

Abstract
The emerging links between breast cancer and metabolic dysfunctions spawned by the obesity pandemic predict a disproportionate early disease onset in successive generations. Moreover, sensitivity to chemotherapeutic agents may be influenced by the patient’s metabolic status to affect disease outcome. Maternal metabolic stress as a determinant of drug response in progeny is not well-defined. Here, we evaluated mammary tumor response to doxorubicin in female mouse mammary tumor virus-Wnt1-transgenic offspring exposed to a metabolically-compromised environment imposed by maternal high-fat diet; control progeny were from dams consuming diets with regular fat content. Maternal high-fat diet exposure increased tumor incidence and reduced tumor latency but did not affect tumor volume response to doxorubicin, when compared to control diet exposure. However, doxorubicin-treated tumors from high-fat diet-exposed offspring demonstrated higher proliferation status (Ki-67), mammary stem cell-associated gene expression (Notch1, Aldh1), and basal stem cell-like (CD29hiCD24+) epithelial subpopulation frequencies, than tumors from control diet progeny. Notably, all epithelial subpopulations [CD29hiCD24+, CD29loCD24+, CD29hiCD24+Thy1+] in tumors from high-fat diet-exposed offspring were refractory to doxorubicin. Further, sera from high-fat diet-exposed offspring promoted sphere formation of mouse mammary tumor epithelial cells and of human MCF7 cells. Untargeted metabolomics analyses identified higher levels of kynurenine and 2-hydroxyglutarate in plasma of high-fat diet than control diet offspring. Kynurenine/doxorubicin co-treatment of MCF7 cells enhanced mammosphere-formation ability and decreased apoptosis, relative to doxorubicin only-treated cells. Maternal metabolic dysfunctions during pregnancy and lactation may be targeted to reduce breast cancer risk and improve early drug response in progeny and may inform clinical management of disease.
Keywords: breast cancer, doxorubicin, high-fat diet, kynurenine metabolite, stem cells
Introduction
Breast cancer is the most common malignancy of women worldwide, with a lifetime risk approximating 12% in the Western world (DeSantis et al., 2013). While disease initiation and progression result from genetic and epigenetic changes (Hanahan & Weinberg, 2011), the heterogenous nature of breast cancer categorized by different clinical and molecular subtypes (Prat & Perou, 2011) and manifested as subclonal heterogeneity within tumors (Martelotto LG et al., 2014) presents an enormous challenge for its clinical management. The increasing disease incidence in younger women coincident with the global obesity pandemic suggests that a disproportionate early disease onset is conceivable in successive generations, consistent with Barker’s fetal origins of adult disease hypothesis (Barker, 1996). Whereas support for precision medicine with consideration of patient’s genetic make-up has gained substantial momentum (Narod SA, 2015), this remains a highly costly proposition and currently unattainable for the general population. Understanding the causes, prognosis, and prediction of breast cancer heterogeneity can significantly impact breast cancer prevention and therapy (Brooks et al., 2015).
Metabolic stress in the early life environment that is imposed by maternal over-nutrition and obesity is a predisposing factor for increased risk of adult metabolic syndrome in human, primate, and rodent offspring (Srinivasan et al., 2006; Dyer & Rosenfeld, 2011). Moreover, increasing evidence supports a possible link between metabolic syndrome and breast cancer (Simmen & Simmen, 2011; Hauner & Hauner, 2014). A recent study from our group (Montales et al., 2014) provided a proof of this concept by using the Wnt1-Transgenic (Tg) mouse model of human breast cancer (Li et al, 2000). In this study, female offspring of dams consuming high-fat diet (HFD) and who were weaned to a diet with regular fat content (control diet, CD) which continued through adulthood, exhibited higher mammary tumor incidence and shorter tumor latency, than offspring of CD-fed dams. The metabolic stress status of HFD dams, manifested as elevated levels of serum glucose and oxidative stress biomarkers at the completion of lactation, was mimicked by pups at adulthood, who also exhibited dysregulated insulin signaling (Montales et al., 2014). Nevertheless, the duration, magnitude, and significance of maternal metabolic stress in influencing offspring tumor outcomes remain unclear and in population-based studies, are difficult to delineate, due to confounding environment and lifestyle risk factors between generations.
The success of currently available anthracycline- and taxane-based drugs in improving the outcome of early breast cancer remains limited to a small proportion of patients due to varying responses and toxicities caused by these drugs at high doses. To circumvent these limitations, potential genomic predictors of drug sensitivity are now being examined in patients with different tumor types. In one such study (Martin et al., 2011), resistance to doxorubicin was correlated with estrogen receptor-negative tumor status and with basal-like tumor subtype. While potentially valuable in enhancing treatment options for breast cancer patients, these analyses did not address the etiology of tumor subtypes that may underlie drug response. In the present study, we utilized the Wnt1-Tg mouse model and the maternal HFD paradigm to assess the effects of maternal metabolic history on chemotherapeutic sensitivity to doxorubicin in primary mammary tumors of offspring and to elucidate underlying mechanism(s).
Materials and Methods
Animals and Diets
Animal studies were conducted in accordance with approved protocols by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee. Mice were housed in polycarbonate cages under conditions of 24 °C, 40% humidity and a 12-h light/12-h dark cycle. Food and water were provided ad libitum. Male MMTV-Wnt1-Tg mice (B6SJL-Tg(Wnt1)1Hev/J) and wildtype (WT) females of the same strain were obtained from Jackson Laboratories (Bar Harbor, Maine). To generate the offspring used for the study, WT females were randomly assigned to one of two American Institute of Nutrition-93G-based pelleted diets (Harlan, Indianapolis, IN) beginning at weaning [post-natal day (PND) 21]. The composition of the individual diets differed largely by fat content [control diet (CD) = 17% vs. high fat diet (HFD) = 45% total kcal from lard fat]; HFD also had higher maltodextrin and sucrose content (the latter by 1.5-fold) when compared to CD (Supplementary Table 1), thus, closely recapitulating a typical ‘Western diet’ (Wilson et al., 2007). After 12 weeks on their assigned diets, females were bred with CD-fed Wnt1-Tg males and plug-positive dams were continued on their respective diets throughout pregnancy and lactation (Fig. 1A). At weaning, offspring from CD or HFD dams (designated hereafter CDO or HFDO) were genotyped for the presence or absence of Wnt1 transgene by PCR of genomic DNA from tail snips (Rahal et al, 2013a). Female pups of both genotypes were weaned to CD and used for the analyses described below. A total of 33 CDO and 23 HFDO Wnt1-Tg mice were monitored for spontaneous mammary tumor formation by weekly palpation beginning at four weeks and continued until 6 months of age (Fig. 1A). Random blood glucose levels were measured from tail vein blood by glucometer (One Touch; Lifescan, Milpitas, CA) using glucose strips. Mice with no detectable tumors at age 6 months were euthanized.
