

Abstract
We investigated the impact of chronic hypercorticosteronemia (due to neonatal monosodium L-glutamate, MSG, and treatment) on liver oxidative stress (OS), inflammation, and carbohydrate/lipid metabolism in adult male rats. We evaluated the peripheral concentrations of several metabolic and OS markers and insulin resistance indexes. In liver we assessed (a) OS (GSH and protein carbonyl groups) and inflammatory (IL-1b, TNFa, and PAI-1) biomarkers and (b) carbohydrate and lipid metabolisms. MSG rats displayed degenerated optic nerves, hypophagia, low body and liver weights, and enlarged adipose tissue mass; higher peripheral levels of glucose, triglycerides, insulin, uric acid, leptin, corticosterone, transaminases and TBARS, and peripheral and liver insulin resistance; elevated liver OS, inflammation markers, and glucokinase (mRNA/activity) and fructokinase (mRNA). Additionally, MSG liver phosphofructokinase-2, glucose-6-phosphatase (mRNA and activity) and glucose-6-phosphate dehydrogenase, Chrebp, Srebp1c, fatty acid synthase, and glycerol-3-phosphate (mRNAs) were increased. In conclusion adult MSG rats developed an insulin-resistant state and increased OS and serious hepatic dysfunction characterized by inflammation and metabolic signs suggesting increased lipogenesis. These features, shared by both metabolic and Cushing's syndrome human phenotypes, support that a chronic glucocorticoid-rich endogenous environment mainly impacts on hepatic glucose cycle, displacing local metabolism to lipogenesis. Whether correcting the glucocorticoid-rich environment ameliorates such dysfunctions requires further investigation.
1. Introduction
Several studies have described the effects of neonatal monosodium L-glutamate (MSG) administration in rodents [1, 2]. It is openly accepted that MSG treatment induces morphological, behavioral, and endocrine abnormalities, such as growth disturbances, hyperadiposity, and hypogonadism [1, 3]. Other studies reported severe loss of catecholaminergic and peptidergic neurons in the retina and the hypothalamic arcuate nucleus (ARC) [3–5]. This neuronal loss alters several endocrine-metabolic functions, such as energy balance [6–8] and anterior pituitary hormone secretion [9, 10]. A conspicuous effect of MSG-induced hypothalamic damage is enhanced overall response of median eminence neurons terminals [9, 10]. The extensive brain damage has been used to explain several of the altered neuroendocrine functions in this model [5, 11]. ARC plays a pivotal role in regulation of energy storage and expenditure, and adipose tissue leptin secretion and hypothalamic leptin signaling system play key roles for maintaining homeostasis [12].
Among others, neonatal MSG treatment also affects white adipocyte function. MSG rats develop hyperleptinemia due to both enlarged adipose tissue mass and adipocyte size [13]. Moreover, chronic excess of endogenous glucocorticoids (GC) has been considered a main factor of the hypertrophic adiposity developed by the adult MSG rat [14]. Indeed, MSG animals are partly refractory to leptin inhibition (leptin resistance) of food intake and body weight gain [15] and, importantly, to the leptin negative control on adrenocortical function [16–18]. Moreover, the adult MSG rat phenotype is also characterized by an enhanced proinflammatory peripheral environment [19].
Because of the overall endocrine-metabolic characteristics, namely, hypercorticosteronemia and adipose tissue mass excess, the MSG rat is an excellent experimental model for basic investigation of different dysfunctions representative of the human Cushing's syndrome (hypothalamic obesity) phenotype [13, 14, 16–19]. Interestingly, it has been addressed that correcting their high circulating levels of GC various neuroendocrine and adiposity dysfunctions can be reversed [16, 18], including the hypertrophy of abdominal white adipocytes [14].
Endocrine and metabolic dysfunctions characterizing the hypothalamic obese rat phenotype have been largely revisited by several authors including us; however, whether a liver shift from carbohydrate to lipid metabolism sustaining liver/ectopic lipid deposition exists in MSG rats has not been addressed yet. Therefore, in the present study we examined whether neonatal MSG treatment later results in hepatic metabolism dysfunction by analysing local carbohydrate and lipid metabolic pathways. In addition, the peripheral concentrations of several metabolic-endocrine markers and tissue insulin sensitivity, oxidative stress (OS), and inflammation were evaluated.
2. Materials and Methods
2.1. Chemicals and Drugs
Reagents of the purest available grade were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Primary antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, California, USA). The secondary antibody, peroxidase-conjugated AffiniPure donkey anti-rabbit IgG, was provided by Dianova (Hamburg, Germany).
2.2. Experimental Animals
Adult male and female Wistar rats were allowed to mate in colony cages in a light (lights on from 07:00 to 19:00 h) and temperature (20–22°C) controlled room. Rat chow and water were available ad libitum. Pregnant rats were transferred to individual cages. Beginning on day 2 after parturition, newborn male pups were injected i.p. with either 4 mg/g BW MSG (Sigma Chemical Co., St. Louis, MO) dissolved in sterile 0.9% (w/v) NaCl or 10% (w/v) NaCl (littermate controls; C) once every two days up to 10 days of age [14]. Rats were weaned at 21 days of age and housed (3 rats per cage) in a controlled environment (20–22°C and lights on from 07:00 to 19:00 hs) with free availability to rat Purina chow and water. Thereafter, individual daily body weight and food intake were recorded until the experiment day (150 days of age). In each experiment, control (C) and MSG rats were members of the same litters; however, when accumulating experiments, each different experiment was performed with animals from different litters. On the morning (between 08:00 and 10:00 h) of the experiment day, overnight-fasting animals were weighed and euthanized and trunk blood was collected (into EDTA-coated tubes). The brain was immediately dissected out in order to check effectiveness of MSG treatment by macroscopic observation of degenerated optic nerves (inclusion criteria). Thereafter, visceral adipose tissue (VAT) pad and the liver were dissected and weighed. Next, hepatic medial lobes were excised to perform several other biochemical assays (see below). Animals were killed by decapitation according to protocols for animal use, in agreement with NIH Guidelines for care and use of experimental animals. All experimentation was approved by our Institutional Animal Care Committee.
2.3. Circulating Metabolites and Insulin Sensitivity Indexes
Glycemia was measured by the glucose-oxidase GOD-PAP method (Roche Diagnostics, Mannheim, Germany) and plasma concentrations of triglycerides, uric acid, and transaminases aspartate aminotransferase (GOT) and alanine aminotransferase (GPT) were determined by commercial (enzymatic-colorimetric) assays (Wiener Lab, Argentina). Peripheral immunoreactive insulin [13], leptin, and corticosterone [16] levels were determined by previously described specific radioimmunoassays. Lipid peroxidation was estimated by measuring TBARS (thiobarbituric acid-reactive substance) [20]. The amount of TBARS formed was calculated by extinction coefficient of the MDA (malondialdehyde)-TBA complex of 1.56 × 105 (mol/L)−1·cm−1 and expressed as pmol of MDA/mg of plasma protein per mL of plasma, measured with the Bio-Rad Protein Assay kit.
