–60 h (Fig.4j,k). The small 8% amplitude (compared with a 10% baseline) of biphasic α-helical changes reaching a maximum at 6–8 h is likely entirely associated with the oligomeric fraction. Since the Aβ segment accounts for 40% of the K2Q23Aβ25–42 residues, and since 20% of the molecules in the reaction mixture are assembled into sedimentable oligomers at 6–8 h (Fig. 4k, black square), the entire reaction mixture at this time should exhibit 8% α-helix attributable to transient oligomer formation—as observed.
Figure 4. Aggregation of K2Q23Aβ25–42.

(a) Aggregation assessed by disappearance of monomer for 37 °C incubation in PBS of 42 μM K2Q23Aβ25–42 (red circle) compared with 103 μM K2Q23K2 (black square). (b) Log–log plot of initial aggregation rates of K2Q23Aβ25–42 at various starting concentrations (Table 1 and Supplementary Fig. 1c). (c) Particle histograms from DLS analysis at different incubation times of 25 μM K2Q23Aβ25–42 in PBS at 37 °C (Supplementary Fig. 2b). (d–i) TEM images (scale bars, 50 nm) of incubation of K2Q23Aβ25–42 at 37 °C in PBS at either 25 μM (d,e 60 min; f, 1,200 min) or 3.4 μM (g, 15 min; h, 45 min; i, 2.7 h). (j) Time-dependent changes in secondary structure content from CD analysis (Supplementary Fig. 3c) of reaction mixtures of the incubation of 3.4 μM K2Q23Aβ25–42 at 37 °C in PBS (92 h, anti-parallel β-sheet=41%, parallel β-sheet=0%). (k) Aggregation kinetics of the reaction described in j as assessed by changes in monomer concentration (black square) and aggregate weight-normalized ThT fluorescence (AWN-ThT) (red square). The grey-shaded bar spanning j and k highlights the time domain in which the α-helix content increases while ThT fluorescence is nil.
Robustness of α-helix detection
The behaviour of these three peptides is consistent with a model in which the Aβ30–42 segment, but not the Aβ30–40 segment, triggers formation of amyloid fibrils by mediating rapid and transient formation of α-helical oligomers. The data supporting transient α-helix, however, derives from deconvolution of CD spectra, which can be prone to a number of possible fitting artefacts, such as from inaccurate concentration determination and from shifts in CD spectra due to light scattering (LS). It is therefore important to examine the robustness of the evidence for transient α-helix in the hybrid peptides.
We believe our method for analytical HPLC determination of peptide concentrations41 used to set-up the aggregation reactions is highly accurate. Even if the concentrations of the starting reaction mixtures were imprecise, however, it could not produce the kind of waxing and waning of α-helix we see in the peptide aggregation time courses, since the same (starting) concentration value is used to calculate molar ellipticities at all reaction time points.
Although severe LS can distort CD curves and make deconvolution unreliable, we also do not believe LS is producing artifacts in our experiments. First, we have observed that on a weight concentration basis mature polyQ amyloid gives relatively low turbidity/LS compared with other amyloids. In fact, it is sufficiently low that it allows accurate deconvolution of secondary structure in mature polyQ amyloid that reveals its independently characterized26 anti-parallel β-sheet structure42. Similarly, the anti-parallel basis of the polyQ cores is consistently revealed in the mature amyloids of the three hybrid Aβ-polyQ peptides in spite of some LS in the sample (Figs 2, 3, 4, legends). Second, the burst of α-helix observed here occurs very early in the aggregation reactions at times when LS is much lower than in the final reaction mixtures. Third, increases in β-structure estimated from CD deconvolution closely parallel increases indicated by AWN-ThT values (Figs 2, 3, 4). Fourth, estimates of the percentages of β-structure in the mature amyloids are very consistent with calculated percentages based on independently determined weight concentrations of aggregates and percentages of peptide sequences found in the polyQ amyloid cores. Fifth, samples of the freshly dissolved Aβ C-terminal fragment K4G2Aβ30–42 at intermediate concentrations exhibit a mixture of disorder and α-helix in the CD, consistent with the ability of C-terminal Aβ peptides to readily access mixtures of disordered monomer and α-oligomers (Supplementary Fig. 4). Sixth, at higher concentrations where α-oligomer formation leads to nucleation of amyloid-like structure, K4G2Aβ30–42 exhibits similar mixtures of α-helix and β-sheet as assessed by both CD and FTIR (Supplementary Fig. 4), consistent with accurate assessment of secondary structure levels from CD deconvolution even in the presence of LS. Seventh, CD deconvolution of this peptide is so accurate that it reveals that the β-sheet component is anti-parallel β-sheet, as expected from FTIR analysis of mature Aβ31–42 fibrils (Supplementary Fig. 4). Eighth, FTIR spectra of inclusion bodies from bacterial expression of Aβ indicate substantial α-helical content, and solid state NMR spectrometry and hydrogen–deuterium exchange indicate coiled coil helix involving the Aβ C terminus43.
Aggregate structure
According to our hypothesis and designs of the peptide models, we expected the Aβ segments of the hybrid peptides to not be involved in amyloid core formation. FTIR spectroscopy showed that the mature amyloid fibrils of all three hybrid peptides exhibit typical polyQ amyloid second derivative spectra (Fig. 5a), and CD deconvolution showed that the β-sheets in the mature fibrils are anti-parallel (Figs 2, 3, 4 legends), as expected for a polyQ core. Since the typical α-helix band in FTIR overlaps the characteristic polyQ amyloid band at ∼1,657 cm−1 (ref. 23), it is not expected that we might observe any α-helical structure in these fibrils that might be analogous to the HTTNT α-helix in HTT exon1 fibrils25. To address whether a portion of the Aβ segment might be involved in stable β-structure in the fibrils, we used hydrogen–deuterium exchange evaluated by mass spectrometry (HX-MS). As shown previously18, in mature amyloid fibrils of Aβ42, the C-terminal fragment starting at residue 35 resists exchange, leading to a 35–42 fragment that, after exposure of fibrils to D2O, has a mass distribution (Fig. 5d, blue) that is much more like that from protonated Aβ42 monomers (Fig. 5d, black) and quite unlike the fragment from fully deuterated Aβ42 monomers (Fig. 5d, red). This behaviour contrasts with that of Aβ40 fibrils exposed to D2O, which release a C-terminal 35–40 fragment that is essentially fully exchanged (Fig. 5e) (ref. 18). We found that in the mature fibrils of all three polyQ-Aβ hybrid peptides, the 35–42/35–40 fragments released from fibrils after exchange (blue) resemble the corresponding fragment from deuterated monomers (red), and therefore are essentially fully exchanged (Fig. 5b,c,f). This suggests that the Aβ segments are not involved in amyloid core structure. The absence of a large amount of β-structure within the Aβ segment of the polyQ-Aβ hybrid fibrils is consistent with the total β-structure content of about 50% seen in the CD spectra of the final amyloid suspensions (Figs 2j, 3h and 4j) of peptides containing about 50% Gln residues. Given the ability of Aβ C-terminal peptide fragments ending at residue 42 to assemble into amyloid fibrils (Supplementary Fig. 4), it is remarkable that the same sequence appended to polyQ resists adopting highly protective β-secondary structure within the Aβ component in these hybrid peptide fibrils. This is may be due to topological or other structural restrictions imposed by details of the polyQ amyloid core.
