

Abstract
Inflammation is a response to infections or tissue injuries. Inflammation was once defined by clinical signs, later by the presence of leukocytes, and nowadays by expression of “proinflammatory” cytokines and chemokines. But leukocytes and cytokines often have rather anti-inflammatory, proregenerative, and homeostatic effects. Is there a need to redefine “inflammation”? In this review, we discuss the functions of “inflammatory” mediators/regulators of the innate immune system that determine tissue environments to fulfill the need of the tissue while regaining homeostasis after injury.
1. Introduction
Inflammation is one of the major danger control programs of tissue pathology conserved during evolution till date with a major aim to resolve the infection, repair the tissue damage, and regain the state of tissue homeostasis [1, 2]. It is a highly complex but still a very well-coordinated process, classically triggered by infection or tissue injury. Historically, “inflammation” was initially defined based on the clinical representations by Hippocrates as calor, rubor, tumor, and dolor [3]. This definition was challenged by the discovery of microscope in the 19th century, and the microscopic presence of leukocytes at the site of infection or injury was called “inflammation” since then [4]. However, this simplistic definition of “inflammation” does no longer hold true in the 21st century mainly because of the advancements in immunology and leukocyte biology in the last decade. We now know that leukocytes present numerous immunoregulatory phenotypes, for example, M2 macrophages, regulatory T and B cells, and fibrocytes, having anti-inflammatory functions. This implies that the presence of leukocytes observed by pathologists at sites of infection or injury does not necessarily indicate “inflammation,” at least without further characterizing their functional phenotypes. As such, we now define “inflammation” based on the presence of proinflammatory leukocyte phenotypes along with the expression of proinflammatory cytokines.
A successful inflammatory response eliminates the trigger followed by a resolution of inflammation and tissue repair by numerous anti-inflammatory cytokines as well as lipid mediators [5–8]. However, a persistent injurious trigger shifts the homeostatic set points fetching several changes in the initial inflammatory process (chronic inflammation), for example, replacement of neutrophils with macrophages and T cells and subsequent formation of granulomata or tertiary lymphoid tissues. In case these cellular effectors fail to control the injurious trigger, collateral tissue damage occurs [9–11]. Moreover, chronic inflammation can also arise as a result of autoimmune responses [9, 11]. Regardless of the cause, inflammation supposedly evolved to restore homeostasis. In this review, we discuss how different mediators of inflammation, in particular, of the innate immune system, set tissue environments to resolve inflammation and reinforce tissue repair, by promoting either regeneration or fibrosis in order to regain homeostasis after injury (Figure 1).
Figure 1.
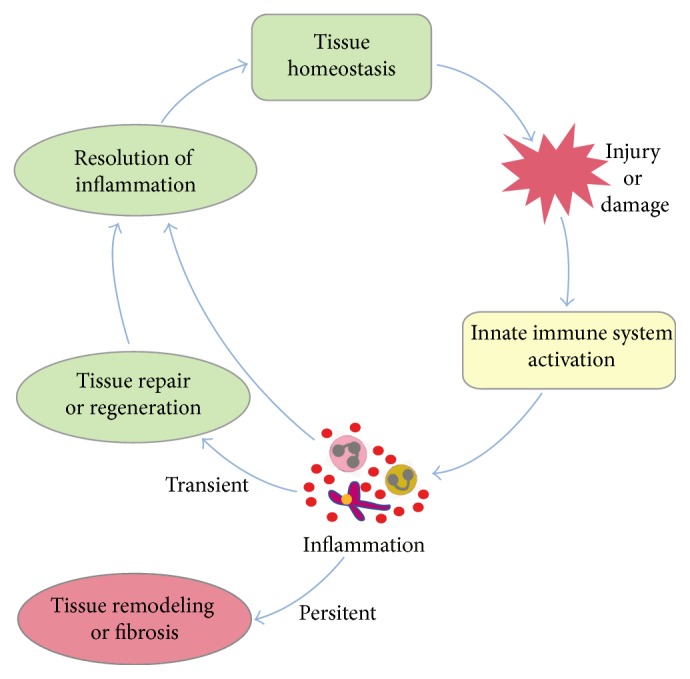
The role of the innate immune system in regaining tissue homeostasis. An injury disturbs the tissue homeostasis and activates the innate immune system leading to the recruitment of several immune cells at the site of injury. These immune cells secrete cytokines, growth factors, and enzymes to establish an inflammatory milieu. They also secrete anti-inflammatory and proregenerative cytokines to promote resolution of inflammation as well as tissue repair. A transient inflammation is often helpful to get rid of the cause of the tissue injury and return to homeostasis. However, an uncontrolled or persistent inflammation promotes tissue remodeling and fibrosis.
2. Resolution of Inflammation
An acute inflammatory response is followed by the resolution phase. The processes to return to tissue homeostasis, that is, catabasis [12], are governed by innate immune cells and specific mediators produced by them. These processes involve neutrophil apoptosis and their phagocytic removal via efferocytosis, clearance of proinflammatory dead cells and cytokines, and recruitment or phenotype switching of macrophages to anti-inflammatory phenotype [13]. Neutrophil-derived microparticles can also trigger the resolution of inflammation [13, 14]. Factors that mediate resolution include interleukin- (IL-) 10 and TGF-β, as well as lipid mediators, for example, lipoxins, resolvins, protectins, and maresins, collectively termed as specialized proresolving mediators (SPMs) [6, 15]. Within minutes after tissue injury prostaglandin and leukotriene synthesis from arachidonic acid metabolism occurs at the site of inflammation leading to the recruitment of neutrophils as a result of the chemotactic gradient, increased blood flow, and vascular permeability [16]. This is often followed by the class switching of lipid mediators, in which arachidonic acid metabolism switches from the production of leukotrienes to anti-inflammatory lipoxins, thus sending the “stop” signal to neutrophils recruitment and begins the end of the acute inflammatory response [17]. Lipoxins and resolvins stimulate the nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages [18]. SPMs counterregulate the proinflammatory mediators and thus reduce the magnitude and duration of inflammation and tissue regeneration [12, 19].