Fasting glycemia and insulin values were used to estimate peripheral insulin resistance by homeostasis model assessment-insulin resistance (HOMA-IR) [insulin (μUI/mL) × glycemia (mM)/22.5]. Liver insulin sensitivity index (LISI) was calculated with the following formula: k/(fasting plasma insulin) × fasting glycemia, where k = 22.5 × 18 [21].
2.4. Liver Protein Carbonyl Groups and Reduced Glutathione (GSH)
Protein carbonyl and GSH liver contents were determined as described elsewhere [22]. Briefly, both components were spectrophotometrically measured (at 366 nm for protein carbonyl groups and 414 nm for GSH) and results were expressed in nmol of carbonyl residues per mg of protein based on the molar extinction coefficient of 21,000 M−1 cm−1. The results of GSH content were expressed in μmol of –SH per g of tissue.
2.5. Total RNA Isolation
Total liver RNA was isolated using the TRIzol Reagent (Gibco-BRL, Rockville, MD, USA) as previously described [22]. The integrity and quality of the isolated RNA were checked by agarose-formaldehyde gel electrophoresis and by measuring the 260/280 nm absorbance ratio. DNA contamination was avoided by using DNase I digestion reagent (Gibco-BRL). Reverse transcription-PCR was performed with SuperScript III (Gibco-BRL) and total RNA (50 ng) as a template.
2.6. Liver mRNA Expression Levels Using Real-Time PCR (qPCR)
qPCR was performed with a Mini Opticon Real-Time PCR Detector Separate MJR (Bio-Rad), using SYBR Green I as fluorescent dye. cDNA (10 ng) was amplified in a qPCR reaction mixture containing 0.36 μM of each specific primer, 3 mM MgCl2, 0.2 mM dNTPs, and 0.15 μL Platinum Taq DNA Polymerase (6 U/μL) (Invitrogen). Samples were first denatured at 95°C for 3 min followed by 40 PCR cycles. Each cycle comprised a melting step at 95°C for 30 sec, an annealing step at 65°C for 30 sec, and an extension step at 72°C for 45 sec followed by a final extension at 72°C for 10 min. Specific oligonucleotide primers (Invitrogen) are shown in Table 1. Amplicons were designed in a size range of 90 to 250 bp, with Actinb used as housekeeping gene. Purity and specificity of amplified PCR products were verified by performing melting curves generated at the end of each PCR. Data are expressed as relative gene expression after normalization to Actinb using Qgene96 and LineRegPCR software.
Table 1.
Rat specific primers used for real-time PCR analyses.
| GBAN | bp | ||
|---|---|---|---|
| Actb | F, 5′-AGAGGGAAATCGTGCGTGAC-3′ | NM_031144 | 138 |
| R, 5′-CGATAGTGATGACCTGACCGT-3′ | |||
| Chrebp | F, 5′-CAGATGCGGGACATGTTTGA-3′ | NM_133552.1 | 205 |
| R, 5′-AATAAAGGTCGGATGAGGATGCT-3′ | |||
| Fas | F, 5′-GTCTGCAGCTACCCACCCGTG-3′ | NM_017332.1 | 214 |
| R, 5′-CTTCTCCAGGGTGGGGACCAG-3′ | |||
| Fk | F, 5′-ACGGATCGCAGGTGCCTAT-3′ | NM_031855.3 | 68 |
| R, 5′-AGCACAGTGCAGGAGTTGGA-3′ | |||
| Gck | F, 5′-GTGTACAAGCTGCACCCGA-3′ | NM_012565.1 | 156 |
| R, 5′-CAGCATGCAAGCCTTCTTG-3′ | |||
| Gpat | F, 5′-GACGAAGCCTTCCGAAGGA-3′ | AF_021348 | 68 |
| R, 5′-GACTTGCTGGCGGTGAAGAG-3′ | |||
| G6pase | F, 5′-GATCGCTGACCTCAGGAACGC-3′ | NM_013098.2 | 198 |
| R, 5′-AGAGGCACGGAGCTGTTGCTG-3′ | |||
| G6pdh | F, 5′-TTCCGGGATGGCCTTCTAC-3′ | NM_017006.2 | 81 |
| R, 5′-TTTGCGGATGTCATCCACTGT-3′ | |||
| Il1b | F, 5′-ACAAGGAGAGACAAGCAACGAC-3′ | NM_031512.2 | 140 |
| R, 5′-TCTTCTTTGGGTATTGTTTGGG-3′ | |||
| Pai1 | F, 5′-CCACGGTGAAGCAGGTGGACT-3′ | NM_012620.1 | 195 |
| R, 5′-TGCTGGCCTCTAAGAAGGGG-3′ | |||
| Pepck | F, 5′-TGCCCCAGGAAGTGAGGAAG-3′ | NM_198780.3 | 177 |
| R, 5′-GGTCAGTGAGAGCCAGCCAAC-3′ | |||
| Pfk2 | F, 5′-CGATCTATCTACCTATGCCGCCAT-3′ | NM_012621.4 | 256 |
| R, 5′-ACACCCGCATCAATCTCATTCA-3′ | |||
| Srebp1c | F, 5′-TTTCTTCGTGGATGGGGACT-3′ | XM_213329.5 | 208 |
| R, 5′-CTGTAGATATCCAAGAGCATC-3′ | |||
| Tnfa | F, 5′- GGCATGGATCTCAAAGACAACC-3′ | NM_012675.3 | 130 |
| R, 5′- CAAATCGGCTGACGGTGTG-3′ |
F: forward primer; R: reverse primer; GBAN: GenBank accession number; amplicon length, in bp.
2.7. Western Blot Analysis
Immunodetection of glucokinase (GCK), fructokinase (FK), phosphofructokinase-2 (PFK2), and ACTINB was performed with liver homogenates from each experimental group. Protein concentration was quantified by the Bio-Rad Protein Assay kit. Thereafter, dithiothreitol and bromophenol blue were added (final concentrations 100 mM and 0.1% w/v, resp.). Aliquots of 20 μg for GCK and 100 μg for PFK2 of whole protein were placed in reducing 10% (w/v) SDS-PAGE and electroblotted to polyvinylidene difluoride membranes. ACTINB density was used to normalize protein content: the relative content of target protein was divided by the relative ACTINB protein level in each group. Nonspecific binding sites of membranes were blocked by overnight incubation with nonfat dry milk at 4°C. Enzyme identification and quantification were performed with specific primary antibodies against FK and GCK (final dilution of 1 : 2000) [22] for 90 min, PFK2 antibody (final dilution of 1 : 10000) for 16 hours [23], and ACTINB antibody (final dilution of 1 : 10000) for 60 min. After the respective incubation period, membranes were rinsed in TBS and further incubated (1 h) with the corresponding secondary antibody: for GCK and PFK2, anti-sheep IgG streptavidin-peroxidase conjugate and anti-chicken IgY peroxidase-labelled were used, respectively, and for ACTINB biotinylated anti-mouse IgG was used. Diaminobenzidine (DAB, Sigma Co.) was used for color development. Bands were quantified by densitometry using Gel-Pro Analyser software.