Figure 5. Structural features of mature amyloid products.

(a) Second derivative FTIR spectra of amyloid products showing the triplet of bands characteristic of polyQ amyloid23. (b–f) Mass/charge distribution from HX-MS analysis of the C-terminal fragment generated by pepsin cleavage between Leu34 and Met35 of various peptides (protonated monomer, black line; deuterated monomer, red line; Gdn-HCl dissolved deuterated fibrils, blue line; aggregates formed in presence of K10G2Aβ31–42, magenta line): (b). K2Q23K2Aβ30–42; (c) K2Q23K2Aβ30–40; (d) Aβ42; (e) Aβ40; (f) K2Q23Aβ25–42.
We also addressed whether the Aβ appendages of these hybrid peptides provide any additional stability to the Q23 amyloid cores, in analogy to the stabilizing effect of HTTNT on HTT exon1 amyloid27. Stabilities were assessed by determining the concentration of monomer present when the amyloid assembly reaction reaches equilibrium (Supplementary Fig. 5)41. The lower the Cr value, the more stable the fibrils with respect to monomers in solution. The results (Table 1) indicate some interesting trends. First, we found that the Cr value of 3.6 μM for K2Q23K2Aβ30–40 is essentially the same as the value of 3.0 μM previously reported for K2Q23K2, suggesting that the Aβ30–40 sequence does not provide stabilizing interactions within the fibrils. This is in stark contrast to the Aβ peptides ending at residue 42. The Cr for K2Q23K2Aβ30–42 is 0.42, or about sevenfold lower than that of K2Q23K2, and the Cr for K2Q23Aβ25–42 is 0.08, almost 40-fold lower than that of K2Q23K2. Interestingly, these stabilizations by Aβ fragment flanking sequences are similar to those obtained for Q23 peptides in HTT exon1 flanking sequence contexts (Table 1)27, and the ∼9-fold difference between Cr values of K2Q23K2Aβ30–40 and K2Q23K2Aβ30–42 is remarkably similar to the ∼10-fold difference in the Cr values of Aβ40 (1.06±0.18 μM;38) and Aβ42 (0.11±0.01 μM;18) fibrils. These similar relative stabilities of mature fibrils are achieved in spite of radically different structural roles for the C-terminal segments in fibrils of Aβ compared with polyQ-Aβ hybrids.
A probe of mechanism using the Aβ31–42 fragment
In the HTT system, addition of isolated HTTNT peptides to aggregation reactions modestly delays the onset of amyloid formation by HTT exon1-like fragments28. This appears to involve dilution of local polyQ concentrations when mixed α-oligomers are formed between HTT exon1-like fragments and the HTTNT sequence (compare Fig. 1d with Fig. 1c). We set out to investigate whether an appropriate Aβ C-terminal fragment might act similarly to delay aggregation onset by the hybrid Aβ-polyQ peptides. First, we attempted to improve the poor kinetic solubility of the Aβ31–42 fragment44 by adding a series of Lys residues to its N terminus to generate K10G2Aβ31–42 (Fig. 1f).
On physical characterization of this peptide, we found that at all concentrations tested, K10G2Aβ31–42 incubated alone leads to rapid formation of a small percentage of sedimentable aggregates, after which there is a concentration-dependent further aggregation that is negligible, over the first 75 h, at concentrations of 10 μM or less (Supplementary Fig. 6a). At 22 μM after 24 h, aggregate suspensions exhibit negligible ThT fluorescence (Supplementary Fig. 6b), overall 22% α-helix (Supplementary Fig. 6c) and a uniform distribution of spherical oligomers of 20–30 nm radius (Supplementary Fig. 6d; 2 h). After 24 h, the AWN-ThT signal increases steadily until reaching a maximum at 7 days (Supplementary Fig. 6b), suggesting a homogeneous β-aggregate which, at 23 days, is completely fibrillar (Supplementary Fig. 6d; 23 days). Since after 35 days ∼13 μM of monomer remain in the reaction mixture (Supplementary Fig. 6b), this peptide's fibrils likely have a Cr value in the 10–13 μM range. The data suggest that K10G2Aβ31–42 itself undergoes an oligomer-mediated amyloid initiation mechanism that is unfavourable at concentrations of 10 μM or less but which advances slowly at ∼20 μM. This solubility behaviour revealed a limited window of concentrations that can be used to test K10G2Aβ31–42 as an inhibitor, above which the inhibitor itself forms fibrils.
We found that K10G2Aβ31–42 induces a modest delay in the onset of aggregation in both K2Q23K2Aβ30–42 and K2Q23Aβ25–42, but not K2Q23K2Aβ30–40. Thus, at molar ratios of 1.2 and 1.8, K10G2Aβ31–42 shifts the aggregation curves of K2Q23K2Aβ30–42 and K2Q23Aβ25–42 by 15 h (Fig. 6a) and 7 h (Fig. 6c), respectively. Consistent with our model, these delays are associated with initial formation of sedimentable aggregates that exhibit very low AWN-ThT signals (Supplementary Fig. 7a,d). The ability to delay, but not eliminate, onset of amyloid formation is very similar to the action of HTTNT against HTT exon1-like peptides28. In contrast to the hybrid peptides ending at Aβ residue 42, a 1.4:1 ratio of K10G2Aβ31–42 to K2Q23K2Aβ30–40 neither significantly shifts the latter's aggregation curve (Fig. 6b) nor eliminates the immediate ThT-positive content of its aggregates (Supplementary Fig. 7b). Although the inhibition we observed in these experiments was modest, the observed inhibition further supports a role for α-helical, ThT-negative oligomers in fibril formation by K2Q23K2Aβ30–42 and K2Q23Aβ25–42.