Apart from limiting neutrophil recruitment, SPMs also help to increase natural killer (NK) cells mediated neutrophil apoptosis and subsequent efferocytosis by macrophages [20]. They potently inhibit the release of proinflammatory cytokines from the group 2 innate lymphoid cells (ILCs) [20] and increase IL-10 production by macrophages as well as induce M1 to M2 macrophage phenotype switch [21]. In addition to SPMs, the complement system also contributes to the resolution of inflammation by enhancing efferocytosis of apoptotic cells [22, 23]. The continuous phagocytosis of apoptotic cells, regulated by the mitochondrial membrane protein Ucp2 [24], stimulates monocytes to release IL-10 and TGF-β further promotes the switch toward an anti-inflammatory M2 macrophage phenotype [25, 26]. Recently, resolvin D1 has been demonstrated to trigger GPR32 to polarize macrophages toward the proresolving M2 phenotype [27]. Furthermore, IL-10 is an important cytokine with anti-inflammatory functions [28]. For example, in mouse models of acute kidney injury, IL-10 administration has a beneficial effect by inhibition of leukocyte infiltration and inflammatory renal cell death [29]. It also influences T cells by attenuating proliferation of CD4+ T cells and their cytokine production [30]. The tissue-resident dendritic cells (DCs) also promote the resolution of inflammation by producing pentraxin-3 (PTX3) which inhibits P-selectin on the vascular endothelial cells and thus inhibits immune cell recruitment to sites of injury [31–33]. Moreover, neutrophils released the prestored PTX3 in the early phase of acute myocardial infarction that bind to activated circulating platelets and dampen their proinflammatory response [34], whereas PTX3 also aggregated with histones and protected from histone-mediated endothelial cytotoxicity in sepsis [35, 36]. Furthermore, PTX3 suppressed complement dependent inflammation as well as reduced tumor infiltration by macrophages [37].
Group 3 ILCs gets activated and produces IL-22 after an intestinal epithelial injury suggesting that inflammation can override injury by promoting tissue regeneration [38]. Moreover, IL-22-producing ILCs prevented systemic inflammation during chronic diseases by promoting anatomical containment of lymphoid-resident commensal bacteria [39]. Similarly, the redox modification of high mobility group box 1 (HMGB1), a danger associated molecular pattern (DAMP) released after tissue injury as well as by macrophages and monocytes, regulated its proinflammatory functions during the resolution of inflammation and prevented excessive acetaminophen-induced hepatic injury [40, 41]. Together, immune cells, as well as mediators released by them, promote resolution of inflammation in order to reestablish the homeostasis after injury.
3. Tissue Regeneration and Repair
The immune system is instrumental in supplying growth factors and cytokine signals that orchestrate tissue repair. For example, the tissue-resident macrophages originated from yolk sac-derived erythromyeloid progenitors that possess the capacity to self-replenish [42, 43], while bone-marrow-derived circulating monocytes differentiate into tissue macrophages [44], but both are activated during injury. Blood-derived young monocytes/macrophages have enhanced remyelinating activity compared to old macrophages in the central nervous system [45]. Although M2 macrophages are the main driver of the resolution of inflammation, tissue repair, and scar formation, the M1 macrophages clear cellular debris in order to prevent the persistence of toxic and immunogenic material at the site of injury. Therefore, depletion of M1 macrophages resulted in impaired healing and regeneration after myocardial as well as skeletal muscle injuries [46, 47]. In addition, M1 macrophages also activated proliferative myogenesis via IL-6, TNF-α, and IL-1β whereas M2 macrophages supported myogenic differentiation via TGF-β production during skeletal muscle regeneration [47, 48].
Moreover, infiltrating eosinophils secreted IL-4 to induce proliferation of fibro/adipocyte progenitor cells, which promoted clearance of necrotic debris and skeletal muscle regeneration [49]. The CXCL12-CXCR4 pathway regulates the recruitment of progenitors, the unipotent proliferative cells with a capacity of self-renewal [50], at the site of injury [51]. Other mediators of the innate immune system that induced progenitor cells proliferation and regeneration include leukotriene C4, which activated radial glial cell proliferation and neurogenesis either upon or without an injury [52], oncomodulin derived from neutrophils, and macrophages which promoted the optic nerve regeneration [53, 54]. Furthermore, macrophages derived Wnt suppressed Notch signaling and thus regulated the fate of hepatic as well as renal progenitor cells after liver and kidney injury, respectively [55, 56]. Macrophage-derived Wnt7b also stimulated epithelial responses and, thus, regarded critical for kidney repair and regeneration [57]. In the acidic tissue environments after skin, liver, and lung injury and arterial thrombosis, neutrophils and macrophages derived PTX3 promoted remodeling of the fibrin-rich inflammatory matrix ensuring normal tissue repair [58]. In addition, the complement system is also instrumental in promoting tissue repair and regeneration by inducing growth factors as well as disposal of dead cells [23]. In particular, C3a and C5a activated NF-κB/STAT-3 and enhanced hepatocyte regeneration after liver injury [59]. Recent reports showed that the delayed postinjury administration of C5a inhibited caspase-3 mediated neuron apoptosis leading to improved regeneration and functional recovery after murine spinal cord injury [60], as well as administration of C3a retina regeneration via STAT-3 activation in the progenitor cells present in the eye [61].