2.8. Glucokinase Activity
Freshly removed liver pieces were immediately homogenized in hand-held homogenizers (20 times) containing ice cold phosphate saline buffer, with 0.1 mM PMSF, 0.1 mM benzamidine, 2 mM DTT, 4 μg/mL aprotinin, and 0.3 M sucrose (pH 7.5). Homogenates were then passed through a 23-gauge needle syringe (5 times) to ensure appropriate sample mixing. Aliquots of homogenates were centrifuged at 600 ×g to separate and discard the nuclear fraction. Supernatants were centrifuged at 8,000 ×g and 100,000 ×g at 4°C, collected, and identified as cytosolic fractions. Phosphorylation in the 100,000 ×g soluble cytosolic fractions was measured at 37°C, pH 7.4, by recording increasing absorbance (measured at 340 nm) in a well-established enzyme-coupled photometric assay containing glucose-6-phosphate dehydrogenase (G6PDH), ATP, and NADP [23]. For each assay, five different experiments were performed in triplicate. GCK activity was calculated by subtracting activity measured at 1 mM glucose (hexokinase) from that measured at 100 mM glucose. Enzyme activity was then expressed in mU per milligram of protein. One unit of enzyme activity was defined as 1 μmol glucose-6-phosphate formed from glucose and ATP per minute at 37°C.
2.9. Glucose-6-Phosphatase (G6Pase) Activity
Homogenization of liver samples and isolation of local microsomes were done as described by Nordlie and Arion [24]. Briefly, the homogenization medium was 0.25 M sucrose/5 mM Tris-acetate/0.5 mM Na-EDTA, pH 7.4 (3 mL per g of tissue). Microsomes were washed once with 0.25 M sucrose/5 mM Tris-acetate, pH 7.4, and centrifuged at 100,000 ×g. Untreated microsomes were diluted to the desired final concentration with the sucrose buffered solution and then assayed without any other further treatment. Fully disrupted microsomes were prepared at 0°C by adding 0.1 mL of 0.75% (w/v) Triton X-100 and 0.9 mL of untreated microsomes (containing approximately 10 mg of protein) and allowed to stand in an ice bath for 20 min. The reaction was stopped by adding 250 μL of 10% (w/v) TCA and 2 mL of MoNH4 (diluted in H2SO4 1 N) plus 320 μL of FeSO4 (diluted in H2SO4 0.15 N) to 200 μL of sample. The OD at 660 nm was determined photometrically and results were expressed as “latency.” Latency was calculated by the following formula: 100 × (activity measured in disrupted microsomes minus activity measured in untreated microsomes)/activity measured in disrupted microsomes [25].
2.10. Fructokinase Activity
Pieces of liver were homogenized in buffer containing 25 mM HEPES (pH 7.1), 100 mM KCl, 1 mM DTT, and 0.1 mM EDTA and spun at 10,000 ×g at 4°C for 20 min. We measured FK activity by using a coupled enzymatic assay based on existing methods [26]. Briefly, 20 μL of the sample was added to 200 μL of the reaction mixture containing 25 mM HEPES (pH 7.1), 6 mM MgCl2, 25 mM KCl, 10 mM NaF, 5 mM D-fructose, 0.2 mM NADH, 1 mM phosphoenolpyruvate, 40 U/mL pyruvate kinase, 40 U/mL lactate dehydrogenase, and 50 mM N-acetyl-D-glucosamine (to inhibit hexokinase activity). The reaction was started by adding 10 μL of ATP (5 mM final concentration) and quantitatively measured according to changes in optical density at 340 nm (30 min).
2.11. Statistical Analysis
Statistical analysis was performed by ANOVA, followed by Dunnett's test for multiple comparisons with the Prism analysis program (GraphPad). Bartlett's test was used to assess variance homogeneity. Results were expressed as means (±SEM) of the indicated number of observations; differences were considered significant when P was less than 0.05 [22].
3. Results
3.1. The MSG Rat Phenotype
On the experimental day, MSG rats showed degenerated optic nerves (macroscopic observation) and other characteristics of this model, such as (a) low body weight, (b) high visceral adipose tissue mass, and (c) hypophagia (average of individual daily food intake between 60 and 150 days of age), thus confirming several features of the complete MSG phenotype (Table 2). Moreover, relative wet liver weight (expressed as individual liver weight per 100 g body weight) was also significantly (P < 0.05 versus C rats) reduced in MSG rats (Table 2).
Table 2.
Body weight (BW), daily food intake, wet tissue (visceral adipose tissue, VAT, and liver), and mass and peripheral levels of several biomarkers in control (C) and MSG rats.
| C | MSG | |
|---|---|---|
| Body weight (g) | 404.6 ± 9.1 | 349.3 ± 7.6∗ |
| Food intake (g/day) | 20.81 ± 1.95 | 15.61 ± 0.87∗ |
| VAT mass (g) | 4.47 ± 0.88 | 14.18 ± 1.77∗ |
| Liver weight (g/100 g BW) | 3.19 ± 0.07 | 2.93 ± 0.06∗ |
| Glycemia (g/L) | 1.06 ± 0.02 | 1.17 ± 0.03 |
| Insulin (ng/mL) | 0.81 ± 0.05 | 1.37 ± 0.19∗ |
| Leptin (ng/mL) | 1.39 ± 0.37 | 16.49 ± 2.38∗ |
| Triglycerides (g/L) | 1.06 ± 0.07 | 1.85 ± 0.18∗ |
| Uric acid (mg/dL) | 1.19 ± 0.09 | 1.82 ± 0.22∗ |
| Corticosterone (μg/dL) | 6.19 ± 0.98 | 13.75 ± 1.12∗ |
| GOT (U/L) | 75.5 ± 3.2 | 96.9 ± 5.8∗ |
| GPT (U/L) | 9.51 ± 0.69 | 15.49 ± 1.19∗ |
Values are means ± SEM. ∗ P < 0.05 versus C values (n = 12 rats per group).
3.2. Circulating Metabolites and Insulin Sensitivity Indexes
MSG rats had significantly (P < 0.05 versus C rats) higher plasma insulin and triglyceride levels, despite similar glycemia (Table 2). Uric acid levels in MSG animals also were higher (P < 0.05 versus C values), as well as those of GOT and GPT. These altered parameters are clearly indicative of liver dysfunction (Table 2). Regarding two specific peripheral markers in this animal model, leptin and corticosterone concentrations, we found both hormones several times higher (P < 0.05 versus C values) in MSG rats (Table 2).
Finally, after calculation, HOMA-IR and LISI values were significantly (P < 0.05) higher and lower, respectively, in MSG (11.35 ± 0.98 and 1.99 ± 0.19, resp.) than in C (5.35 ± 0.46 and 3.24 ± 0.31, resp.) animals (n = 12 rats per group).
3.3. Peripheral and Liver Oxidative Stress Markers
Circulating levels of TBARS, a marker of peripheral OS, were significantly (P < 0.05 versus C values) higher in MSG animals (Table 3). Concordant with this finding, liver content of protein carbonyl groups also was significantly (P < 0.05 versus C) higher in MSG rats (Table 3), whereas the local amount of GSH, a peptide highly protective against OS development, was significantly (P < 0.05 versus C) lower in MSG rats (Table 3).