Figure 6. Inhibition of spontaneous amyloid formation by K10G2Aβ31–42.

Results of sedimentation-HPLC assays on mixtures of Aβ-related peptides and K10G2Aβ31–42. (a) K2Q23K2Aβ30–42 (6.4 μM) and K10G2Aβ31–42 (7.6 μM). (b) K2Q23K2Aβ30–40 (16.7 μM) and K10G2Aβ31–42 (23 μM). (c) K2Q23Aβ25–42 (3.9 μM) and K10G2Aβ31–42 (6.9 μM). (d) Aβ42 (3.1 μM) and K10G2Aβ31–42 (8.3 μM). Symbols: amyloidogenic peptide alone (black square); K10G2Aβ31–42 alone (pink inverted triangle); mixture of amyloidogenic peptide (red circle) and K10G2Aβ31–42 (blue triangle).
Spontaneous aggregation of Aβ42
Previously we were unable to tease out details of Aβ42 amyloid assembly at a starting concentration of 10 μM due to rapid onset of amyloid formation18. Here we used a substantially lower concentration that is at the same time still above the peptide's Cr of 0.11 μM (ref. 18) leading to a reaction that exhibits a good lag phase in the development of AWN-ThT, confirming an early non-β oligomeric intermediate (Fig. 7a). Under these conditions, we inquired whether K10G2Aβ31–42 might also inhibit spontaneous amyloid formation by Aβ42. In a reaction of 3.1 μM Aβ42 and 8.3 μM K10G2Aβ31–42, however, we observed little effect on aggregation kinetics as assessed by either the sedimentation-HPLC assay or by AWN-ThT (Fig. 7b). At the same time, Aβ42 grown in the presence of K10G2Aβ31–42 showed a markedly (∼10-fold) lower final AWN-ThT value (Fig. 7b), suggesting a substantially different end-stage aggregate morphology compared with normally produced fibrils. We confirmed this in a repeat study at 7.4 μM Aβ42 plus 11.5 μM K10G2Aβ31–42, which showed—compared with Aβ42 alone—larger initial oligomers (Fig. 7f versus Fig. 7c), a prolonged protofibril stage (Fig. 7g versus Fig. 7d), and a different mature fibrillar morphology (Fig. 7h versus Fig. 7e). The different structure in the mature fibrils was confirmed by FTIR, which showed a new, anti-parallel β-sheet band (Supplementary Fig. 8), and by HX-MS. For global HX, Aβ42 aggregated alone showed a typical18 incorporation of 14.8±1.2 deuteriums, while Aβ42 aggregates grown in the presence of K10G2Aβ31–42 showed 25.6±1.2 deuteriums. Many of the ∼11 newly unprotected amide hydrogens in the latter are located in the peptide's C terminus, as shown by segmental HX-MS (Fig. 5d, magenta line). We confirmed that the aggregates subjected to FTIR and HX-MS contained only Aβ42 (and not K10G2Aβ31–42 inhibitor) by HPLC of dissolved aggregates (Supplementary Fig. 8c).
Figure 7. Effect on Aβ42 amyloid formation by K10G2Aβ31–42.

(a,b) Kinetics of Aβ42 self-assembly at 37° in PBS, as assessed by decreases in monomer concentration (black square) and changes in aggregate weight-normalized ThT fluorescence (AWN-ThT) (red square), in reactions of 2.7 μM Aβ42 alone (a) or 3.1 μM Aβ42 mixed with 8.3 μM K10G2Aβ31–42 (b). (c–h). TEM images (scale bars, 50 nm) of reaction time points of 7.4 μM Aβ42 incubated alone (c–e) or with 11.5 μM K10G2Aβ31–42 (f–h), with time points at 1 h (c,f), 4 h (d,g) and 24 h (e,h).
Discussion
Our results are consistent with a HTT exon1-like amyloid assembly mechanism for the Q23-Aβ42 hybrid peptides (Fig. 8) that features rapid formation of transient oligomeric intermediates held together by non-amyloid-type contacts involving the Aβ segments. The resulting high local concentration of the tethered Q23 segments overcomes the high-concentration barrier for polyQ amyloid nucleation to initiate fibril growth. Although it can be challenging to obtain accurate secondary structures by deconvolution of CD spectra of protein aggregates, there are a number of reasons, outlined in the Results section, to trust the experimental indications that these transient oligomers are held together as bundles of α-helices composed of the Aβ C terminus.
Figure 8. Model for a modular role for the Aβ C terminus in amyloid initiation.

Monomeric peptides containing an Aβ C terminus ending at residue 42 (a) undergo rapid formation of a small, metastable population of oligomers held together by packing of α-helical Aβ C termini (b). The resulting high local concentration of amyloidogenic segments in these oligomers overcomes the concentration barrier leading to formation of β-rich amyloid intermediates (c). These progress, through a poorly defined combination of structural rearrangements and seeded elongation, to mature amyloid (d,e). For polyQ-Aβ hybrids, the resulting amyloid has an anti-parallel β-sheet polyQ core decorated with non-β Aβ segments that appear to interact within the fibril (d). For Aβ42, the resulting amyloid adopts parallel, in-register β-sheet that includes the Aβ C-terminal segment (e). Aβ C terminus (green), amyloidogenic polyQ and Aβ16–20 (orange), hydrophilic segments (see Fig. 1f) (blue).