Apoptotic cells released after tissue injury promoted angiogenic properties of macrophages by releasing prostaglandin E2, which induced endothelial-derived progenitors to angiogenesis and vascular repair during skeletal muscle regeneration [62–64]. In addition, proliferation and differentiation of renal progenitor cells were also enhanced by the Toll-like receptor- (TLR-) 2-agonistic DAMPs released after tissue injury [65–67]. Several recent data suggests additional mechanisms of DAMP-driven tissue regeneration. For example, TLR4-agonistic DAMPs activated the interstitial mononuclear phagocytes to secrete a proregenerative cytokine IL-22 [68, 69] to promote tubular cell regeneration after injury by activating the JAK/STAT3 and ERK1/2 signaling pathway [68, 70]. Group 3 ILCs also produced IL-22 after an intestinal injury to promote the intestinal stem cell-mediated epithelial regeneration [71]. IL-22 mediated protection and regeneration were also observed in experimental models of hepatic, pancreatic, and thymic injuries [72–75]. In addition, a mast cell-specific tryptase, mouse mast cell protease (mMCP) 6, directly cleaves fibronectin and collagen IV and, therefore, suppressed scars and promoted functional recovery after spinal cord injury [76]. Platelets contributed to liver regeneration by secreting serotonin in mice as well as humans [77, 78]. Group 2 ILCs also promoted lung-tissue homeostasis after infection with influenza virus by producing a growth factor Amphiregulin [79]. After an injury to skeletal muscles, IL-33 recruited a special population of regulatory T cells (Tregs) to the injured muscles that produced Amphiregulin and improved the muscle repair [80–82]. Together, this illustrates that the innate immune system and its mediators do not only contribute to the immune injury but also to the immune-mediated repair or regeneration after injury as a part of a danger control response [10, 83] (Figure 2).
Figure 2.

Mediators of innate immune system in regaining tissue homeostasis. Innate immune cells secrete several cytokines, growth factors, and enzymes, which promotes either resolution of inflammation and tissue repair/regeneration or tissue remodeling/fibrosis. PTX3: pentraxin 3, ROS: reactive oxygen species, IL: interleukin, TGF: transforming growth factor, MMP: matrix metalloproteinase, TIMP: tissue inhibitor of matrix metalloproteinase, TNF: tumor necrosis factor, MIP: macrophage inhibitory protein, MBP: major basic protein, mMCP: mouse mast cell protease, and PDGF: platelet-derived growth factor.
4. Tissue Remodeling and Fibrosis
The well-defined chronology of inflammatory events is essential for optimal repair. However, an overactivated immune response leads to tissue remodeling rather than tissue regeneration, which is clinically termed as tissue fibrosis. Fibrosis is characterized by excess deposition of extracellular matrix (ECM) due to the accumulation and activation of fibroblasts and myofibroblasts. Inflammatory cells of the immune system, as well as factors released by them, facilitate fibrosis. For example, tissue injury is always followed by altered vascular permeability to enhance the neutrophils recruitment to the site of injury. The delayed clearance of neutrophils from the site of injury further exacerbates the injury [84]. Neutrophils count is in fact used as a prognostic marker for cardiac remodeling [85]. Neutrophils are known to increase oxidative stress as well as release a number of enzymes like matrix metalloproteinases (MMPs), elastase, and cathepsins which contribute significantly to the process of fibrosis [86–88]. Apart from neutrophils, platelets can also respond to the state of infection or inflammation through activation of TLRs [89, 90]. The factors derived from platelets, for example, platelet-derived growth factor (PDGF), is a potent chemotactic agent, whereas TGF-β drives fibroblast proliferation and activation [91]. Moreover, the factors involved in coagulation can also contribute significantly to fibrosis, for example, factors VII, IX, and X [92–94]. The coagulation system and complement system are linked very closely, often involving a cross talk, to maintain the tissue homeostasis [23, 95]. For example, in the absence of C3, thrombin replaces the C3 dependent C5 convertase and directly cleaves C5 to generate the biologically active C5a [96], which induced fibrosis in lungs, liver, pancreas, and kidney after injury [97–100]. In addition to C5a, C3a has also been implicated in renal fibrosis [101].
Other cells of the innate immune system, for example, eosinophils and mast cells, also contribute to fibrosis. Eosinophils produce TGF-β, major basic protein (MBP)-1, eosinophilic peroxidase as well as granule proteins, and lysosomal hydrolytic enzymes which are implicated to be a part of fibrosis process [102]. Mast cells produce various proteases, vasoactive factors like histamine, cytokines, and TGF-β during the tissue injury and fibrosis [103]. IL-4, one of the major products of mast cell activation, contributes to the cardiac fibrosis [104]. Moreover, mast cell deficient mice are protected against pulmonary as well as cardiac fibrosis [105, 106]. Among the various immune cells, the macrophages are essential for efficient wound healing [107–111]. Macrophages are the main source of MMPs and tissue inhibitor of metalloproteinases (TIMPs) [109, 112, 113]. The balance between MMPs and TIMPs is crucial for maintaining the composition of ECM. Apart from MMPs and TIMPs, macrophages also contribute to the production of TGF-β, the most significant factor involved in fibrosis [114]. TGF-β regulates fibroblast activation, differentiation, and proliferation [115]. It also upregulates ECM genes and suppresses genes associated with MMPs, thus causing increased deposition of matrix. TGF-β promotes collagen synthesis and the expression of profibrotic genes such as type I collagen and connective tissue growth factor (CTGF) [15, 116]. Furthermore, TGF-β is also an anti-inflammatory factor; therefore, its early inhibition is associated with increased mortality, increased chemokine expression, and leukocyte infiltration while its inhibition during the resolution phase resulted in improved survival and reduced tissue fibrosis [117–122]. Activated macrophages induce production of various cytokines and factors like interleukins (e.g., IL-1β) and TNF-α, which drive further inflammation and fibrosis by enhancing ECM production as well as upregulating expression of TGF-β [123, 124]. Depletion of macrophages during the early phase of tissue injury ameliorated fibrosis, while delayed depletion of macrophages during the resolution phase exaggerated fibrosis with persistence of profibrotic cellular and matrix components [110, 111, 125]. Although recent studies demonstrated an association between macrophages derived PTX3 and tissue fibrosis in nonalcoholic fatty liver disease as well as in lung fibrosis, whether PTX3 causes fibrosis or not is still unclear [126, 127].