Table 3.
Peripheral and liver (n = 12 and 6 specimens per group, resp.) oxidative stress markers in control (C) and MSG rats.
| C | MSG | |
|---|---|---|
| Peripheral TBARS (pmol/mg of protein per mL of plasma) | 40.3 ± 3.6 | 85.1 ± 15.2∗ |
| Liver protein carbonyl groups (nmol/mg of tissue protein) | 4.43 ± 0.15 | 7.81 ± 1.32∗ |
| Liver GSH (nmol/g tissue) | 2.71 ± 0.03 | 2.05 ± 0.04∗ |
Values are means ± SEM. ∗ P < 0.05 versus C values.
3.4. Neonatal MSG Treatment Resulted in Liver Inflammation and Dysmetabolism
The livers from MSG rats displayed a significant (P < 0.05 versus C values) increase in the mRNA levels of Il1b, Tnfa, and Pai-1 (Figures 1(a), 1(b), and 1(c), resp.), all key signals indicative of local inflammatory process.
Figure 1.

Liver mRNA levels of Il1b, Tnfa, and Pai-1 (panels (a), (b), and (c), resp.) in control (white bars) and MSG rats (black bars). Results are means ± SEM (n = 8 rats per group). ∗ P < 0.05 versus control values. Statistical analysis was performed by ANOVA, followed by Dunnett's test for multiple comparisons.
Although no group difference was observed in hepatic glycogen content (data not shown), MSG livers showed a significantly higher Gck mRNA expression (1.77 ± 0.31 versus 1.01 ± 0.26 AU; P < 0.05 versus C; n = 8 rats per group). Moreover, GK protein content and activity were also significantly (P < 0.05 versus C) higher in MSG livers (Figures 2(a) and 2(b), resp.). Tissues from MSG rats displayed a robust increase (P < 0.05 versus C) in PFK2 mRNA expression and protein content (Figures 2(c) and 2(d), resp.); similarly, G6Pase mRNA expression and activity also were significantly (P < 0.05 versus C) higher in MSG tissues (Figures 2(e) and 2(f), resp.).
Figure 2.
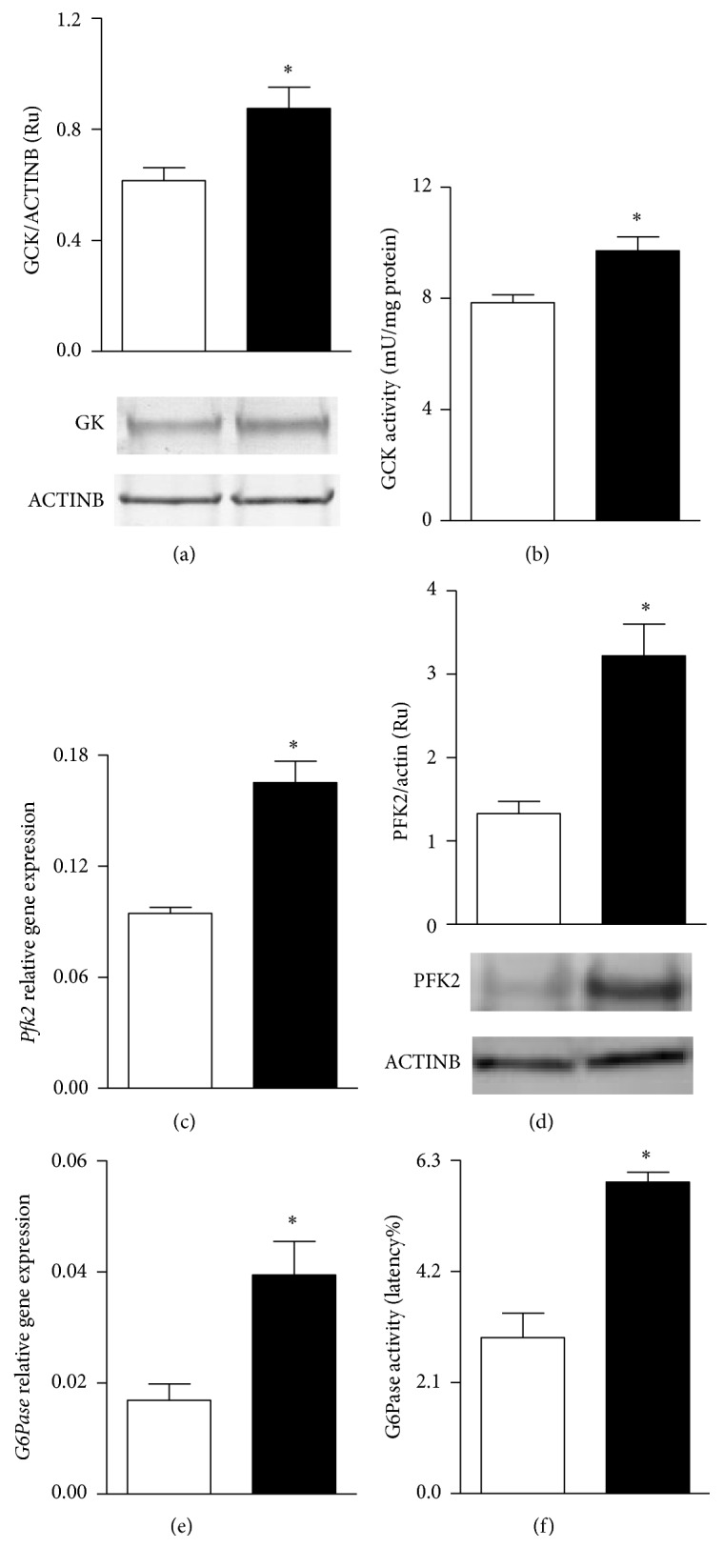
Hepatic protein content and activity levels of GCK (panels (a) and (b), resp.), mRNA levels and protein content of PFK2 (panels (c) and (d), resp.), and mRNA levels and enzyme activity of G6Pase (panels (e) and (f), resp.) in control (white bars) and MSG-treated rats (black bars). Results are means ± SEM (n = 8 rats per group). ∗ P < 0.05 versus control values. Statistical analysis was performed by ANOVA, followed by Dunnett's test for multiple comparisons.
The mRNA levels of G6pdh and Chrebp were also significantly (P < 0.05 versus C) increased in MSG tissues (Figures 3(a) and 3(b), resp.), despite no group difference in that of phosphoenolpyruvate carboxykinase (Pepck) (Figure 3(c)).
Figure 3.

Hepatic G6pdh (panel (a)), Chrebp (panel (b)), and Pepck (panel (c)) gene expression in control (white bars) and MSG-treated rats (black bars). Results are means ± SEM (n = 8 rats per group). ∗ P < 0.05 versus control values. Statistical analysis was performed by ANOVA, followed by Dunnett's test for multiple comparisons.