Figure 1. Maternal HFD promotes mammary tumorigenesis in the Wnt1-Tg mouse model of human breast cancer.

A. Schematic of dietary regimen. Wild-type (WT) dams were fed with AIN-93G based diets (CAS as sole protein source) containing 17% kcal from fat (Control diet, CD) or 45% kcal from fat (High Fat diet, HFD) beginning at postnatal day 21 (PND21; weaning) all through adulthood and subsequent pregnancy and lactation. Dams were mated with CD-fed Wnt1-Tg males to yield either WT or Wnt1-Tg progeny. At PND21, Wnt1-Tg female offspring were weaned to CD and followed for mammary tumor formation. B. Mammary tumor incidence of Wnt1-Tg offspring of dams fed CD (CDO; n=33) or HFD (HFDO; n=23). Mammary tumor formation was followed weekly in offspring from 4 weeks to 6 months of age by palpation. * P< 0.05. C. Age of tumor onset was calculated as the age of initial tumor appearance (by palpation) up to 6 months of age. CDO (n=16 mice) and HFDO (n=13 mice). D. Body weights of CDO and HFDO at weaning (PND21; n=33 for CDO, n=23 for HFDO) and for tumored mice (n=16 for CDO, n=13 for HFDO), one week after tumor onset. *P< 0.05 for CDO vs. HFDO. E. Blood glucose levels measured at PND21 (weaning) and for tumored mice, one week after tumor onset. *P< 0.05 for CDO vs. HFDO. For D and E, data are presented as the mean ± SEM.
Doxorubicin injection and tumor collection
To evaluate if early exposure to HFD alters response to the chemotherapeutic drug doxorubicin (Dox), tumor-bearing Wnt1-Tg females of both diet groups were twice-administered Dox (Pfizer, New York, NY) at a dose of 8 mg/kg body weight by intraperitoneal injection (Fig. 2A). The first Dox-treatment occurred one week after initial tumor detection, with the second injection administered one week thereafter. Tumor size and left ventricular (LV) function were assessed via high-frequency ultrasound biomicroscopy (UBM) using a Vevo2100 system (VisualSonics, Canada) immediately prior to the first Dox administration and one week after the second Dox treatment at tissue harvest. Briefly, mice were anesthetized with 1.5% isoflurane (Thermo Fisher Scientific, Waltham, MA) and then quickly placed in dorsal recumbency on a temperature-controlled platform under 0.8–1 % isoflurane anesthesia with four legs taped onto ECG electrodes. The body temperature was monitored throughout with a rectal thermomenter. Hair in chest and tumor areas were removed and a prewarmed ultrasound gel was applied to the cleaned area. UBM imaging was acquired using a high frequency transducer (MS550D with 40 MHz), and data were analyzed with VisualSonics software. Imaging of tumor size was started from the long-axis view followed by the short-axis view to obtain the maximum sagittal and transactional diameter, respectively. The total volume of the tumor was assessed by 3-dimensional imaging modality. The M-mode images from the left parasternal long-axis view with the 2-D B-mode image were used to measure LV function (Liu, 2014). Repeated measures of LV function (4–6 cardiac cycles) were performed for each mouse.
Figure 2. Dox-treatment effects on CDO vs. HFDO mammary tumor volume.
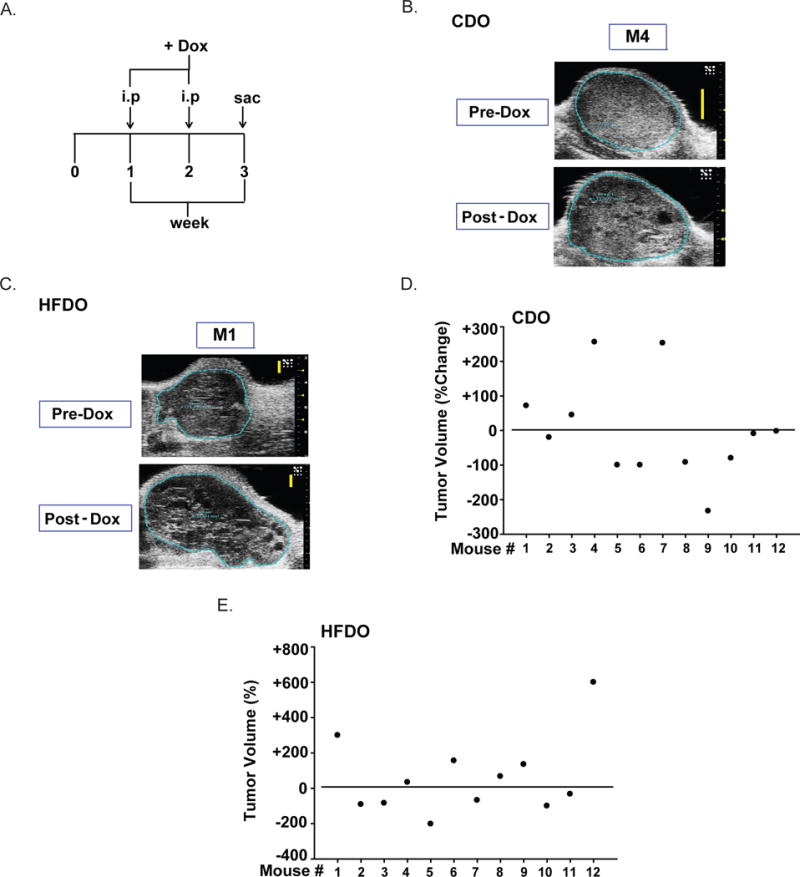
A. Treatment regimen. Dox (8 mg/kg body weight) was administered intraperitoneally to mice one week after tumor detection (designated as 0) and mice were sacrificed one week after the second Dox-treatment (week 3). Tumors were measured as described under Materials and Methods. Representative tumor scans for Dox-insensitive CDO (B) and HFDO (C) tumors. D, E. Percent changes in tumor volume for CDO (D) and HFDO (E) with Dox-treatments. Data for each mouse (n=12 individual mice) are shown. Basal value (pre-Dox) is designated as horizontal line across the entire graph. Percentages above and below the horizontal line refer to an increase (positive) or a decrease (negative), respectively in tumor volumes with Dox.