Along with macrophages, DCs are the primary determinants of the cytokine and chemokine milieu during fibrogenesis [128, 129]. For example, IL-6 and TNF-α produced by DCs have pleiotropic effects on liver fibrosis [128]. Furthermore, they also activated NK cells to produce TNF-α and, therefore, elevate the inflammatory environment in fibrotic livers [128]. Group 3 ILCs promoted bleomycin-induced pulmonary fibrosis by secreting IL-17 [130, 131], whereas IL-25 induced expansion of the group 2 ILCs within the lungs, which promoted pulmonary fibrosis via IL-13 dependent mechanism [132]. Together, the innate immune system and its mediators contribute to tissue remodeling and fibrosis (Figure 2).
5. Conclusions and Future Perspectives
Maintaining tissue morphology is essential to maintain tissue function, that is, homeostasis. An injury or damage affects the structural integrity of the tissue implying a loss of tissue function, and, therefore, the structural and functional recovery, that is, regaining homeostasis after injury, is the ultimate goal. The inflammatory mediators of the innate immune system are important regulators of tissue homeostasis. They modulate tissue environments at all phases of the homeostatic imbalance, for example, promotion as well as the resolution of inflammation, tissue regeneration, and tissue remodeling/fibrosis.
Our understanding of inflammation biology has increased over the last few decades and has gone far beyond the basic concept of inflammation that was originally introduced by Hippocrates. The advancements in microscopic as well as flow associated cell sorting (FACS) techniques have allowed us to understand and redefine the “inflammation.” This progress has also raised several questions, for example, what are the molecules or signals that regulate the function of the innate immune cells, what are the critical mechanisms that regulate the balance of different populations of these cells in the specific phase after injury, and how to modulate the behavior as well as balance of these cells in each phase after injury to enhance tissue regeneration and reduce fibrosis. The advancements in the new genomic technologies such as CRISPR-Cas9 have transformed the field of immunology research and will certainly speed up novel discoveries in regulatory and signaling components of inflammation biology.
The newly obtained knowledge will translate into novel therapeutic strategies for inflammatory diseases. For example, recent studies have identified proinflammatory and proregenerative potential of a cytokine IL-22 and a regulatory oncoprotein murine double minute- (MDM-) 2 in the pathogenesis of ischemic renal injury (IRI) and have demonstrated the therapeutic potential of recombinant IL-22 and MDM-2 inhibitor, nutlin-3a, in IRI and other inflammatory diseases [68, 133–135]. Moreover, other studies have identified a pattern recognition molecule PTX3 as a potential target for therapeutic manipulation in damaged tissues as well as a variety of diseases [37, 58]. Therefore, in-depth understanding of the functions of inflammatory cells as well as mediators of inflammation will be instrumental in the identification of novel therapeutic targets and treatment strategies for several inflammatory diseases. As written by a Scottish surgeon in 1974 “Inflammation in itself is not to be considered as a disease but as a salutary operation consequent to some violence or some disease” [136].
Acknowledgments
The work presented here was supported by the Department of Science and Technology- (DST-) YSS/2015/001969, India, to Onkar P. Kulkarni; by the Deutsche Forschungsgemeinschaft (DFG), Germany (AN372/11-2, AN372/12-2, AN372/14-3, AN372/16-1, AN372/17-1, AN372/23-1, and MU3906/1-1), to Hans-Joachim Anders and Shrikant R. Mulay; and by the European Union's Seventh Framework Program for Research, Technological Development and Demonstration under Grant Agreement no. 305736 (“REDDSTAR”) and by the European Union's Horizon 2020 Research and Innovation Program under Grant Agreement no. 634086 (“NEPHSTROM”).
Competing Interests
The authors declare no conflict of interests.
References
- 1.Nathan C. Points of control in inflammation. Nature. 2002;420(6917):846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 2.Hagemann J. H., Haegele H., Müller S., Anders H.-J. Danger control programs cause tissue injury and remodeling. International Journal of Molecular Sciences. 2013;14(6):11319–11346. doi: 10.3390/ijms140611319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams F. The Genuine Works of Hippocrates. London, UK: The Sydenham Society; 1849. [Google Scholar]
- 4.Cohnheim J. F. Ueber entzündung und eiterung. Virch Arch Path Anat. 1867;40:1–79. [Google Scholar]
- 5.Pinegin B., Vorobjeva N., Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmunity Reviews. 2015;14(7):633–640. doi: 10.1016/j.autrev.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Serhan C. N. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annual Review of Immunology. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 7.Ariel A., Serhan C. N. Resolvins and protectins in the termination program of acute inflammation. Trends in Immunology. 2007;28(4):176–183. doi: 10.1016/j.it.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Weidenbusch M., Anders H.-J. Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair and fibrosis. Journal of Innate Immunity. 2012;4(5-6):463–477. doi: 10.1159/000336717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 10.Suárez-Álvarez B., Liapis H., Anders H. J. Links between coagulation, inflammation, regeneration, and fibrosis in kidney pathology. Laboratory Investigation. 2016;96(4):378–390. doi: 10.1038/labinvest.2015.164. [DOI] [PubMed] [Google Scholar]
- 11.Drayton D. L., Liao S., Mounzer R. H., Ruddle N. H. Lymphoid organ development: from ontogeny to neogenesis. Nature Immunology. 2006;7(4):344–353. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 12.Basil M. C., Levy B. D. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nature Reviews Immunology. 2016;16(1):51–67. doi: 10.1038/nri.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabas I., Glass C. K. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339(6116):166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalli J., Serhan C. N. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15):e60–e72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varga J., Jimenez S. A. Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-β . Biochemical and Biophysical Research Communications. 1986;138(2):974–980. doi: 10.1016/s0006-291x(86)80591-5. [DOI] [PubMed] [Google Scholar]