Finally, regarding the lipogenic pathway, FK mRNA expression and protein content were significantly (P < 0.05 versus C values) augmented in MSG tissues (1.93 ± 0.16 versus 1.02 ± 0.12 AU and 4.69 ± 0.58 versus 2.11 ± 0.49 AU, resp.; n = 8 rats per group). Interestingly, FK activity was drastically (P < 0.05 versus C) reduced in livers from MSG animals (Figure 4(a)). Moreover, liver lipogenesis-related genes expressions were modified by the neonatal MSG treatment; indeed the liver mRNA expression levels of Srebp1c, Fas, and Gpat were 2-fold higher (P < 0.05 versus C) in MSG tissues (Figures 4(b), 4(c), and 4(d), resp.).
Figure 4.

Liver FK activity (panel (a)), mRNA levels of Srebp1c (panel (b)), Fas (panel (c)), and Gpat (panel (d)) in control (white bars) and MSG rats (black bars). Results are means ± SEM (n = 8 rats per group). ∗ P < 0.05 versus control values. Statistical analysis was performed by ANOVA, followed by Dunnett's test for multiple comparisons.
4. Discussion
The hypothalamus is a key brain structure in charge of the control of energy balance, and its injury is a major cause of neuroendocrine (hypothalamic) obesity development [27]. Conspicuous neuronal loss at the hypothalamic level can certainly be induced by the neonatal administration of MSG [28]. As mentioned above, in adult age, MSG-damaged rats have already installed hypophagy, low body weight, hyperinsulinemia, hyperleptinemia, hyperuricemia, hyperadiposity, inflammation, and increased plasma levels of corticosterone, TBARS, and lipids [19, 29]. In the present study we were able to find that neonatal MSG treatment induced a wide neuroendocrine-metabolic alteration once adult age was reached. It must be borne in mind that one of the expected consequences of neonatal MSG treatment in rats is the development of stunted growth, thus reaching adult age with a paradoxical phenotype characterized by hyperadiposity [13, 14, 16–19] and decreased body and several tissues weights [3]. Chronic excess of endogenous GC, in turn, will induce protein breakdown (chronic hypercatabolic state), thus resulting in hyperuricemic phenotype [30]. Both metabolic and Cushing's syndrome human phenotypes [30, 31] share hyperuricemia, and it has been claimed that this high uric acid peripheral level may have a key pathogenic role in several metabolic syndrome phenotypes [32], because, among others, uric acid peripheral levels correlate well with those of insulin and triglycerides in fructose-induced metabolic syndrome and, interestingly, correcting the IR state the hypertriglyceridemia and hyperuricemia can be overridden [32]. Aside from the disturbances mentioned above, we determined in this rat phenotype a deeply dysfunctional liver, as indicated by wet tissue weight, carbohydrate and lipid metabolisms, and local increase in OS and inflammatory markers. It should be stressed that in the adult male MSG rat no previous integrative hepatic studies on the carbohydrate and lipid metabolic functions and the inflammatory and OS processes have been examined.
As shown by the endogenous OS status, the higher HOMA-IR and lower LISI values, this being the first work demonstrating the latter, recorded in MSG animals clearly indicate that these rats developed OS-related peripheral and liver IR. However, it remains to be precisely ascertained whether long-term exposure to high circulating leptin levels [14] could be a key cooperative factor for the establishment of an overall IR state [13, 28], namely, at the liver level. It is also plausible that, as occurred at the adrenal level [13, 16], liver IR could be reversed by correcting endogenous overproduction of glucocorticoid and leptin.
High glucocorticoid treatment is known to induce cell inflammation and to enhance OS in hepatocytes by increasing and decreasing total oxidative and antioxidant capacities, respectively [33]. High OS in the MSG liver could result from endogenous enhanced reactive oxygen species production and/or deficient antioxidant mechanism [34, 35], consequently designating high OS as a key pathogenic factor in the initiation and progression of metabolic diseases, including hepatic steatosis [36] and type 2 diabetes [34, 37]. Strong evidence also indicates that intracellular signals generated by OS may also induce chronic inflammation together with insulin resistance in vivo [38, 39]. High fat diet intake in rodents is able to increase OS at both liver and adipose tissue levels [40], and Bloch-Damti et al. [41] claimed that local OS increased IRS-1 and IRS-2 serine phosphorylations, decreased IRS-1 protein content, and impaired insulin sensitivity in 3T3-L1 adipocytes. Interestingly, Park et al. [42] also observed that MSG administration in rodents reduces liver antioxidant biomarkers, such as glutathione level and activities of various antioxidant enzymes, also enhancing local TBARS content.
We found that our MSG rats are dyslipidemic, tallying with the stimulated liver lipogenic process revealed by elevated expression levels of their master regulator gene, Srebp1c, as well as their target genes, Fas and Gpat [43]. Although other authors have reported similar changes in lipogenic genes [44], we also found an enhanced gene expression and reduced activity in liver FK, enzyme providing fructose-1-P that, in turn, renders trioses for entering into triglyceride synthesis. This phenomenon clearly indicates that when needed substrates are available, local synthesis of triacylglycerols is expected [45, 46].
It should be considered that, within an insulin-resistant state scenario, we were able to determine that, in chronic hypercorticosteronemic MSG rats, similarly to that previously found in Cushing's patients [47], the liver high activity of the two key enzymes, GCK and G6Pase, assures (within a very active futile cycle) a drastic increase of high glucose liver content. Although a previous study has reported an enhanced activity of G6Pase in MSG animals [48], to our knowledge this is the first report of an active involvement of GCK, the hepatic glucose sensor, in the changes induced by MSG. These data are in agreement with and supported by those indicating that hypercortisolemic-hyperinsulinemic (IR) patients did not develop hyperglycemia due to their reduced peripheral glucose uptake [47]. Indeed, our MSG rats, sharing with those patients the high GC and insulin peripheral levels among other conditions, were fully able to maintain a normal glycemia. These data probably indicate that GC, through a liver glucocorticoid receptor- (GR-) mediated effect [49], could be responsible for further distribution of the intrahepatic glucose store. In this regard, if glucose did not enter in the MSG liver glycogenogenic process, indeed we and other researchers [50] did not notice any group difference in liver glycogen content. The fact that this cycle is highly active in our normoglycemic MSG rats suggests that glucose cycling could be a common feature of the metabolic syndrome and in nonhyperglycemic Cushing's syndrome phenotypes [47, 51]; moreover, the enhanced glucose cycle activity has been claimed to counteract hyperglycemia in hyperglycemic Cushing's syndrome patients [47], ob/ob mice [52], and diabetic rats [53]. Because glucose-6-phosphate (G6P) is a branching point for glucose fate, it is also plausible to expect that glucose is metabolized through either or both of the following pathways: (a) being channeled into the hexose monophosphate shunt pathway due to enhanced G6PDH activity, thereby producing cell reducing power (as NADPH) later used for hepatocyte lipogenesis and replenishing GSH pool, hence providing an additional mechanism to counteract OS [54], and/or (b) G6P being isomerized to fructose-6-phosphate (F6P), then oxidized to pyruvate and acetyl-CoA, and finally entering into the lipid anabolic pathway. This/these possibility/ies could be reinforced by our finding of a robust increase in PFK2, a potent cytosolic activator of GCK [23] in the MSG liver, thus suggesting that, in this model, there is an active role of the posttranslational regulatory mechanisms of GCK activity. It is known that GCK is also expressed in neurons, pituitary cells, pancreatic B-cells, and enteroendocrine K and L cells [55], an overall cell network acting as a glucose sensor for maintaining glucose homeostasis. Therefore, further research focused on the fact that whether or not the neonatal MSG treatment broadly impairs this glucose sensor net is required.