Analyses of mammary tumors from Dox-treated mice
Excised tumors from Dox-treated CDO and HFDO were classified as Dox-sensitive or Dox-insensitive, based on changes in tumor volumes after Dox-treatments (above). Dox-insensitive tumors (i.e., those whose volumes were increased or did not change with Dox-administration) for each diet group were analyzed by quantitative real-time PCR (QPCR) for expression of tumor suppressor (Pten, Egr1), anti-apoptotic (Bcl2), tumor-inducer (Stat1, Il6), and stem cell marker (Notch1, Notch2, Aldh1) genes and by immunohistochemistry for PTEN, EGR1 and Ki-67 proteins. Tumor sections were prepared as previously described (Rahal et al., 2013a, b). Immunostaining with rabbit anti-human PTEN antibody (Cell Signaling Technology, Danvers, MA; 1:200 dilution), rabbit anti-EGR1 antibody (Cell Signaling Technology; 1:1000 dilution) and rabbit anti-Ki-67 antibody (Abcam, Cambridge, MA; 1:100) followed previously described protocols (Heard et al, 2014; Montales et al. 2014). Five randomly selected fields per tumor section per mouse were analyzed, and percent (%) immunostaining was calculated by counting the number of nuclear-immunostained cells over the total number of cells counted (×100) using Aperio ImageScope and Aperio-associated software (Vista, CA). Procedures for RNA isolation, cDNA synthesis and primer design were as previously described (Montales et al, 2014). Real-time QPCR was performed on an ABI Prism 7000 Detection System (Applied Biosystems, Foster City, CA). Target messenger RNA expression was normalized to a factor that was derived from the geometric mean of expression for TATA-box binding protein, beta-actin and cyclophilin A, using GeNorm excel file software (Al-Dwairi et al., 2012). A total of 4 (immunostaining) and 4–6 (QPCR) Dox-insensitive tumors, each harvested from a different mouse within each diet group, were analyzed.
In vitro assays of epithelial mammary tumor cells
Tumors were isolated from CDO and HFDO (3 individual mice per diet group), one-week after initial tumor detection. The isolation of epithelial mammary tumor cells (designated T-MEC) was previously described (Montales et al., 2012). Cells were plated in appropriate culture medium for each assay and treated or not with Dox (100 nM) with or without added sera (5% final concentration). Sera were harvested from PND85 WT CDO or PND85 WT HFDO (n=6 mice per diet group) and pooled in equal volumes for the treatments. For mammosphere formation assay, cells (2.5 × 103 cells/well) were plated in 6-well low-attachment plates after 24h Dox treatment and then evaluated for numbers of spheroids (mammospheres) 5 days later, in the absence of additional treatments, as previously described (Montales et al. 2012). Cell viability using cells plated at an initial density of 2 × 105 per well was measured by the trypan blue exclusion method using the Vi-Cell cell viability analyzer (Beckman Coulter Inc., Atlanta, GA) (Montales et al. 2015). The percent of apoptotic cells was evaluated 48h post-treatment by Annexin V staining (Trevigen, Gaithersburg, MD), followed by analyses using a Becton-Dickinson LSRFortessa Flow Cytometer (BD Biosciences, San Jose, CA) (Montales et al. 2015). For all assays, treatment effects were determined from three independent experiments in triplicates, with each experiment representing a distinct T-MEC isolation.
Fluorescence-activated cell sorting (FACS)
Mammary tumors isolated from CDO and HFDO prior to and after Dox treatments were evaluated for presence of basal stem cell-like, luminal progenitor and tumor-initiating epithelial subpopulations as described for Wnt1-Tg mice (Cho et al., 2008; Rahal et al., 2013b). Tumors whose volumes did not change or decrease in response to Dox were used for isolation of T-MECs after Dox treatments. Briefly, freshly isolated T-MECs were labeled with selected antibodies (Supplementary Table 2) for 30 min on ice. Cells were washed in HBSS+ buffer (Invitrogen, Carlsbad, CA), incubated with streptavidin-APC for 20 min in ice, washed briefly with the same buffer, and then subjected to FACS on a LSRFortessa Flow Cytometer. Dead cells were excluded using 4′, 6-diamidino-2-phenylindole (DAPI; 1 μg/ml; Sigma-Aldrich, St. Louis, MO). For each tumor sample (4–5 independent tumors for each diet and/or treatment groups), the percentages of basal stem-like (CD29hiCD24+), luminal progenitor (CD29loCD24+), and tumor-initiating (CD29hiCD24+Thy1+) cells within the Lin(–) epithelial population (Cho et al., 2008) were analyzed using FACS Diva Software (BD Biosciences).
Tissue oxidative stress biomarker and plasma metabolite levels
Mammary tissues and sera were obtained from PND85 WT CDO and HFDO (littermates of Wnt1-Tg CDO and HFDO) generated as described in Fig. 1A (n=6 mice/group). The content of free aminothiols (glutathione-reduced and glutathione-oxidized) in tissues were measured by High-Performance Liquid Chromatography and Coloumetric Electrochemical Detection (HPLC-ED) method utilizing CoulAssay System (Thermo Fisher Scientific) and C18 (3.6 μm × 150 mm × 2.1 mm) reverse-phase columns (Phenomenex Inc., Torrence, CA) as previously described (Melnyk et al., 1999). Plasma levels of 2-hydroxyglutaric acid (2-OHG), tryptophan (Trp) and kynurenine (Kyn) were measured following published protocols (Gibson et al., 1993; Medana et al., 2003), with slight modifications. Briefly, 100 μl of plasma was mixed with an equal volume of 10% metaphosphoric acid and incubated on ice for 30 min. Supernatants were retrieved by centrifugation (14000×g for 15 min at 4°C), and aliquots (10–50 μl) were subjected to LC-MS analysis using the UltiMate 3000 system from Dionex and LTQ XL Linear Ion Trap Mass Spectrometer (Thermo Fisher Scientific) and Kinetic C18 columns (2.6 μm × 50 mm × 2.1 mm) (Phenomenex Inc., Torrence, CA) with a Security Guard Ultra Cartridge for analytical column protection. Samples were eluted at 0.4 ml/min with Acetonitrile/Water (50:50) mobile phase (pH 6.2) using a LC-MS method with Electrospray ionization in positive mode. Metabolites were quantified by peak area comparisons using commercially available standards.