The primary nucleation phase for amyloid formation by Aβ peptides is particularly important to understand for two reasons. First, and foremost, without primary nucleation amyloid assembly can never occur. Second, non-fibrillar, oligomeric intermediates forming early in Aβ amyloid assembly have been implicated as particularly toxic species associated with the mechanism of Alzheimer's disease45. Through the design and study of Aβ-polyQ hybrid peptides we specifically investigated the early steps of Aβ assembly leading to the initiation of amyloid formation. By replacing the LVFF segment of Aβ with a polyQ segment we expected the amyloid initiation phase of Aβ itself to be replicated, but the downstream consolidation of additional amyloid structure in the C terminus of Aβ to not to be replicated. In fact, the data on the three hybrid peptides support a model for the earliest steps of Aβ42 assembly that is largely identical to what we see for the two hybrid peptides ending at position 42 of Aβ. Thus, for both 6 μM K2Q23K2Aβ30–42 and for 3.4 μM K2Q23Aβ25–42 we observe formation of sedimentable aggregates that exhibit no ThT signal (Figs 2k and 4k) in exact analogy to the ThT-negative aggregates formed in the early phase of Aβ42 assembly (Fig. 7a). For the reactions of the two hybrid peptides this ThT-negative phase occurs precisely when CD analysis of the reaction mixtures reveal transient formation of α-helix (Figs 2j and 4j) and transient formation of spherical oligomers (Figs 2h and 4h). Similarly, transient α-helix16 and formation of spherical oligomers2 (Fig. 7c) have been observed early in the self-assembly of Aβ peptides. As expected for the proposed nucleation mechanism28, we also observed modulation of the self-assembly of both Aβ42 (Fig. 7) and the hybrid peptides (Fig. 6) by the presence of an Aβ31–42-related peptide. Finally, the markedly different assembly kinetics of the model hybrid peptides K2Q23K2Aβ30–40 (Fig. 3a) and K2Q23K2Aβ30–42 (Fig. 2a) beautifully reproduce the well-known kinetic difference in the amyloid assembly of Aβ40 and Aβ42 (ref. 39), while suggesting that these two highly related Aβ peptides undergo substantially different mechanisms for initiation of amyloid formation.
Thus, our results for Aβ-polyQ hybrids support an analogous nucleation mechanism for longer versions of Aβ itself, in which the Aβ C terminus initially plays a similar role of mediating α-helical oligomer formation before this segment later becomes swallowed up in the amyloid core. Based on solution NMR analysis of Aβ42 monomers that found no evidence of any residual secondary structure in this peptide14, Bax and colleagues recently proposed that early aggregation intermediates must be assembled via hydrophobically driven self-interactions of peptide segments in irregular structure. While our data on Aβ42 concur that the first formed oligomers do not involve amyloid-like β-structure18 (Fig. 7), based on the polyQ-Aβ hybrid data provided here as well as previous data on Aβ assembly16 we think it is likely that the early oligomers are clusters of α-helical elements. In fact, there is a precedent in our HTT exon1 results for a peptide exhibiting very little α-structure as a monomer that nonetheless forms robust α-helix rich assembly intermediates22,23,28. Such counter-intuitive phenomena seem possible, for example, if there are levels of helical peptides in the monomer ensemble that are below NMR detection limits but sufficient to support helical bundle assembly. Alternatively, helical bundles might develop by concerted mechanisms in which peptides acquire helical structure during self-assembly46. In analogy to the proposed involvement of α-helix rich oligomers in an amyloid nucleation mechanism, some β-sheet rich globular proteins are known to acquire their native structures via α-helix-rich folding intermediates47,48.
Our data strongly suggest that the two C-terminal residues of Aβ42 enhance amyloid formation kinetics compared with Aβ40 by favouring the formation of α-helical oligomeric intermediates that serve as the phase within which a particularly efficient amyloid nucleation occurs. A separate segment of Aβ—probably centred in the LVFF region—acts as the driving amyloidogenic segment involved in this second step. In our polyQ-Aβ model peptides, the mature amyloid core structure appears to be confined to the polyQ segment, while the Aβ portion does not possess highly protective backbone H-bonded structure. We propose that in Aβ42 itself, a similar early β-intermediate is formed featuring amyloid-like structure only in the central region of the peptide including the LVFF segment (Fig. 8). Interestingly, a highly similar structural model featuring β-structure in the LVFF segment and α-structure in the C terminus (Fig. 1e) has been deduced for Aβ in inclusions bodies formed in bacteria, which lead to the suggestion that such a structure might be relevant to the normal amyloid assembly mechanism43. The mechanism and timing of the ‘finishing steps' required to move from such early β-intermediates to mature amyloid, however, are not immediately obvious.
Some insights into these finishing steps are offered by the AWN-ThT kinetics. In the model peptide K2Q23Aβ25–42, the period featuring the initial rise and fall of α-structure and the lag in ThT is over at 7 h, after which both total β-structure and AWN-ThT begin to rise inexorably (Fig. 4j,k). However, the subsequent trend in AWN-ThT values does not resemble that expected for a simple nucleated growth process. That is, in a classical nucleated growth polymerization mechanism, where the product of nucleus formation and initial elongation is immediately amyloid-like, the AWN-ThT is expected to jump to a high value immediately after nucleation (Supplementary Fig. 7b,e; Fig. 3i). In contrast, the AWN-ThT for K2Q23Aβ25–42 trends upward very slowly and gradually during amyloid assembly, beginning after α-structure is lost. Thus, although there is a rapid loss of α-helix from 4 to 8 h (Fig. 4j), the commensurate immediate gains in total β-structure (Fig. 4j) and AWN-ThT (Fig. 4k) are small and inconsistent with the formation of mature fibrils in the ∼40% of Aβ molecules that are aggregated at this time (Fig. 4k). In fact, AWN-ThT develops very slowly over the entire course of the reaction, only reaching a maximum value after ∼90% of the monomers have been consumed. At the same time, the coincidence between the sharp drop in α-helix and the onset of AWN-ThT at ∼7 h, in addition to the response by the hybrid peptides to the inhibitor K10G2Aβ31–42, supports the view that the α-intermediates play an early on-pathway role in amyloid assembly.
Amyloid assembly of Aβ42 itself follows a similar trend. In the first 2 h of incubation, before AWN-ThT begins to rise, ∼45% of monomers have been converted to aggregates (Fig. 7a). At the end of the AWN-ThT lag, there are no rapid bursts either in the rate of overall aggregation or in the magnitude of AWN-ThT as would be expected from a group-wise conversion to mature amyloid nuclei. Instead, from 2 to 8 h another 45% of the monomer pool is converted relatively slowly to the aggregate fraction, with a corresponding very gradual rise in AWN-ThT. There are several ways these data can be rationalized. It is possible that a small percentage of the conversion of α-oligomers marking the end of the null AWN-ThT phase might be associated with the formation of mature amyloid nuclei, whose elongation then takes place ‘under the radar' of the ensemble AWN-ThT fluorescence. This low level elongation activity might occur via growth supported either by the still substantial monomer pool (that is, a classical nucleated growth polymerization) or by some population of the multitude18 of oligomer types that persist after the decay of α-oligomers (that is, nucleated conformational conversion). Alternatively, the group-wise conversion of α-helical oligomers to aggregates with modest levels of β-structure might be the first in a series of group-wise changes, each of which is associated with a stabilizing increase in amyloid-like β-structure and therefore AWN-ThT, culminating in a final remodelling step into mature amyloid. The latter mechanism is consistent with reports that protofibrillar intermediates exhibit apparent β-structure in the same Aβ segments that are in β-sheet in mature fibrils21,49. Finally, the complex waxing and waning of various oligomeric intermediates might even be an off-pathway smokescreen masking the operation of a classical nucleated growth polymerization mechanism10—a mechanism that might become especially important only at lower Aβ concentrations.