MSG treatment also yielded increased liver Chrebp gene expression, a parameter well correlating with increased expression of glycolysis-related genes and a key factor in the activation of promoter genes involved in the lipogenic process. In fact, Chrebp has been designated as a central link between these two pathways in hepatocytes [56]. Kabashima et al. [57] reported that enhanced activity of the hexose monophosphate shunt pathway leads to intrahepatocyte increase in xylulose-5-phosphate, which in turn activates phosphoprotein phosphatase A2, a main activator of Chrebp. Our results showing high gene expression of G6pdh, a rate limiting enzyme of the hexose monophosphate shunt pathway, demonstrate for the first time that the abovementioned mechanisms remained fully operative in MSG animals. Additionally, as mentioned above, we noticed that FK protein and activity were drastically augmented and reduced, respectively, in livers from MSG rats. It must be borne in mind that the liver lipid metabolism in MSG rats is clearly displaced to enhanced lipogenesis; thus it could be speculated that, in the postprandial condition any further liver entry of hexoses, through FK downstream reactions, could result in dramatic local lipid overproduction because carbons enter through this pathway bypass glycolysis regulatory steps and are then used in the lipogenic process in a direct and an uncontrolled fashion [45–47]. Therefore, it could be hypothesized that inhibited liver FK activity could represent a novel adaptive (protective feedback) mechanism in the MSG liver against further detrimental overlipogenic process in hepatocytes (e.g., hepatic steatosis). In spite of chronic endogenous glucocorticoid excess in MSG rats, it has been clearly established that white adipocytes isolated from these rats are partly refractory to dexamethasone-elicited leptin secretion and that adipocyte GC resistance can be fully reversed shortly after correction of endogenous GC overproduction [13]. Hence, it is also possible that endogenous GC-rich environment in MSG rats could be responsible for liver dysmetabolism [58]. In fact, we recently noticed that, after three weeks of bilateral adrenalectomy, adult male MSG rats normalized their plasmatic concentrations of leptin, triglyceride, uric acid, and transaminases (data not shown). However, because MSG animals developed hyperleptinemia early in life (30 days of age) [14], a detrimental leptin effect on liver metabolism must not be discarded. In this regard, it has previously been assessed that livers from DIO mice are highly susceptible to inflammation, OS, and nonalcoholic hepatic steatosis development; conversely, leptin KO-DIO mice are protected from these dysfunctions [59]. It must be considered that MSG rats also developed an early peripheral leptin resistance [13], a dysfunction fully reversed after normalization of endogenous GC environment [13]; therefore, this leptin-resistance condition could be contributing to liver carbohydrate dysmetabolism. Altogether, our data strongly suggest that adult, neonatally damaged, MSG rats are able to remain prediabetic (normal basal glycemia) due to enhanced entry of glucose into the liver (high glucose cycle activity), resulting in a relevant glucose contribution to increased liver lipogenesis and thus local and ectopic lipid deposition.
5. Conclusions
In conclusion, our study clearly supports that, in adult age, neonatally damaged MSG male rats are overall insulin-resistant and with increased OS (at both peripheral and liver levels). These features are accompanied by a dramatic dysfunction in hepatic metabolism: enhanced glucose cycling (GCK/G6Pase activities) and increased pentose phosphate shunt, together with adaptive changes in FK activity. Indeed, MSG rats displayed several features characteristic of the human Cushing's and metabolic syndrome phenotypes, including a decreased liver weight, high tissue inflammation, and local metabolism highly displaced to an increase in lipid production.
It remains to be determined whether the MSG liver dysfunction can be ameliorated by correcting endogenous glucocorticoid [14, 18], insulin [18, 60], and leptin [14, 18] overproduction. Finally, the risk factor of MSG liver imbalance in the acyl-ceramidase/ceramide [61] cycle, a major cooperative mechanism responsible for development of hepatic steatosis, requires further investigation.
Acknowledgments
The authors are grateful to Mrs. S. H. Rogers and Rebecca Doyle for careful paper edition/correction. Eduardo Spinedi, María Laura Massa, and Flavio Francini are Research Career Awardees from National Research Council of Argentina (CONICET). This work was partially supported by grants from CONICET (PIP-0371-2012) and the Swiss Foundation for Studies on Endocrinology, Diabetology and Metabolism (FPREDM 062013).
Competing Interests
All authors declare there is no conflict of interests.
References
- 1.Olney J. W., Sharpe L. G. Brain lesions in an infant rhesus monkey treated with monosodium glutamate. Science. 1969;166(3903):386–388. doi: 10.1126/science.166.3903.386. [DOI] [PubMed] [Google Scholar]
- 2.Burde R. M., Schainker B., Kayes J. Acute effect of oral and subcutaneous administration of monosodium glutamate on the arcuate nucleus of the hypothalamus in mice and rats. Nature. 1971;233(5314):58–60. doi: 10.1038/233058a0. [DOI] [PubMed] [Google Scholar]
- 3.Redding T. W., Schally A. V., Arimura A., Wakabayashi I. Effect of monosodium glutamate on some endocrine functions. Neuroendocrinology. 1971;8(3):245–255. doi: 10.1159/000122011. [DOI] [PubMed] [Google Scholar]
- 4.Nemeroff C. B., Konkol R. J., Bissette G., et al. Analysis of the disruption in hypothalamic pituitary regulation in rats treated neonatally with monosodium l glutamate (MSG): evidence for the involvement of tuberoinfundibular cholinergic and dopaminergic systems in neuroendocrine regulation. Endocrinology. 1977;101(2):613–622. doi: 10.1210/endo-101-2-613. [DOI] [PubMed] [Google Scholar]
- 5.Holzwarth McBride M. A., Sladek J. R., Jr., Knigge K. M. Monosodium glutamate induced lesions of the arcuate nucleus. II. Fluorescence histochemistry of catecholamines. Anatomical Record. 1976;186(2):197–205. doi: 10.1002/ar.1091860206. [DOI] [PubMed] [Google Scholar]
- 6.Krieger D. T., Liotta A. S., Nicholsen G., Kizer J. S. Brain ACTH and endorphin reduced in rats with monosodium glutamate-Induced arcuate nuclear lesions. Nature. 1979;278(5704):562–563. doi: 10.1038/278562a0. [DOI] [PubMed] [Google Scholar]
- 7.Pelletier G. Ultrastructural localization of neuropeptide Y in the hypothalamus. Annals of the New York Academy of Sciences. 1990;611:232–246. doi: 10.1111/j.1749-6632.1990.tb48935.x. [DOI] [PubMed] [Google Scholar]