In vitro assays of human breast cancer MCF7 cells
The human breast cancer cell line MCF7 was obtained from American Type Culture Collection (Manassas, VA) and authenticated by the company using short-tandem repeat DNA profiling. Cells were used between passage numbers 5 to 15. Cells were propagated in Dulbecco’s modified Eagle medium (Invitrogen) in 5% CO2:95% air at 37°C (Montales et al., 2012) and were evaluated for apoptosis, viability and mammosphere-formation ability in response to Kyn (10 μM) or 2-OHG (10 μM) (Sigma-Aldrich) in the presence of Dox (100 nM). Treatments with Kyn and 2-OHG were carried out for 24h under previously described culture conditions (Montales et al., 2012, 2014) in three independent experiments.
Data Analyses
Data are presented as the mean ± standard error of the mean (SEM) and were compared by t-test or one-way ANOVA using the SigmaStat version 3.5 software (SPSS, Chicago, Illinois, USA. A P value < 0.05 was considered to be statistically significant.
Results
Maternal diet influenced mammary tumor formation in adult Wnt1-Tg offspring
We previously demonstrated that Wnt1-Tg offspring, exposed to HFD through their dams during gestation and lactation only, without further exposure after weaning, had increased tumor incidence and decreased tumor latency when evaluated at age 6 months (Montales et al., 2014). We confirmed those findings in the present study and used the same experimental paradigm to generate mice used in subsequent studies (Fig. 1A). Consistent with previous results, tumor incidence in HFDO at age ≤6 months was significantly greater than in CDO (Fig. 1B). HFDO also developed tumors at a significantly younger age than CDO (Fig. 1C). The higher tumor incidence and shorter tumor latency for HFDO were associated with higher body weights at weaning (PND21) and as adults, since HFDO at sacrifice (one-week after tumor detection) were ~one-month younger than CDO (Fig. 1C, 1D). Glucose levels were higher at weaning for HFDO than CDO (which could be due to the higher maltodextrin and sucrose content in maternal diet) but were comparable for the two groups at sacrifice (Fig. 1E).
Tumor volume response to Dox was comparable for adult CDO and HFDO offspring
Dox is a broad spectrum anti-cancer drug used for many cancer types including breast cancer but its application is limited by cardiotoxicity, dependent on accumulative dose, and the potential for development of drug resistance in patients (Rochette et al., 2015). To evaluate whether metabolic history may be a contributing factor to Dox responsiveness, we assessed Dox effects on tumors of CDO and HFDO, following the regimen shown in Fig. 2A. Mice of both diet groups, one week after initial tumor detection (by palpation) were intraperitoneally administered with Dox (8 mg/kg body weight) twice at one week-interval and the study was terminated one week after the second Dox injection. Dox effects on cardiac parameters in CDO and HFDO pre- and post-Dox treatments were compared to determine if the accumulative dose (16 mg/kg body weight) causes cardiotoxicity. The heart rate (HR) and stroke volume (SV) of Left ventricle (LV) tended to be higher for HFDO than CDO before Dox treatment (Pre-Dox), albeit these differences did not reach statistical significance (Table 1). Dox treatment non-significantly reduced both HR and SV from pre-Dox values in the HFDO, but had no effect in the CDO group (Table 1). Tumor volumes were evaluated for CDO and HFDO pre- and post-Dox treatment; representative images for tumors that were insensitive to Dox treatments are shown (Fig. 2B, C). Of the 12 CDO mice with tumors, 7 responded to Dox with tumor volume reduction; the rest showed either increased (4/12) or no (1/12) tumor volume changes (Fig. 2D). For HFDO tumors, 6 of 12 had reduced tumor volumes with Dox, while the rest (6/12) showed increased tumor volumes (Fig. 2E). Thus, no differences in the response to Dox regarding tumor volumes were noted between CDO and HFDO.
Table 1.
Comparison of Left-Ventricular Function in Mice with Doxorubicin Treatments1
| Heart Function | CDO (n=12)2 | HFDO (n= 12)2 | ||
|---|---|---|---|---|
| Treatment Groups | Pre-Dox | Post-Dox | Pre-Dox | Post-Dox |
| Heart rate (bpm) | 469 ± 12 | 473 ± 31 | 491 ± 12 | 470 ± 14 |
| Stroke volume (ul) | 32.5 ± 1.3 | 33.0 ± 2.9 | 35.6 ± 3.0 | 27 ± 1.9 |
| Ejection fraction (%) | 87.1 ± 3.5 | 79.3 ± 4.5 | 84.0 ± 2.4 | 87.8 ± 2.5 |
| Cardiac output (ml/min) | 15.3± 0.6 | 15.5 ± 1.5 | 17.4 ± 1.3 | 12.7 ± 0.8 |
Doxorubicin cumulative dose= 16 mg/kg body weight
CDO= control diet offspring; HFDO = high-fat diet offspring; values are Mean +/− SEM.
Differences in gene expression of CDO and HFDO tumors
Tumors from CDO and HFDO were further assessed for molecular markers of early pathologic response to Dox, given the comparable numbers of CDO (5 of 12) and HFDO (6 of 12) whose tumors did not decrease in volume with Dox treatment for two weeks (hence, Dox-insensitive). Expression of select tumor suppressor (Pten, Egr1), anti-apoptotic (Bcl2), tumor-inducer (Stat1, Il6), and stem cell marker (Notch1, Notch2, Aldh1) genes were evaluated by QPCR in Dox-insensitive HFDO and CDO tumors. Transcript levels for Pten, Egr1, Notch1 and Aldh1 were significantly elevated while those for Notch2 were reduced in HFDO tumors, compared to those of CDO tumors (Fig. 3A). The levels of Bcl2, Il6 and Stat1 did not differ between the two groups. The higher Pten and Egr1 transcript levels in HFDO relative to CDO tumors were confirmed by their respective protein levels (Fig. 3B, 3C). HFDO tumors also showed higher percentage of cells immunostaining for proliferative marker Ki-67 (Fig. 3D) but did not differ in apoptotic status (by TUNEL; data not shown), when compared to CDO tumors.