- 16.Malawista S. E., Chevance A. D. B., Van Damme J., Serhan C. N. Tonic inhibition of chemotaxis in human plasma. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(46):17949–17954. doi: 10.1073/pnas.0802572105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy B. D., Clish C. B., Schmidt B., Gronert K., Serhan C. N. Lipid mediator class switching during acute inflammation: signals in resolution. Nature Immunology. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 18.Godson C., Mitchell S., Harvey K., Petasis N. A., Hogg N., Brady H. R. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. Journal of Immunology. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 19.Serhan C. N. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnig C., Cernadas M., Dutile S., et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Science Translational Medicine. 2013;5(174) doi: 10.1126/scitranslmed.3004812.174ra26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalli J., Zhu M., Vlasenko N. A., et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. The FASEB Journal. 2013;27(7):2573–2583. doi: 10.1096/fj.13-227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flierman R., Daha M. R. The clearance of apoptotic cells by complement. Immunobiology. 2007;212(4-5):363–370. doi: 10.1016/j.imbio.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Ricklin D., Hajishengallis G., Yang K., Lambris J. D. Complement: a key system for immune surveillance and homeostasis. Nature Immunology. 2010;11(9):785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park D., Han C. Z., Elliott M. R., et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477(7363):220–224. doi: 10.1038/nature10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lucas M., Stuart L. M., Savill J., Lacy-Hulbert A. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. The Journal of Immunology. 2003;171(5):2610–2615. doi: 10.4049/jimmunol.171.5.2610. [DOI] [PubMed] [Google Scholar]
- 26.Lech M., Gröbmayr R., Weidenbusch M., Anders H.-J. Tissues use resident dendritic cells and macrophages to maintain homeostasis and to regain homeostasis upon tissue injury: the immunoregulatory role of changing tissue environments. Mediators of Inflammation. 2012;2012:15. doi: 10.1155/2012/951390.951390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmid M., Gemperle C., Rimann N., Hersberger M. Resolvin D1 polarizes primary human macrophages toward a proresolution phenotype through GPR32. The Journal of Immunology. 2016;196(8):3429–3437. doi: 10.4049/jimmunol.1501701. [DOI] [PubMed] [Google Scholar]
- 28.Couper K. N., Blount D. G., Riley E. M. IL-10: the master regulator of immunity to infection. The Journal of Immunology. 2008;180(9):5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 29.Deng J., Kohda Y., Chiao H., et al. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney International. 2001;60(6):2118–2128. doi: 10.1046/j.1523-1755.2001.00043.x. [DOI] [PubMed] [Google Scholar]
- 30.Sabat R., Grütz G., Warszawska K., et al. Biology of interleukin-10. Cytokine and Growth Factor Reviews. 2010;21(5):331–344. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Deban L., Russo R. C., Sironi M., et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nature Immunology. 2010;11(4):328–334. doi: 10.1038/ni.1854. [DOI] [PubMed] [Google Scholar]
- 32.Lech M., Römmele C., Kulkarni O. P., et al. Lack of the long pentraxin PTX3 promotes autoimmune lung disease but not glomerulonephritis in murine systemic lupus erythematosus. PLoS ONE. 2011;6(5, article e20118) doi: 10.1371/journal.pone.0020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lech M., Römmele C., Gröbmayr R., et al. Endogenous and exogenous pentraxin-3 limits postischemic acute and chronic kidney injury. Kidney International. 2013;83(4):647–661. doi: 10.1038/ki.2012.463. [DOI] [PubMed] [Google Scholar]
- 34.Maugeri N., Rovere-Querini P., Slavich M., et al. Early and transient release of leukocyte pentraxin 3 during acute myocardial infarction. Journal of Immunology. 2011;187(2):970–979. doi: 10.4049/jimmunol.1100261. [DOI] [PubMed] [Google Scholar]
- 35.Daigo K., Nakakido M., Ohashi R., et al. Protective effect of the long pentraxin PTX3 against histone-mediated endothelial cell cytotoxicity in sepsis. Science Signaling. 2014;7(343, article ra88) doi: 10.1126/scisignal.2005522. [DOI] [PubMed] [Google Scholar]
- 36.Daigo K., Hamakubo T. Host-protective effect of circulating pentraxin 3 (PTX3) and complex formation with neutrophil extracellular traps. Frontiers in Immunology. 2012;3, article 378 doi: 10.3389/fimmu.2012.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonavita E., Gentile S., Rubino M., et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160(4):700–714. doi: 10.1016/j.cell.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Sawa S., Lochner M., Satoh-Takayama N., et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nature Immunology. 2011;12(4):320–328. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 39.Sonnenberg G. F., Monticelli L. A., Alenghat T., et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336(6086):1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H., Lundbäck P., Ottosson L., et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Molecular Medicine. 2012;18(2):250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Magna M., Pisetsky D. S. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Molecular Medicine. 2014;20(1):138–146. doi: 10.2119/molmed.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomez Perdiguero E., Klapproth K., Schulz C., et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yona S., Kim K.-W., Wolf Y., et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wynn T. A., Chawla A., Pollard J. W. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruckh J. M., Zhao J.-W., Shadrach J. L., et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10(1):96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nahrendorf M., Swirski F. K., Aikawa E., et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. Journal of Experimental Medicine. 2007;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arnold L., Henry A., Poron F., et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. The Journal of Experimental Medicine. 2007;204(5):1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saclier M., Yacoub-Youssef H., Mackey A. L., et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. STEM CELLS. 2013;31(2):384–396. doi: 10.1002/stem.1288. [DOI] [PubMed] [Google Scholar]