In additional support of the model for how the Aβ C terminus mediates α-oligomer formation to initiate the amyloid assembly pathway, we found that K10G2Aβ31–42 delays onset of β-aggregation by K2Q23K2Aβ30–42 and K2Q23Aβ25–42 (which exhibit transient α-helix) but not by K2Q23K2Aβ30–40 (which does not). Against Aβ42 itself, K10G2Aβ31–42 has little effect on the kinetics of aggregation (Figs 6d and 7b). The reaction mixture passes through a stage with intermediate AWN-ThT values apparently derived from protofibril-like structures (Fig. 7g). The reaction proceeds to a final aggregate structure resembling the previously described D polymorph of Aβ40 fibrils38 in its EM morphology (Fig. 7h), low AWN-ThT value (Fig. 7b), and decreased protection against HX (Fig. 5d). Given our model for how K10G2Aβ31–42 might interact with Aβ42 during assembly, this result suggests that the ability of a peptide to assemble into a particular polymorphic form might be determined very early in the assembly process50, and thus that mechanisms such as the one investigated here might account for some polymorphic Aβ fibrils and not for others. The Aβ42 aggregates produced in the presence K10G2Aβ31–42 also resemble the assembly product of Aβ42 co-incubated with randomly generated peptides designed to form amphipathic α-helices51. This suggests a degree of promiscuity in the assembly of α-helical oligomeric intermediates that is very similar to what we previously described for HTTNT inhibition of HTT exon1 aggregation28. C-terminal fragments of Aβ have been shown previously to inhibit Aβ cytotoxicity52, and these effects may well be related to the influence of such fragments on amyloid assembly kinetics and/or product structures.
Segmental mobility within intrinsically disordered proteins offers the theoretical possibility that through-space, long-range interactions between segments might play a role in defining intrinsically disordered protein global physical properties. Although it is difficult to establish such relationships experimentally, computations suggest that just such a process—rather than the modular effect invoked here—contributes to some aspects of the ability of HTTNT to influence polyQ aggregation behaviour53. Further, the failure of one experimental test54 of the modular ‘local concentration' mechanism might be viewed as indirect support for such an integrative interaction mechanism. At the same time, the mechanistic alternative—the ability of a protein segment to behave as a quasi-independent unit to act in a modular manner to impact an aggregation mechanism—continues to attract strong experimental support. Besides the data on the Aβ C terminus reported here, supportive data have been previously reported for modular roles of HTTNT (refs 22, 23, 24, 27, 28, 55, 56, 57), the Josephin domain of ataxin 3 (ref. 58) and the CRABP domain59 in protein aggregation mechanisms. The ability of HTTNT, as well as Aβ30–42 in a polyQ-Aβ hybrid, to bestow a large rate enhancement on spontaneous amyloid formation without itself being incorporated into the final amyloid core26, is relatively unusual in the amyloid literature. However, local concentration effects within rapidly formed non-β oligomers quite likely also contribute to some amyloid assembly reactions in which the segment mediating non-β oligomer assembly ultimately becomes incorporated into the amyloid core60, as suggested here and elsewhere19 for Aβ42.
In addition to implicating the C terminus of Aβ in the formation of on-pathway α-helical oligomeric intermediates that initiate amyloid formation, our data provide additional insights into the self-assembly process. For example, the relatively rapid initial formation of α-oligomers in both the polyQ-Aβ42 hybrids and in Aβ42 itself suggests that these initial aggregated structures are formed by a non-nucleated, energetically downhill process6. Our AWN-ThT data also suggest that α-intermediates do not directly convert en masse into rapidly elongating amyloid nuclei, but rather convert into immature protofibrillar intermediates exhibiting partial amyloid-like β-structure which continue to mature over much of the reaction coordinate in parallel with the continuing loss of monomeric Aβ from solution. That is, elongation seeded by mature fibrils, which normally is viewed as contributing the bulk of the magnitude of the ThT fluorescence of spontaneous amyloid growth, likely only accounts for the last ∼10% of the aggregation time course (Figs 2k, 4k and 7k). This would appear to rule out a major role for secondary nucleation processes5,61 in spontaneous Aβ amyloid assembly, at least under our experimental conditions. As recently suggested18, the AWN-ThT time course of Aβ42 reveals a rich menagerie of transient and rapidly interconverting aggregated species, from ThT-negative oligomers through a number of ThT-positive intermediates, any one of which might prove to be the critical aggregate responsible for AD toxicity. While β-oligomer forms of Aβ previously have received much attention in this regard, the possible contribution to cytotoxicity by more ephemeral α-oligomers remains to be explored.
Methods
Peptides
The peptides K2Q23K2Aβ30–42, K2Q23K2Aβ30–40 and K2Q23Aβ25–42 (calculated and observed monoisotopic masses of 4,670.467/4,670.466, 4,486.346/4,486.338 and 4,857.490/4,857.520, respectively) were obtained from Keck Biotechnology Center at Yale University ( http://info.med.yale.edu/wmkeck/). K4G2Aβ31–42 and K10G2Aβ31–42 (calculated and observed MWs of 1,768.27/1,768.35 and 2,537.31 and 2,537.40, respectively) were obtained from GenScript, Inc. These were obtained in crude form and purified as follows. After dissolution in 98% formic acid (Sigma) with sonication for 2 min in a bath sonicator, peptides were injected in a reverse phase HPLC column (9.4 × 250 mm Agilent Zorbax SB-C3) and eluted with 30–70% gradient of acetonitrile in water, with 0.05% trifluoroacetic acid (TFA, Pierce). Fractions with purities (LC-MS detector) exceeding 90% were pooled and lyophilized. Aβ42 (calculated and observed monoisotopic mass of 4,514.08/4,513.81) was obtained pre-purified from the Keck Center. Peptides were routinely disaggregated before every experiment using either the Gdn-HCl/SEC method18 (Aβ42) or the mixed TFA:hexofluoroisopropanol (HFIP, Acros Organics) method41 (all other peptides).