Figure 3. Gene expression in Dox-insensitive mammary tumors of CDO and HFDO.
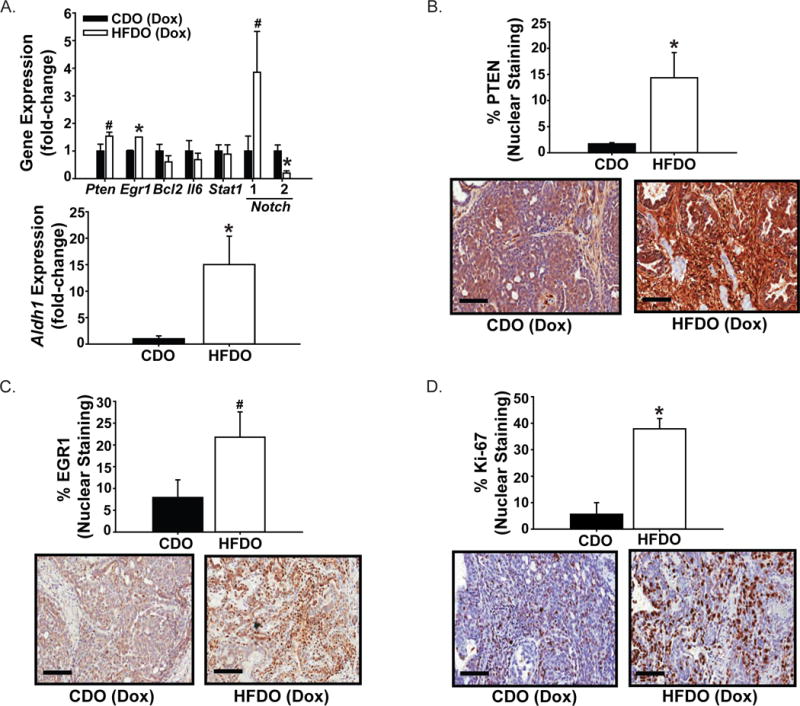
A. Transcript levels were evaluated by QPCR and normalized to a factor from the geometric mean of expression of TATA-box binding protein, beta-actin and cyclophilin A, as described under Materials and Methods. Results (mean ± SEM; n=4 CDO and n=6 HFDO individual tumors) are presented as fold-change of gene expression. *P< 0.05 for CDO vs. HFDO. Representative sections immunostained for PTEN (B), EGR1 (C) and Ki-67 (D) are shown for CDO and HFDO tumors. The % of immunopositive cells for CDO and HFDO tumors (n=4 individual tumors/group) were analyzed a described under Materials and Methods. Values are reported as mean± SEM. *P< 0.05 for CDO vs. HFDO; #P=0.08. Magnification = ×20; scale bar = 100 μm.
Epithelial cell subpopulations in HFDO tumors are insensitive to Dox
The histopathological and molecular subtypes of breast cancer are well-acknowledged to arise from distinct epithelial lineages in the mammary glands (Pratt & Perou, 2011; Anderson et al., 2014). Mammary tumors in Wnt1-Tg mice were shown previously to contain basal stem cell-like, luminal progenitor, and tumor-initiating cells, based on their distinct expression of specific cell-surface antigens CD29, CD24 and Thy1 (Cho et al., 2008). Subsequently, we also showed that the percentages of these subpopulations in pre-neoplastic mammary tissues of Wnt1-Tg mice were altered by dietary factors (Rahal et al., 2013b). To determine whether epithelial subpopulations from HFDO mammary tumors differ from those of CDO and further, whether Dox treatment alters the frequencies of these cell populations, the luminal progenitor, basal, and Thy1-positive epithelial cell subpopulation in non-Dox treated and in Dox-insensitive CDO and HFDO mammary tumors were isolated (Fig. 4A) and their relative frequencies quantified by FACS (Fig. 4B, C). In the absence of Dox, HFDO tumors showed higher % basal (CD29hiCD24+) epithelial subpopulation than CDO tumors (HFDO = 9.03±3.62% vs. CDO = 2.72±0.84%, P<0.05). Non-Dox CDO and HFDO tumors showed comparable % luminal (CD29loCD24+) and %Thy1+ (CD29hiCD24+Thy1+) epithelial subpopulations. Dox treatments decreased the frequencies of all three epithelial subpopulations in CDO tumors but had no effect on those in HFDO tumors (Fig. 4B, 4C).
Figure 4. Frequencies of mammary epithelial subpopulations differ in mammary tumors of Dox-treated CDO and HFDO.