- 49.Heredia J. E., Mukundan L., Chen F. M., et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153(2):376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seaberg R. M., van der Kooy D. Stem and progenitor cells: the premature desertion of rigorous definitions. Trends in Neurosciences. 2003;26(3):125–131. doi: 10.1016/s0166-2236(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 51.Anders H.-J., Romagnani P., Mantovani A. Pathomechanisms: homeostatic chemokines in health, tissue regeneration, and progressive diseases. Trends in Molecular Medicine. 2014;20(3):154–165. doi: 10.1016/j.molmed.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 52.Kyritsis N., Kizil C., Zocher S., et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338(6112):1353–1356. doi: 10.1126/science.1228773. [DOI] [PubMed] [Google Scholar]
- 53.Yin Y., Henzl M. T., Lorber B., et al. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nature Neuroscience. 2006;9(6):843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- 54.Kurimoto T., Yin Y., Habboub G., et al. Neutrophils express oncomodulin and promote optic nerve regeneration. The Journal of Neuroscience. 2013;33(37):14816–14824. doi: 10.1523/jneurosci.5511-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boulter L., Govaere O., Bird T. G., et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nature Medicine. 2012;18(4):572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lasagni L., Ballerini L., Angelotti M. L., et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. STEM CELLS. 2010;28(9):1673–1685. doi: 10.1002/stem.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin S.-L., Li B., Rao S., et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(9):4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doni A., Musso T., Morone D., et al. An acidic microenvironment sets the humoral pattern recognition molecule PTX3 in a tissue repair mode. The Journal of Experimental Medicine. 2015;212(6):905–925. doi: 10.1084/jem.20141268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strey C. W., Markiewski M., Mastellos D., et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. The Journal of Experimental Medicine. 2003;198(6):913–923. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo Q., Cheng J., Zhang J., et al. Delayed post-injury administration of C5a improves regeneration and functional recovery after spinal cord injury in mice. Clinical and Experimental Immunology. 2013;174(2):318–325. doi: 10.1111/cei.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haynes T., Luz-Madrigal A., Reis E. S., et al. Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nature Communications. 2013;4, article 2312 doi: 10.1038/ncomms3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brecht K., Weigert A., Hu J., et al. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB Journal. 2011;25(7):2408–2417. doi: 10.1096/fj.10-179473. [DOI] [PubMed] [Google Scholar]
- 63.Hsu C.-W., Poché R. A., Saik J. E., et al. Improved angiogenesis in response to localized delivery of macrophage-recruiting molecules. PLoS ONE. 2015;10(7) doi: 10.1371/journal.pone.0131643.e0131643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zordan P., Rigamonti E., Freudenberg K., et al. Macrophages commit postnatal endothelium-derived progenitors to angiogenesis and restrict endothelial to mesenchymal transition during muscle regeneration. Cell Death and Disease. 2014;5, article e1031 doi: 10.1038/cddis.2013.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sallustio F., De Benedictis L., Castellano G., et al. TLR2 plays a role in the activation of human resident renal stem/progenitor cells. The FASEB Journal. 2010;24(2):514–525. doi: 10.1096/fj.09-136481. [DOI] [PubMed] [Google Scholar]
- 66.Sallustio F., Costantino V., Cox S. N., et al. Human renal stem/progenitor cells repair tubular epithelial cell injury through TLR2-driven inhibin-A and microvesicle-shuttled decorin. Kidney International. 2013;83(3):392–403. doi: 10.1038/ki.2012.413. [DOI] [PubMed] [Google Scholar]
- 67.Anders H.-J., Schaefer L. Beyond tissue injury—damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. Journal of the American Society of Nephrology. 2014;25(7):1387–1400. doi: 10.1681/asn.2014010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kulkarni O. P., Hartter I., Mulay S. R., et al. Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. Journal of the American Society of Nephrology. 2014;25(5):978–989. doi: 10.1681/ASN.2013050528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weidenbusch M., Rodler S., Anders H.-J. Interleukin-22 in kidney injury and regeneration. American Journal of Physiology—Renal Physiology. 2015;308(10):F1041–F1046. doi: 10.1152/ajprenal.00005.2015. [DOI] [PubMed] [Google Scholar]
- 70.Xu M.-J., Feng D., Wang H., Guan Y., Yan X., Gao B. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. Journal of the American Society of Nephrology. 2014;25(5):967–977. doi: 10.1681/ASN.2013060611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lindemans C. A., Calafiore M., Mertelsmann A. M., et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. 2015;528(7583):560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dudakov J. A., Hanash A. M., van den Brink M. R. M. Interleukin-22: immunobiology and pathology. Annual Review of Immunology. 2015;33:747–785. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pan H., Hong F., Radaeva S., Gao B. Hydrodynamic gene delivery of interleukin-22 protects the mouse liver from concanavalin A-, carbon tetrachloride-, and Fas ligand-induced injury via activation of STAT3. Cellular & Molecular Immunology. 2004;1(1):43–49. [PubMed] [Google Scholar]
- 74.Dudakov J. A., Hanash A. M., Jenq R. R., et al. Interleukin-22 drives endogenous thymic regeneration in mice. Science. 2012;336(6077):91–95. doi: 10.1126/science.1218004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hill T., Krougly O., Nikoopour E., et al. The involvement of interleukin-22 in the expression of pancreatic beta cell regenerative Reg genes. Cell Regeneration. 2013;2, article 2 doi: 10.1186/2045-9769-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vangansewinkel T., Geurts N., Quanten K., et al. Mast cells promote scar remodeling and functional recovery after spinal cord injury via mouse mast cell protease 6. The FASEB Journal. 2016;30(5):2040–2057. doi: 10.1096/fj.201500114r. [DOI] [PubMed] [Google Scholar]