- 8.Billington C. J., Briggs J. E., Harker S., Grace M., Levine A. S. Neuropeptide Y in hypothalamic paraventricular nucleus: a center coordinating energy metabolism. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 1994;266(6):R1765–R1770. doi: 10.1152/ajpregu.1994.266.6.R1765. [DOI] [PubMed] [Google Scholar]
- 9.Miller R. J., Dawson G. Neuoreceptors: an overview. In: Pepeu G., Kuhar M. J., Enna S. J., editors. Receptor for Neurotransmitters and Peptide Hormones. New York, NY, USA: Raven Press; 1979. pp. 11–19. [Google Scholar]
- 10.DePaolo L. V., Negro-Vilar A. Neonatal monosodium glutamate treatment alters the response of median eminence luteinizing hormone-releasing hormone nerve terminals to potassium and prostaglandin E2. Endocrinology. 1982;110(3):835–841. doi: 10.1210/endo-110-3-835. [DOI] [PubMed] [Google Scholar]
- 11.Badger T. M., Millard W. J., Martin J. B., Rosenblum P. M., Levenson S. E. Hypothalamic-pituitary function in adult rats treated neonatally with monosodium glutamate. Endocrinology. 1982;111(6):2031–2038. doi: 10.1210/endo-111-6-2031. [DOI] [PubMed] [Google Scholar]
- 12.Kalra S. P., Dube M. G., Pu S., Xu B., Horvath T. L., Kalra P. S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocrine Reviews. 1999;20(1):68–100. doi: 10.1210/er.20.1.68. [DOI] [PubMed] [Google Scholar]
- 13.Moreno G., Perelló M., Camihort G., et al. Impact of transient correction of increased adrenocortical activity in hypothalamo-damaged, hyperadipose female rats. International Journal of Obesity. 2006;30(1):73–82. doi: 10.1038/sj.ijo.0803109. [DOI] [PubMed] [Google Scholar]
- 14.Zubiría M. G., Vidal-Bravo J., Spinedi E., Giovambattista A. Relationship between impaired adipogenesis of retroperitoneal adipose tissue and hypertrophic obesity: role of endogenous glucocorticoid excess. Journal of Cellular and Molecular Medicine. 2014;18(8):1549–1561. doi: 10.1111/jcmm.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson R., Pelleymounter M. A., Millard W. J., Liu S., Eppler B. Attenuation of leptin-mediated effects by monosodium glutamate-induced arcuate nucleus damage. American Journal of Physiology—Endocrinology and Metabolism. 1997;273(1):E202–E206. doi: 10.1152/ajpendo.1997.273.1.E202. [DOI] [PubMed] [Google Scholar]
- 16.Perelló M., Gaillard R. C., Chisari A., Spinedi E. Adrenal enucleation in MSG-damaged hyperleptinemic male rats transiently restores adrenal sensitivity to leptin. Neuroendocrinology. 2003;78(3):176–184. doi: 10.1159/000072799. [DOI] [PubMed] [Google Scholar]
- 17.Perelló M., Moreno G., Camihort G., et al. Nature of changes in adrenocortical function in chronic hyperleptinemic female rats. Endocrine. 2004;24(2):167–175. doi: 10.1385/ENDO:24:2:167. [DOI] [PubMed] [Google Scholar]
- 18.Perelló M., Moreno G., Gaillard R. C., Spinedi E. Glucocorticoid-dependency of increased adiposity in a model of hypothalamic obesity. Neuroendocrinology Letters. 2004;25(1-2):119–126. [PubMed] [Google Scholar]
- 19.Castrogiovanni D., Gaillard R. C., Giovambattista A., Spinedi E. Neuroendocrine, metabolic, and immune functions during the acute phase response of inflammatory stress in monosodium l-glutamate-damaged, hyperadipose male rat. Neuroendocrinology. 2008;88(3):227–234. doi: 10.1159/000124131. [DOI] [PubMed] [Google Scholar]
- 20.Fariña J. P., Garćia M. E., Alzamendi A., et al. Antioxidant treatment prevents the development of fructose-induced abdominal adipose tissue dysfunction. Clinical Science. 2013;125(2):87–97. doi: 10.1042/cs20120470. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda M., DeFronzo R. A. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22(9):1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 22.Castro M. C., Francini F., Schinella G., et al. Apocynin administration prevents the changes induced by a fructose-rich diet on rat liver metabolism and the antioxidant system. Clinical Science. 2012;123(12):681–692. doi: 10.1042/CS20110665. [DOI] [PubMed] [Google Scholar]
- 23.Massa L., Baltrusch S., Okar D. A., Lange A. J., Lenzen S., Tiedge M. Interaction of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) with glucokinase activates glucose phosphorylation and glucose metabolism in insulin-producing cells. Diabetes. 2004;53(4):1020–1029. doi: 10.2337/diabetes.53.4.1020. [DOI] [PubMed] [Google Scholar]
- 24.Nordlie R. C., Arion W. J. Glucose-6-phosphatase. In: Wood W. A., editor. Methods in Enzymology. IX. New York, NY, USA: Academic Press; 1996. pp. 619–625. [Google Scholar]
- 25.Lange A. J., Arion W. J., Beaudet A. L. Type Ib glycogen storage disease is caused by a defect in the glucose-6-phosphate translocase of the microsomal glucose-6-phosphatase system. The Journal of Biological Chemistry. 1980;255(18):8381–8384. [PubMed] [Google Scholar]
- 26.Ouyang X., Cirillo P., Sautin Y., et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. Journal of Hepatology. 2008;48(6):993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macho L., Ficková M., Ježová D., Zórad Š. Late effects of postnatal administration of monosodium glutamate on insulin action in adult rats. Physiological Research. 2000;49(1):S79–S85. [PubMed] [Google Scholar]
- 28.Nakayama D., Magami Y., Azuma T., et al. Turnover of acinar and islet cells in the pancreas of monosodium glutamate-treated obese mice. Obesity Research. 2003;11(1):87–94. doi: 10.1038/oby.2003.14. [DOI] [PubMed] [Google Scholar]
- 29.Seiva F. R. F., Chuffa L. G. A., Braga C. P., Amorim J. P. A., Fernandes A. A. H. Quercetin ameliorates glucose and lipid metabolism and improves antioxidant status in postnatally monosodium glutamate-induced metabolic alterations. Food and Chemical Toxicology. 2012;50(10):3556–3561. doi: 10.1016/j.fct.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Faggiano A., Pivonello R., Melis D., et al. Nephrolithiasis in Cushing's disease: prevalence, etiopathogenesis, and modification after disease cure. The Journal of Clinical Endocrinology & Metabolism. 2003;88(5):2076–2080. doi: 10.1210/jc.2002-021494. [DOI] [PubMed] [Google Scholar]
- 31.Tsouli S. G., Liberopoulos E. N., Mikhailidis D. P., Athyros V. G., Elisaf M. S. Elevated serum uric acid levels in metabolic syndrome: an active component or an innocent bystander? Metabolism: Clinical and Experimental. 2006;55(10):1293–1301. doi: 10.1016/j.metabol.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa T., Hu H., Zharikov S., et al. A causal role for uric acid in fructose-induced metabolic syndrome. American Journal of Physiology—Renal Physiology. 2006;290(3):F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 33.Aboelwafa H. R., Yousef H. N. The ameliorative effect of thymol against hydrocortisone-induced hepatic oxidative stress injury in adult male rats. Biochemistry and Cell Biology. 2015;93(4):282–289. doi: 10.1139/bcb-2014-0154. [DOI] [PubMed] [Google Scholar]