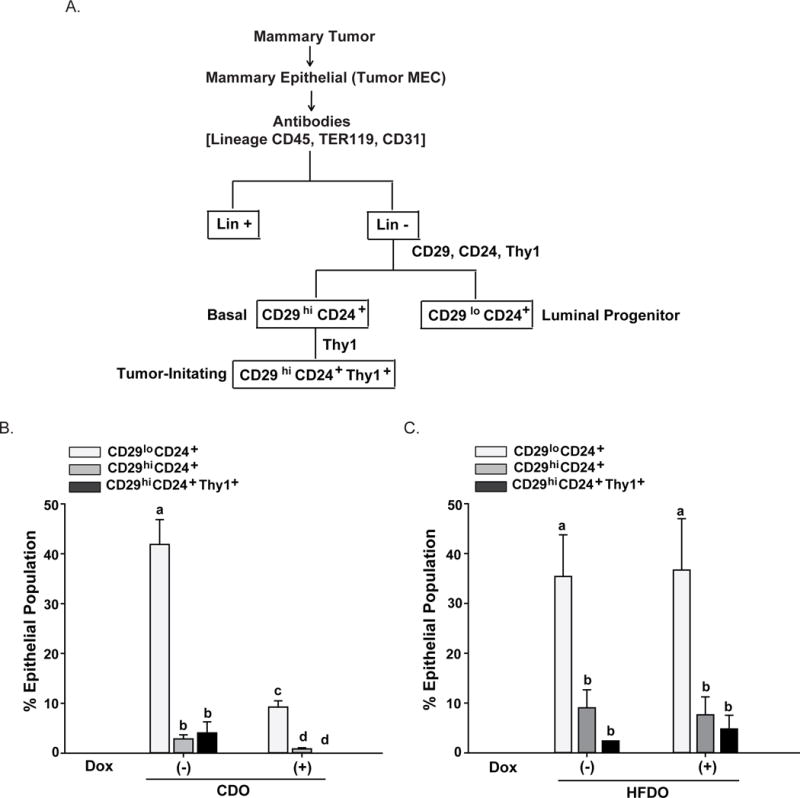
A. Schematic for gating strategy to identify mammary epithelial subpopulations from tumors using FACS. Cell subpopulations were designated based on their cell surface-markers following published studies for Wnt1-Tg mice (Cho et al., 2008; Rahal et al., 2013). B, C. Mammary tumors from CDO (B) and HFDO (C) prior to (−Dox) and post (+Dox) treatments were subjected to the procedures described in (A, above). Results shown are from 4 (CDO) and 5 (HFDO) independent experiments, with each experiment representing individual tumors from CDO and HFDO without (−Dox) and with (+Dox) treatment. Values are reported as mean ± SEM; different letter subscripts differed at P<0.05.
Systemic Factors mediate Dox-resistance of HFDO mammary tumors
To investigate if the lack of response to Dox of epithelial subpopulations present in HFDO tumors is mediated directly by systemic factors that were altered by early exposure to maternal HFD, we isolated mammary epithelial cells from CDO and HFDO tumors (designated T-MEC) one week after tumor detection and evaluated their response to Dox in vitro in the presence and absence of mouse sera harvested from PND85 WT CDO and PND85 WT HFDO. We reasoned that sera from the WT offspring more closely recapitulate systemic factors elicited by maternal HFD in the general population (i.e., without inborn genetic dysfunctions as in Wnt1-Tg mice). The schematic of the in vitro treatments is shown in Fig. 5A. Mammosphere formation is a well-accepted in vitro marker for stem cell activity (Dontu et al., 2003); hence, the ratio of mammospheres formed per number of epithelial cells plated (× 100; designated as % mammosphere-forming units, MFU) was used as a functional measure of the basal stem cell-like epithelial subpopulation. In the absence of Dox, HFDO T-MEC displayed higher % MFU than CDO T-MEC (Fig. 5B). Dox treatment reduced the % MFU in CDO T-MEC but not in HFDO T-MEC (Fig. 5B). Interestingly, mouse sera harvested from PND85 WT HFDO (HFDO sera) had higher ability than PND85 WT CDO sera (CDO sera) (both added at 5% final concentration) to induce mammosphere-formation in CDO T-MEC in the presence of Dox (Fig, 5C). In contrast, HFDO sera and CDO sera elicited comparable mammosphere-forming activity in Dox-treated HFDO T-MEC (Fig. 5C), suggesting that prior in vivo exposure to a HFD environment elicited maximal stem cell-like phenotype potential in mammary epithelial cells. The human MCF7 cell line has been previously shown to display a basal stem-like subpopulation (Filmore & Kuperwasser, 2008) similar to Wnt1-Tg mammary tumors. Dox-treated MCF7 cells showed significantly reduced apoptotic status (Fig. 5D) and enhanced mammosphere-formation (Fig. 5E) with HFDO sera than with CDO sera (P<0.05).
Figure 5. Systemic factors alter response of epithelial mammary tumor cells to Dox.
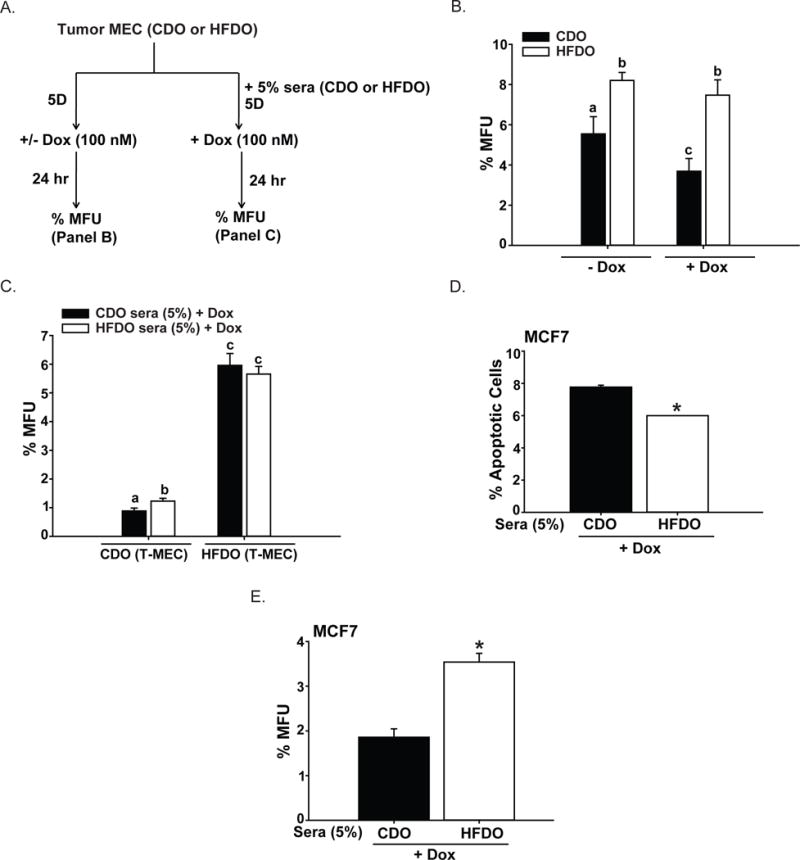
A. Treatment strategy for mammary epithelial cells isolated from CDO and HFDO tumors to evaluate in vitro effects of added Dox and Sera. Sera were pooled in equal volumes from adult (postnatal day 85) WT CDO (n=6) and WT HFDO (n=6) littermates of Wnt1-Tg mice. Isolated epithelial mammary tumor cells were plated and treated with Dox with or without added sera. Treated cells were evaluated for mammosphere-forming activity (measured as percent of mammospheres formed per number of epithelial cells plated; % MFU). B. Mammosphere-formation activity of epithelial mammary tumor cells grown in mammosphere plating medium without (−Dox) and with (+Dox) Dox-treatment. Results (mean ± SEM) are from 3 independent experiments, with each experiment representing an individual tumor isolated from CDO and HFDO one week after initial tumor detection. Values with different letter subscripts differed at P<0.05. C. Mammosphere-formation activity of Dox-treated epithelial mammary tumor cells (T-MEC) grown in mammosphere plating medium with CDO sera or HFDO sera added at 5% final concentration. Sera were pooled from adult WT CDO (n=6) and WT HFDO (n=6) in equal volumes. Results (mean ± SEM) are from 3 independent experiments, with each experiment representing individual tumors isolated from CDO and HFDO one week after initial tumor detection. Values with different letter subscripts differed at P<0.05. D, E. Human MCF7 breast cancer cells were treated with Dox + sera from either CDO or HFDO (added at 5% final concentration) and evaluated for % apoptotic cells (by annexin V-FACS) (D) and mammosphere-formation ability (E). Results (mean ± SEM) are from 3 independent experiments. For mammosphere-formation assays (% MFU), each experiment was carried out in quadruplicates. * P<0.05.