Aggregation kinetics
Disaggregated peptide stock solutions were first checked for approximate concentration. All concentrations, for setting up the reaction and for assaying kinetics time points, were determined from integrated A214 HPLC (Agilent XDB-C18 4.6 × 50 mm column) peaks using standard curves individually constructed for each peptide from standard stock solutions calibrated via the A215 absorption23,41. Kinetics reaction mixtures were prepared by adding HPLC grade water to an aliquot of the peptide stock solution, plus a 1/9 volume of 10 × PBS buffer to give a final 1 × PBS. Sodium azide was added to 0.05% (w/v) and pH adjusted to 7.4. After filtration through a 0.2 μm filter unit (Anotop 10) to get rid of aggregates, the solution was held at 4 °C while the t=0 concentration was determined (∼30 min). The reaction mixture was moved to 37 °C and kinetics monitored by the sedimentation and ThT assays on reaction aliquots and by CD.
Sedimentation assay
Duplicate time points were removed periodically, centrifuged 30 min at 436,000g followed by HPLC analysis of monomer concentration from the carefully removed supernatant41. Nucleation kinetics determination was carried out by evaluating initial time points of aggregation reactions with varied starting concentrations, as described7,8. Critical concentrations were determined from the average of the end point monomer concentrations of amyloid formation and dissociation reactions as described41. (See Supplementary Fig. 5).
AWN-ThT assay
The ThT assay was performed by taking aliquots from the aggregation reaction mixtures containing 1.0 μg of total peptide and transferring to a 4 mm × 4 mm cuvette containing 20 μM ThT (Sigma) in 1 × PBS. Fluorescence was measured over a 2 min integration period using excitation wavelength at 445 nm (5 nm slit width) and emission at 489 nm (10 nm slit width) in a Fluoromax-4 spectrofluorometer (Horiba Jobin Yvon). AWN-ThT values were calculated for each time point by (a) subtracting the buffer+ThT blank, then (b) dividing this value by the mole fraction of peptides that were independently (by the HPLC sedimentation assay above) determined to be aggregated, according to the equation:
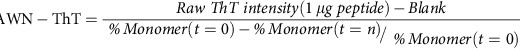 |
Circular dichroism spectroscopy
Reactions to be studied in parallel by CD, sedimentation, and ThT were prepared in sufficient starting volume to allow aliquots to be taken for CD measurements. We chose to incubate these under exactly the same conditions as the other kinetics (rather than in the CD cuvette) in order to avoid differences due to possible surface effects. Aliquots were removed at different times and transferred to a 0.1 cm path length cuvette, and then the far-ultraviolet CD spectrum obtained on either a JASCO J-810 or OLIS 17 spectropolarimeter. Spectra were collected at 0.5 nm resolution with a scan rate of 100 nm min−1 (averaged over three scans) and corrected for the buffer signal. Secondary structure percentages were calculated from these spectra over the range 190–250 nm using the freely accessible algorithm BeStSel (bestsel.elte.hu), which includes independent basis spectra for both parallel and anti-parallel β-sheet42.
Dynamic light scattering
DLS measurements were performed using a DynaPro plate reader (Wyatt Technology Corporation). An aliquot (∼80 μl) was transferred, after gentle mixing, from the ongoing aggregation reaction periodically to a fresh well of a 384-well microtitre plate, after which scattering data were obtained immediately. The intensity autocorrelation functions were analysed using a Dynamics V6 software to obtain the hydrodynamic radii (Rh).
Electron microscopy
Aliquots (∼8 μl) from the aggregation reaction at various time points were placed in a freshly glow-discharged carbon-coated 400 mesh size copper grids and allowed to adsorb for 90 s. The grid was washed with deionized water, stained with freshly filtered 8 μl of 1% (w/v) uranyl acetate for 30 s and then washed again with water. Each time, excess of sample, water and stain were blotted away with filter paper. Imaging was done using a Technai T12 microscope (FEI Company, Hillsboro, OR) operating at 120 kV with a magnification of × 30,000 and equipped with an Ultrascan 1000 CCD camera with post-column magnification of × 1.4.
FTIR spectroscopy
Aggregates were collected at the end of aggregation reaction by centrifugation at 14,000 r.p.m. for 30 min. The pellet is washed once (PBS plus centrifugation), resuspended in ∼50 μl of 1 × PBS, and the suspension placed between two polished CaF2 windows using a BioCell module (BioTools, Inc.) on an ABB Prota-2 × MB 3000 FTIR instrument. Spectra were collected at 4 cm−1 resolution (average of 400 scans) and corrected for buffer absorption until a flat baseline was obtained in the 1,700–1,800 cm−1 region. Second derivative spectra for the amide-I region were calculated using PROTA software.
Hydrogen exchange-mass spectrometry
All HX-MS data were analysed using an Agilent 1100 series single quadruple ESI mass spectrometer. Aggregates were washed and incubated in D2O and the exchange reactions analysed after an appropriate exchange time. Global HX was analysed by simultaneous dissolution and MS analysis of the exchange reaction using a T-tube front end to the MS62; data were not corrected for back-exchange. Segmental exchange was performed as described63. Briefly, fibrils were recovered from the exchange reaction by centrifugation (20,800g, 30 min), and in a 4 °C room to 20 μl ice-cold 8 M Gdn.HCl plus 0.1% formic acid was added to the pellets and mixed for 10 s, then 100 μl pepsin agarose (Sigma-Aldrich) in 0.1% cold formic acid was added and mixed for 10 s. The sample was immediately centrifuged to remove the immobilized pepsin (20,800g, 30 s) and the supernatant subjected to 4 °C LC-MS analysis.
Reproducibility and data analysis
In amyloid kinetics analyses, the greatest determinant of lab-to-lab reproducibility lies in the details of peptide purity and source, preparation8, concentration determination23 and kinetics analysis41. In particular, it is critical to carry out a completely fresh disaggregation reaction on the peptide immediately before initiating each reaction, and to follow recommended disaggregation protocols8,18,41 exactly and completely. If a protocol must be modified or streamlined, it is critical, before carrying out detailed analyses, to determine that the modification does not compromise the desired monomeric state by introducing oligomeric forms or otherwise affect overall peptide behaviour.