- 77.Lesurtel M., Graf R., Aleil B., et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312(5770):104–107. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- 78.Starlinger P., Assinger A., Haegele S., et al. Evidence for serotonin as a relevant inducer of liver regeneration after liver resection in humans. Hepatology. 2014;60(1):257–266. doi: 10.1002/hep.26950. [DOI] [PubMed] [Google Scholar]
- 79.Monticelli L. A., Sonnenberg G. F., Abt M. C., et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nature Immunology. 2011;12(11):1045–1054. doi: 10.1038/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burzyn D., Kuswanto W., Kolodin D., et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuswanto W., Burzyn D., Panduro M., et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity. 2016;44(2):355–367. doi: 10.1016/j.immuni.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Castiglioni A., Corna G., Rigamonti E., et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS ONE. 2015;10(5) doi: 10.1371/journal.pone.0128094.e0128094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anders H.-J. Four danger response programs determine glomerular and tubulointerstitial kidney pathology: clotting, inflammation, epithelial and mesenchymal healing. Organogenesis. 2012;8(2):29–40. doi: 10.4161/org.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stramer B. M., Mori R., Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. Journal of Investigative Dermatology. 2007;127(5):1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 85.Meissner J., Irfan A., Twerenbold R., et al. Use of neutrophil count in early diagnosis and risk stratification of AMI. American Journal of Medicine. 2011;124(6):534–542. doi: 10.1016/j.amjmed.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 86.Barnes T. C., Anderson M. E., Edwards S. W., Moots R. J. Neutrophil-derived reactive oxygen species in SSc. Rheumatology. 2012;51(7):1166–1169. doi: 10.1093/rheumatology/ker520. [DOI] [PubMed] [Google Scholar]
- 87.Ma Y., Yabluchanskiy A., Lindsey M. L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis and Tissue Repair. 2013;6(1, article 11) doi: 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takemasa A., Ishii Y., Fukuda T. A neutrophil elastase inhibitor prevents bleomycin-induced pulmonary fibrosis in mice. European Respiratory Journal. 2012;40(6):1475–1482. doi: 10.1183/09031936.00127011. [DOI] [PubMed] [Google Scholar]
- 89.Shiraki R., Inoue N., Kawasaki S., et al. Expression of Toll-like receptors on human platelets. Thrombosis Research. 2004;113(6):379–385. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 90.Andonegui G., Kerfoot S. M., McNagny K., Ebbert K. V. J., Patel K. D., Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106(7):2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 91.Sinno H., Prakash S. Complements and the wound healing cascade: an updated review. Plastic Surgery International. 2013;2013:7. doi: 10.1155/2013/146764.146764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Scotton C. J., Krupiczojc M. A., Königshoff M., et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. The Journal of Clinical Investigation. 2009;119(9):2550–2563. doi: 10.1172/jci33288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ameri A., Kurachi S., Sueishi K., Kuwahara M., Kurachi K. Myocardial fibrosis in mice with overexpression of human blood coagulation factor IX. Blood. 2003;101(5):1871–1873. doi: 10.1182/blood-2002-05-1581. [DOI] [PubMed] [Google Scholar]
- 94.Xu H., Noria F., Sandoval-Cooper M. J., et al. Severe deficiency of coagulation Factor VII results in spontaneous cardiac fibrosis in mice. Journal of Pathology. 2009;217(3):362–371. doi: 10.1002/path.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kenawy H. I., Boral I., Bevington A. Complement-coagulation cross-talk: a potential mediator of the physiological activation of complement by low pH. Frontiers in Immunology. 2015;6, article 215 doi: 10.3389/fimmu.2015.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huber-Lang M., Sarma J. V., Zetoune F. S., et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nature Medicine. 2006;12(6):682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 97.Addis-Lieser E., Köhl J., Chiaramonte M. G. Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. The Journal of Immunology. 2005;175(3):1894–1902. doi: 10.4049/jimmunol.175.3.1894. [DOI] [PubMed] [Google Scholar]
- 98.Hillebrandt S., Wasmuth H. E., Weiskirchen R., et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nature Genetics. 2005;37(8):835–843. doi: 10.1038/ng1599. [DOI] [PubMed] [Google Scholar]
- 99.Boor P., Konieczny A., Villa L., et al. Complement C5 mediates experimental tubulointerstitial fibrosis. Journal of the American Society of Nephrology. 2007;18(5):1508–1515. doi: 10.1681/asn.2006121343. [DOI] [PubMed] [Google Scholar]
- 100.Sendler M., Beyer G., Mahajan U. M., et al. Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology. 2015;149(3):765–776.e10. doi: 10.1053/j.gastro.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bao L., Wang Y., Haas M., Quigg R. J. Distinct roles for C3a and C5a in complement-induced tubulointerstitial injury. Kidney International. 2011;80(5):524–534. doi: 10.1038/ki.2011.158. [DOI] [PubMed] [Google Scholar]
- 102.Aceves S. S., Ackerman S. J. Relationships between eosinophilic inflammation, tissue remodeling, and fibrosis in eosinophilic esophagitis. Immunology and Allergy Clinics of North America. 2009;29(1):197–211. doi: 10.1016/j.iac.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levick S. P., McLarty J. L., Murray D. B., Freeman R. M., Carver W. E., Brower G. L. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53(6):1041–1047. doi: 10.1161/hypertensionaha.108.123158. [DOI] [PubMed] [Google Scholar]
- 104.Kanellakis P., Ditiatkovski M., Kostolias G., Bobik A. A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovascular Research. 2012;95(1):77–85. doi: 10.1093/cvr/cvs142. [DOI] [PubMed] [Google Scholar]