Tumor-related metabolites differ in HFDO and CDO sera
To identify factors in HFDO sera that promoted stem cell-like activity in vitro (Fig. 5), which may underlie the higher % basal stem cell-like subpopulation in HFDO relative to CDO tumors that showed Dox-insensitivity (Fig. 4B, 4C), we first evaluated whether mammary epithelial cells (MEC) isolated from mammary glands of WT CDO and WT HFDO display differences in stem cell-like activity as shown for T-MEC from CDO and HFDO (Fig, 5B). We found that WT MEC from HFDO had greater mammosphere-formation ability than those from CDO (Fig. 6A). This is consistent with recent reports that human and murine tumor subtypes share features with FACS-purified normal cell types (Spike et al., 2012; Pfefferle et al., 2015). Moreover, similar to our previous findings (Montales et al., 2014), mammary glands from WT HFDO displayed greater oxidative stress status, as determined by elevated oxidized glutathione (GSSG) levels and lower ratio of reduced glutathione (GSH) to GSSG, than mammary glands from WT CDO (Fig. 6B). We subjected WT CDO and WT HFDO plasma to untargeted metabolomics LC-MS analyses and selected two candidate metabolites for further study, based on the differences in their plasma levels in WT CDO and WT HFDO and their reported linkages to cancer. 2-Hydroxyglutarate (2-OHG) is considered an oncometabolite and linked to breast cancer subtypes with poorer prognosis (Tang et al., 2014; Terunuma et al., 2014). We found higher plasma levels of 2-OHG in WT HFDO than in WT CDO (Fig. 6C). Increased Tryptophan (Trp) catabolism to kynurenine (Kyn) has been suggested to have prognostic importance in cancers, due to the postulated role of Kyn in immune escape (Suzuki et al., 2010; Gostner et al., 2015). We found that plasma Kyn levels were also higher in WT HFDO relative to WT CDO, while there were no significant differences (P=0.101) in the levels of Trp between WT CDO (23.85±2.61 μM) and WT HFDO (29.03±8.92 μM). However, a higher plasma Kyn/Trp ratio for WT HFDO than WT CDO was observed (Fig. 6C).
Figure 6. Serum metabolites differ in adult CDO and HFDO.
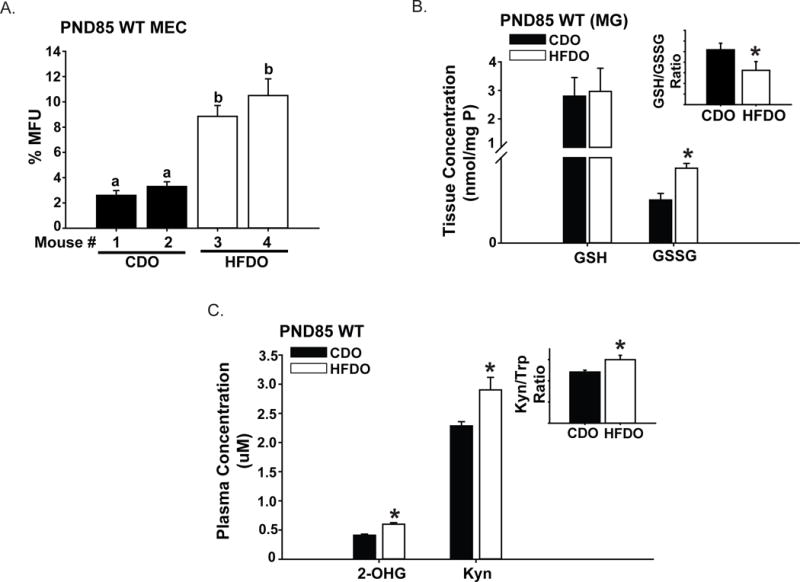
A. Mammary epithelial cells (MEC) from adult [postnatal day (PND) 85] WT CDO (n=2 mice) and WT HFDO (n=2 mice) were isolated and evaluated for mammosphere-formation ability. For independently isolated mouse epithelial cells, results (mean ± SEM) are from quadruplicate wells. Values with different letter subscripts differed at P<0.05. B. Mammary tissues from PND85 WT CDO and PND85 WT HFDO were evaluated for levels of aminothiols glutathione reduced (GSH) and glutathione oxidized (GSSG) by HPLC-ED. Inset, GSH/GSSG ratio for CDO vs. HFDO. Results (mean ± SEM) are from n=6 mice each for CDO and HFDO. *P < 0.05 between groups. C. Levels of 2-OH glutarate (2-OHG) and kynurenine (Kyn) were measured in plasma samples of PND85 WT CDO (n=6) and WT HFDO (n=6), as described under Materials and Methods. Inset: Trp/Kyn ratio for CDO vs. HFDO. Results (mean ± SEM) are from n=6 mice each for CDO and HFDO. *P < 0.05 between groups.