For sedimentation and ThT kinetics, as well as kinetics underlying the n* and Cr determinations and inhibition kinetics studies, each plotted data point is derived from analysis of 2–3 independent reaction aliquots. Each aggregation study was independently carried out twice, with similar results obtained. For the time2 plots underlying the n* log–log plots, only the early phase of the aggregation reaction is considered, as described previously8,41; this corresponds to the first 2 h for Figs 2b and 4b, and the first 48 h for Fig. 3b. DLS autocorrelations were carried out on averages of at least 15 data acquisitions. Analysis of CD curves to produce time courses of secondary structural changes was accomplished on averages of at least three scans. EM images shown are representative of multiple similar images obtained under each experimental condition. FTIR and HX-MS analysis were conducted once on each end-stage aggregate.
Data sets for all experiments were analysed using Origin 7.5 software (OriginLab Corporation). The curves in sedimentation and ThT assay were fit to B-spline fitting function to guide the eye only. Error bars shown are standard errors obtained from analysis of duplicate samples. The log–log plots were fit by linear regression. S.d.'s listed for the n* values in Table 1 were determined for each log–log plot by running linear regressions on a series of data sets, each missing one data point, to produce a set of slope values whose variance was then calculated. Thus, if there are a total of nine values in the full data set, there will be eight modified sets and eight resulting slope values whose s.d. from the mean is listed with the n* in the table.
Data availability
Any additional data that support the findings of this study are available from the corresponding author upon request.
Additional information
How to cite this article: Misra, P. et al. Rapid α-oligomer formation mediated by the Aβ C terminus initiates an amyloid assembly pathway. Nat. Commun. 7:12419 doi: 10.1038/ncomms12419 (2016).
Supplementary Material
Supplementary Figures 1-8
Acknowledgments
We gratefully acknowledge helpful comments and suggestions by Jozsef Kardos and pre-publication access to his secondary structure algorithm. We acknowledge funding support from NIH grants R01GM099718 (R.W.) and R01 AG018416 (R.W.). We acknowledge James Conway and Alexander Makhov for access to the Structural Biology Department's cryo-EM facility. We are grateful to Seth Horne for making available his lab's CD spectropolarimeter.
Footnotes
Author contributions P.M. purified the peptides, determined and analysed the aggregation kinetics by sedimentation-HPLC, ThT, DLS and CD. R.K. obtained the HX-MS, EM and FTIR data and CD data on some Aβ fragment peptides. S.C. contributed to some kinetics experiments and advised on handling of Aβ42. K.K. provided the sedimentation and ThT aggregation kinetics data for K2Q23K2. R.W. and P.M. wrote the paper. All authors contributed to study design, data interpretation and improving the manuscript.
References
- Eisele Y. S. et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 14, 759–780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. D., Wong S. S., Lieber C. M. & Lansbury P. T. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem. Biol. 4, 119–125 (1997). [DOI] [PubMed] [Google Scholar]
- Harper J. D., Lieber C. M. & Lansbury P. T. Jr.. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem. Biol. 4, 951–959 (1997). [DOI] [PubMed] [Google Scholar]
- Walsh D. M., Lomakin A., Benedek G. B., Condron M. M. & Teplow D. B. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 272, 22364–22372 (1997). [DOI] [PubMed] [Google Scholar]
- Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 309, 256–274 (1999). [DOI] [PubMed] [Google Scholar]
- Modler A. J. et al. Polymerization of proteins into amyloid protofibrils shares common critical oligomeric states but differs in the mechanisms of their formation. Amyloid 11, 215–231 (2004). [DOI] [PubMed] [Google Scholar]
- Chen S., Ferrone F. & Wetzel R. Huntington's Disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl Acad. Sci. USA 99, 11884–11889 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar K., Jayaraman M., Sahoo B., Kodali R. & Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 18, 328–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J. K., Jack R. L., Schwantes C. R. & Pande V. S. Dynamical phase transitions reveal amyloid-like states on protein folding landscapes. Biophys. J. 107, 974–982 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrone F. A. Assembly of Aβ proceeds via monomeric nuclei. J. Mol. Biol. 427, 287–290 (2015). [DOI] [PubMed] [Google Scholar]
- Stine W. B. Jr., Dahlgren K. N., Krafft G. A. & LaDu M. J. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622 (2003). [DOI] [PubMed] [Google Scholar]
- Serio T. R. et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321 (2000). [DOI] [PubMed] [Google Scholar]
- Lee J., Culyba E. K., Powers E. T. & Kelly J. W. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J., Shen Y., Lee J. H., Ying J. & Bax A. Monomeric Abeta(1-40) and Abeta(1-42) peptides in solution adopt very similar Ramachandran map distributions that closely resemble random coil. Biochemistry 55, 762–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G. et al. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl Acad. Sci. USA 100, 330–335 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze M. D., Condron M. M. & Teplow D. B. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 312, 1103–1119 (2001). [DOI] [PubMed] [Google Scholar]
- Williams A. D. et al. Structural properties of Abeta protofibrils stabilized by a small molecule. Proc. Natl Acad. Sci. USA 102, 7115–7120 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemuru S., Kodali R. & Wetzel R. C-terminal threonine reduces Aβ43 amyloidogenicity compared with Aβ42. J. Mol. Biol. 428, 274–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A. & Raleigh D. P. A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng. Des. Sel. 22, 453–459 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y. et al. Abeta(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 22, 499–505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov A., Yau W. M., Ghirlando R., Thurber K. R. & Tycko R. Successive stages of amyloid-beta self-assembly characterized by solid-state nuclear magnetic resonance with dynamic nuclear polarization. J. Am. Chem. Soc. 137, 8294–8307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A. K. et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman M. et al. Slow amyloid nucleation via alpha-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J. Mol. Biol. 415, 881–899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo B. et al. Folding landscape of mutant Huntingtin exon1: diffusible multimers, oligomers and fibrils, and no detectable monomer. PLoS ONE 11, e0155747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop C. L. et al. Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance. Biochemistry 53, 6653–6666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop C. L. et al. Huntingtin exon 1 fibrils feature an interdigitated beta-hairpin-based polyglutamine core. Proc. Natl Acad. Sci. USA 113, 1546–1551 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo B., Singer D., Kodali R., Zuchner T. & Wetzel R. Aggregation behavior of chemically synthesized, full-length huntingtin exon1. Biochem 53, 3897–3907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra R. et al. Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting alpha-helix-rich oligomeric intermediates. J. Mol. Biol. 415, 900–917 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland B. P., Kodali R., Mishra R. & Wetzel R. A serendipitous survey of prediction algorithms for amyloidogenicity. Biopolymers 100, 780–789 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardberg A. S. et al. Molecular basis for passive immunotherapy of Alzheimer's disease. Proc. Natl Acad. Sci. USA 104, 15659–15664 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H., Jao S., Ma K. & Zagorski M. G. Solution structures of micelle-bound amyloid beta-(1-40) and beta-(1-42) peptides of Alzheimer's disease. J. Mol. Biol. 285, 755–773 (1999). [DOI] [PubMed] [Google Scholar]
- Kumar S. T. et al. Solvent removal induces a reversible beta-to-alpha switch in oligomeric Abeta peptide. J. Mol. Biol. 428, 268–273 (2016). [DOI] [PubMed] [Google Scholar]
- Fraser P. E., Nguyen J. T., Surewicz W. K. & Kirschner D. A. pH-dependent structural transitions of Alzheimer amyloid peptides. Biophys J. 60, 1190–1201 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjernberg L. O. et al. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 271, 8545–8548 (1996). [DOI] [PubMed] [Google Scholar]
- Williams A. D. et al. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842 (2004). [DOI] [PubMed] [Google Scholar]
- Lazo N. D., Grant M. A., Condron M. C., Rigby A. C. & Teplow D. B. On the nucleation of amyloid beta-protein monomer folding. Protein Sci. 14, 1581–1596 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto A. et al. Analysis of the secondary structure of beta-amyloid (Abeta42) fibrils by systematic proline replacement. J. Biol. Chem. 279, 52781–52788 (2004). [DOI] [PubMed] [Google Scholar]
- Kodali R., Williams A. D., Chemuru S. & Wetzel R. Abeta(1-40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. J. Mol. Biol. 401, 503–517 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett J. T., Berger E. P. & Lansbury P. T. Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 (1993). [DOI] [PubMed] [Google Scholar]
- Landrum E. & Wetzel R. Biophysical underpinnings of the repeat length dependence of polyglutamine amyloid formation. J. Biol. Chem. 289, 10254–10260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Nuallain B. et al. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 413, 34–74 (2006). [DOI] [PubMed] [Google Scholar]
- Micsonai A. et al. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl Acad. Sci. USA 112, E3095–E3103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari M. et al. Bacterial inclusion bodies of Alzheimer's disease beta-amyloid peptides can be employed to study native-like aggregation intermediate states. Chembiochem 12, 407–423 (2011). [DOI] [PubMed] [Google Scholar]
- Li H., Monien B. H., Fradinger E. A., Urbanc B. & Bitan G. Biophysical characterization of Abeta42 C-terminal fragments: inhibitors of Abeta42 neurotoxicity. Biochemistry 49, 1259–1267 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallucci V., D'Amelio M. & Cecconi F. Abeta toxicity in Alzheimer's disease. Mol. Neurobiol. 45, 366–378 (2012). [DOI] [PubMed] [Google Scholar]
- Sugase K., Dyson H. J. & Wright P. E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 (2007). [DOI] [PubMed] [Google Scholar]
- Hamada D., Segawa S. & Goto Y. Non-native alpha-helical intermediate in the refolding of beta-lactoglobulin, a predominantly beta-sheet protein. Nat. Struct. Biol. 3, 868–873 (1996). [DOI] [PubMed] [Google Scholar]
- Li J. et al. An alpha-helical burst in the src SH3 folding pathway. Biochemistry 46, 5072–5082 (2007). [DOI] [PubMed] [Google Scholar]
- Kheterpal I., Chen M., Cook K. D. & Wetzel R. Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange--mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol. 361, 785–795 (2006). [DOI] [PubMed] [Google Scholar]
- Kodali R. & Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 17, 48–57 (2007). [DOI] [PubMed] [Google Scholar]
- Arslan P. E., Mulligan V. K., Ho S. & Chakrabartty A. Conversion of Abeta42 into a folded soluble native-like protein using a semi-random library of amphipathic helices. J. Mol. Biol. 396, 1284–1294 (2010). [DOI] [PubMed] [Google Scholar]
- Fradinger E. A. et al. C-terminal peptides coassemble into Abeta42 oligomers and protect neurons against Abeta42-induced neurotoxicity. Proc. Natl Acad. Sci. USA 105, 14175–14180 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson T. E., Vitalis A., Crick S. L. & Pappu R. V. Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J. Mol. Biol. 396, 1295–1309 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokona B., Rosenthal Z. P. & Fairman R. Role of the coiled-coil structural motif in polyglutamine aggregation. Biochemistry 53, 6738–6746 (2014). [DOI] [PubMed] [Google Scholar]
- Jayaraman M. et al. Kinetically competing huntingtin aggregation pathways control amyloid polymorphism and properties. Biochemistry 51, 2706–2716 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsellier E., Redeker V., Ruiz-Arlandis G., Bousset L. & Melki R. Molecular iInteraction between the chaperone Hsc70 and the N-terminal flank of Huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt J. R. et al. Huntingtin N-terminal monomeric and multimeric structures destabilized by covalent modification of heteroatomic residues. Biochem 54, 4285–4296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisdon A. M., Thomas B. & Bottomley S. P. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem 281, 16888–16896 (2006). [DOI] [PubMed] [Google Scholar]
- Ignatova Z., Thakur A. K., Wetzel R. & Gierasch L. M. In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. J. Biol. Chem. 282, 36736–36743 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser C. A. et al. Natural tri- to hexapeptides self-assemble in water to amyloid beta-type fiber aggregates by unexpected alpha-helical intermediate structures. Proc. Natl Acad. Sci. USA 108, 1361–1366 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. et al. An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 (2009). [DOI] [PubMed] [Google Scholar]
- Kheterpal I., Cook K. D. & Wetzel R. Hydrogen/deuterium exchange mass spectrometry analysis of protein aggregates. Methods Enzymol. 413, 140–166 (2006). [DOI] [PubMed] [Google Scholar]
- Smirnovas V. et al. Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J. Biol. Chem. 284, 24233–24241 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-8
Data Availability Statement
Any additional data that support the findings of this study are available from the corresponding author upon request.