- 105.Veerappan A., O'Connor N. J., Brazin J., et al. Mast cells: a pivotal role in pulmonary fibrosis. DNA and Cell Biology. 2013;32(4):206–218. doi: 10.1089/dna.2013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Levick S. P., Melndez G. C., Plante E., McLarty J. L., Brower G. L., Janicki J. S. Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovascular Research. 2011;89(1):12–19. doi: 10.1093/cvr/cvq272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Novak M. L., Koh T. J. Phenotypic transitions of macrophages orchestrate tissue repair. The American Journal of Pathology. 2013;183(5):1352–1363. doi: 10.1016/j.ajpath.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lech M., Anders H.-J. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2013;1832(7):989–997. doi: 10.1016/j.bbadis.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 109.Murray P. J., Wynn T. A. Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khanna S., Biswas S., Shang Y., et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE. 2010;5(3) doi: 10.1371/journal.pone.0009539.e9539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van Amerongen M. J., Harmsen M. C., Van Rooijen N., Petersen A. H., Van Luyn M. J. A. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. American Journal of Pathology. 2007;170(3):818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Matsumoto Y., Park I.-K., Kohyama K. Matrix metalloproteinase (MMP)-9, but not MMP-2, is involved in the development and progression of C protein-induced myocarditis and subsequent dilated cardiomyopathy. The Journal of Immunology. 2009;183(7):4773–4781. doi: 10.4049/jimmunol.0900871. [DOI] [PubMed] [Google Scholar]
- 113.Westermann D., Savvatis K., Lindner D., et al. Reduced degradation of the chemokine MCP-3 by matrix metalloproteinase-2 exacerbates myocardial inflammation in experimental viral cardiomyopathy. Circulation. 2011;124(19):2082–2093. doi: 10.1161/CIRCULATIONAHA.111.035964. [DOI] [PubMed] [Google Scholar]
- 114.Murray L. A., Chen Q., Kramer M. S., et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. The International Journal of Biochemistry & Cell Biology. 2011;43(1):154–162. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 115.Leask A., Abraham D. J. TGF-β signaling and the fibrotic response. FASEB Journal. 2004;18(7):816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 116.Chen S.-J., Yuan W., Mori Y., Levenson A., Trojanowska M., Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-β: involvement of Smad 3. Journal of Investigative Dermatology. 1999;112(1):49–57. doi: 10.1046/j.1523-1747.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 117.Shah M., Foreman D. M., Ferguson M. W. J. Control of scarring in adult wounds by neutralising antibody to transforming growth factor β . The Lancet. 1992;339(8787):213–214. doi: 10.1016/0140-6736(92)90009-R. [DOI] [PubMed] [Google Scholar]
- 118.Sharma K., Jin Y., Guo J., Ziyadeh F. N. Neutralization of TGF-β by anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45(4):522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 119.Kuwahara F., Kai H., Tokuda K., et al. Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106(1):130–135. doi: 10.1161/01.CIR.0000020689.12472.E0. [DOI] [PubMed] [Google Scholar]
- 120.Kulkarni A. B., Karlsson S. Transforming growth factor-β1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. The American Journal of Pathology. 1993;143(1):3–9. [PMC free article] [PubMed] [Google Scholar]
- 121.Bottinger E. P., Letterio J. J., Roberts A. B. Biology of TGF-β in knockout and transgenic mouse models. Kidney International. 1997;51(5):1355–1360. doi: 10.1038/ki.1997.185. [DOI] [PubMed] [Google Scholar]
- 122.Ikeuchi M., Tsutsui H., Shiomi T., et al. Inhibition of TGF-β signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovascular Research. 2004;64(3):526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 123.Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. Journal of Clinical Investigation. 2012;122(3):787–795. doi: 10.1172/jci59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sullivan D. E., Ferris M., Nguyen H., Abboud E., Brody A. R. TNF-α induces TGF-β 1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. Journal of Cellular and Molecular Medicine. 2009;13(8):1866–1876. doi: 10.1111/j.1582-4934.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Duffield J. S., Forbes S. J., Constandinou C. M., et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. Journal of Clinical Investigation. 2005;115(1):56–65. doi: 10.1172/JCI200522675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ozturk K., Kurt O., Dogan T., et al. Pentraxin 3 is a predictor for fibrosis and arterial stiffness in patients with nonalcoholic fatty liver disease. Gastroenterology Research and Practice. 2016;2016:7. doi: 10.1155/2016/1417962.1417962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pilling D., Cox N., Vakil V., Verbeek J. S., Gomer R. H. The long pentraxin PTX3 promotes fibrocyte differentiation. PLoS ONE. 2015;10(3) doi: 10.1371/journal.pone.0119709.e119709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Connolly M. K., Bedrosian A. S., Mallen-St Clair J., et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-α . The Journal of Clinical Investigation. 2009;119(11):3213–3225. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bantsimba-Malanda C., Marchal-Sommé J., Goven D., et al. A role for dendritic cells in bleomycin-induced pulmonary fibrosis in mice? American Journal of Respiratory and Critical Care Medicine. 2010;182(3):385–395. doi: 10.1164/rccm.200907-1164OC. [DOI] [PubMed] [Google Scholar]
- 130.Kim H. Y., Lee H. J., Chang Y.-J., et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nature Medicine. 2014;20(1):54–61. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wilson M. S., Madala S. K., Ramalingam T. R., et al. Bleomycin and IL-1β-mediated pulmonary fibrosis is IL-17A dependent. The Journal of Experimental Medicine. 2010;207(3):535–552. doi: 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hams E., Armstrong M. E., Barlow J. L., et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(1):367–372. doi: 10.1073/pnas.1315854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mulay S. R., Thomasova D., Ryu M., Anders H.-J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney International. 2012;81(12):1199–1211. doi: 10.1038/ki.2011.482. [DOI] [PubMed] [Google Scholar]
- 134.Mulay S. R., Thomasova D., Ryu M., et al. Podocyte loss involves MDM2-driven mitotic catastrophe. Journal of Pathology. 2013;230(3):322–335. doi: 10.1002/path.4193. [DOI] [PubMed] [Google Scholar]
- 135.Mulay S. R., Romoli S. R., Desai J., et al. Murine double minute-2 inhibition ameliorates established crescentic glomerulonephritis. The American Journal of Pathology. 2016;186(6):1442–1453. doi: 10.1016/j.ajpath.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 136.Majno G. The Healing Hand: Man and Wound in the Ancient World. Cambridge, Mass, USA: Harward University Press; 1975. [Google Scholar]