- 34.Padmalayam I., Hasham S., Saxena U., Pillarisetti S. Lipoic acid: synthase (LASY): a novel role in inflammation, mitochondrial function, and insulin resistance. Diabetes. 2009;58(3):600–608. doi: 10.2337/db08-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazarin M. D. O., Ishii-Iwamoto E. L., Yamamoto N. S., et al. Liver mitochondrial function and redox status in an experimental model of non-alcoholic fatty liver disease induced by monosodium l-glutamate in rats. Experimental and Molecular Pathology. 2011;91(3):687–694. doi: 10.1016/j.yexmp.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Lanaspa M. A., Sanchez-Lozada L. G., Cicerchi C., et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE. 2012;7(10, article e47948) doi: 10.1371/journal.pone.0047948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baynes J. W. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40(4):405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- 38.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 39.Fridlyand L. E., Philipson L. H. Oxidative reactive species in cell injury: mechanisms in diabetes mellitus and therapeutic approaches. Annals of the New York Academy of Sciences. 2005;1066:136–151. doi: 10.1196/annals.1363.019. [DOI] [PubMed] [Google Scholar]
- 40.Matsuzawa-Nagata N., Takamura T., Ando H., et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57(8):1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Bloch-Damti A., Potashnik R., Gual P., et al. Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction of insulin resistance by oxidative stress. Diabetologia. 2006;49(10):2463–2473. doi: 10.1007/s00125-006-0349-6. [DOI] [PubMed] [Google Scholar]
- 42.Park C. H., Kim M. Y., Sok D.-E., Kim J. H., Lee J. H., Kim M. R. Butterbur (Petasites japonicus Max.) extract improves lipid profiles and antioxidant activities in monosodium L-glutamate-challenged mice. Journal of Medicinal Food. 2010;13(5):1216–1223. doi: 10.1089/jmf.2009.1380. [DOI] [PubMed] [Google Scholar]
- 43.Shimomura I., Shimano H., Korn B. S., Bashmakov Y., Horton J. D. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. The Journal of Biological Chemistry. 1998;273(52):35299–35306. doi: 10.1074/jbc.273.52.35299. [DOI] [PubMed] [Google Scholar]
- 44.Bonfleur M. L., Borck P. C., Ribeiro R. A., et al. Improvement in the expression of hepatic genes involved in fatty acid metabolism in obese rats supplemented with taurine. Life Sciences. 2015;135:15–21. doi: 10.1016/j.lfs.2015.05.019.14403 [DOI] [PubMed] [Google Scholar]
- 45.Mayes P. A. Intermediary metabolism of fructose. The American Journal of Clinical Nutrition. 1993;58(supplement 5):754S–765S. doi: 10.1093/ajcn/58.5.754S. [DOI] [PubMed] [Google Scholar]
- 46.Bray G. A., Nielsen S. J., Popkin B. M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. The American Journal of Clinical Nutrition. 2004;79(4):537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- 47.Heaney A. P., Harper R., Ennis C., et al. Insulin action and hepatic glucose cycling in Cushing's syndrome. Clinical Endocrinology. 1997;46(6):735–743. doi: 10.1046/j.1365-2265.1997.2121024.x. [DOI] [PubMed] [Google Scholar]
- 48.Diniz Y. S., Fernandes A. A. H., Campos K. E., Mani F., Ribas B. O., Novelli E. L. B. Toxicity of hypercaloric diet and monosodium glutamate: oxidative stress and metabolic shifting in hepatic tissue. Food and Chemical Toxicology. 2004;42(2):313–319. doi: 10.1016/j.fct.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 49.Lemke U., Krones-Herzig A., Diaz M. B., et al. The glucocorticoid receptor controls hepatic dyslipidemia through Hes1. Cell Metabolism. 2008;8(3):212–223. doi: 10.1016/j.cmet.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Ribeiro E. B., do Nascimento C. M. O., Andrade I. S., Hirata A. E., Dolnikoff M. S. Hormonal and metabolic adaptations to fasting in monosodium glutamate-obese rats. Journal of Comparative Physiology B. 1997;167(6):430–437. doi: 10.1007/s003600050093. [DOI] [PubMed] [Google Scholar]
- 51.Chanson P., Salenave S. Metabolic syndrome in Cushing's syndrome. Neuroendocrinology. 2010;92(1):96–101. doi: 10.1159/000314272. [DOI] [PubMed] [Google Scholar]
- 52.Khan A., Chandramouli V., Östenson C.-G., et al. Glucose cycling is markedly enhanced in pancreatic islets of obese hyperglycemic mice. Endocrinology. 1990;126(5):2413–2416. doi: 10.1210/endo-126-5-2413. [DOI] [PubMed] [Google Scholar]
- 53.Khan A., Chandramouli V., Östenson C.-G., Löw H., Landau B. R., Efendić S. Glucose cycling in islets from healthy and diabetic rats. Diabetes. 1990;39(4):456–459. doi: 10.2337/diab.39.4.456. [DOI] [PubMed] [Google Scholar]
- 54.Spolarics Z., Meyenhofer M. Augmented resistance to oxidative stress in fatty rat livers induced by a short-term sucrose-rich diet. Biochimica et Biophysica Acta. 2000;1487(2-3):190–200. doi: 10.1016/s1388-1981(00)00093-7. [DOI] [PubMed] [Google Scholar]
- 55.Massa M. L., Gagliardino J. J., Francini F. Liver glucokinase: an overview on the regulatorymechanisms of its activity. IUBMB Life. 2011;63(1):1–6. doi: 10.1002/iub.411. [DOI] [PubMed] [Google Scholar]
- 56.Uyeda K., Repa J. J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metabolism. 2006;4(2):107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Kabashima T., Kawaguchi T., Wadzinski B. E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5107–5112. doi: 10.1073/pnas.0730817100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moreno-Aurioles V. R., Sobrino F. Glucocorticoids inhibit fructose 2,6-bisphosphate synthesis in rat thymocytes. Opposite effect of cycloheximide. Biochimica et Biophysica Acta (BBA)—Molecular Cell Research. 1991;1091(1):96–100. doi: 10.1016/0167-4889(91)90227-o. [DOI] [PubMed] [Google Scholar]
- 59.Seth R. K., Kumar A., Das S., et al. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicological Sciences. 2013;134(2):291–303. doi: 10.1093/toxsci/kft104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castrogiovanni D., Ongaro L., Zuburía G., Giovambattista A., Spinedi E. Oral metformin treatment counteracts adipoinsular axis dysfunction in hypothalamic obese rats. International Scholarly Research Notices. 2015;2015:9. doi: 10.1155/2015/284042.284042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia J. Y., Holland W. L., Kusminski C. M., et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metabolism. 2015;22(2):266–278. doi: 10.1016/j.cmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]