To evaluate if elevated plasma Kyn and 2-OHG levels induced by early HFD exposure contribute to breast cancer cells’ response to Dox, we treated MCF7 cells with these metabolites in the presence of Dox and evaluated metabolite effects on apoptosis, cell viability and mammosphere-formation (Fig. 7A). In Dox-treated treated cells, co-treatment with Kyn reduced apoptosis, had no effect on cell viability, and increased mammosphere-formation ability than without added Kyn (Fig. 7B–D). In contrast, 2-OHG co-treatment of Dox-treated cells (Fig. 7A) did not influence any of these parameters relative to Dox-treatment alone (Fig. 7E–G).
Figure 7. In vitro effects of Kynurenine and 2-hydroxyglutarate in human breast cancer MCF7 cells.
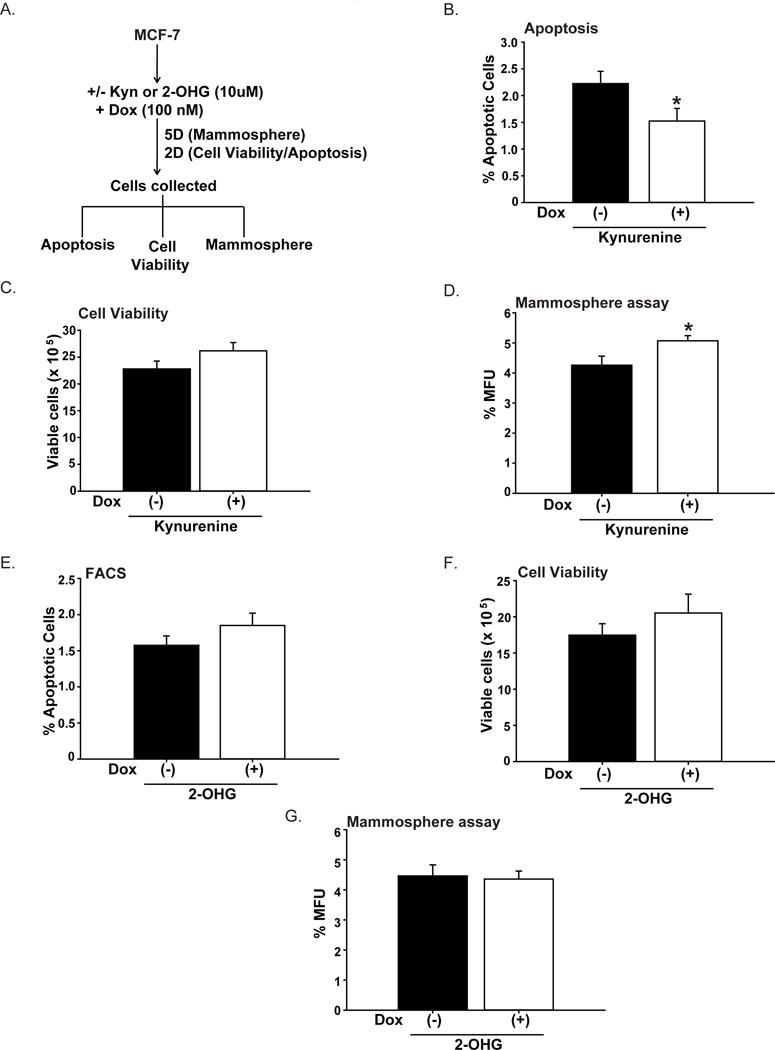
A. Treatment protocols for MCF7 cells with kynurenine (Kyn) or 2-hydroxyglutarate (2-OHG) in Dox-treated cells. Dox-treated cells without (−) and with (+) added Kyn (10 μM) were evaluated for apoptotic status (B), cell viability (C), and mammosphere-formation ability (D). Results (mean ± SEM) are from 3 independent experiments. For C and D, each experiment was conducted in quadruplicates. *P < 0.05 between treatment groups. Dox-treated cells were similarly treated with 2-OHG (10 μM) and analyzed for apoptotic status (E), cell viability (F), and mammosphere-formation ability (G), following described protocols (A). Results (mean ± SEM) are from 3 independent experiments. For F and G, each experiment was conducted in quadruplicates. *P < 0.05 between treatment groups.
Discussion
Increasing evidence supports the many adverse consequences of maternal obesity to the long-term health of progeny (Ruager-Martin R et al., 2010; Houghton et al., 2016). Indeed, studies in mouse models have demonstrated causality between maternal obesity and offspring’s compromised cardiovascular health and increased cancer risks (Blackmore HL et al., 2014; Simmen & Simmen, 2011). While consumption of a high-fat diet does not necessarily result in obesity, there is a growing consensus that maternal dietary intake of high-fat mimicking a typical ‘Western diet’, similar to maternal obesity, elicits metabolic dysfunctions in progeny through early changes in metabolome due in part to deregulated insulin signaling (Cox et al., 2009; Montales et al., 2014; Vogt et al., 2014). Our studies described here for mouse Wnt1-Tg that recapitulates Wnt1-signaling dysfunctions in women with breast cancer (Deming SL et al., 2000, Lin SY et al., 2000) suggest that maternal dietary intake of high fat during pregnancy and lactation can negatively influence early tumor response to chemotherapy. We found that while tumor volume in response to short-term Dox treatment was not a sensitive indicator of effects of maternal metabolic stress, molecular markers associated with proliferation (Ki-67) and stemness (Notch1, Aldh1) can distinguish mammary tumors that originate from an environment with early metabolic perturbations. We show that maternal HFD exposure alters the frequencies of cell subpopulations along the epithelial hierarchy in mammary tumors of offspring, favoring expansion of the self-renewing, less-differentiated subpopulation with mammosphere-forming competence in vitro (Montales et al., 2012; Rahal et al., 2013b; dos Santos et al., 2013) and the ability to initiate tumors in vivo (Cho et al., 2008). Further, we report that Dox-insensitivity of this tumor subpopulation in HFDO may be partly attributed to altered tryptophan metabolism leading to higher systemic levels of its catabolite kynurenine. Using the well-differentiated MCF7 human mammary breast cancer cell line which is estrogen receptor-positive and which displays a limited subpopulation of basal stem-like cells with tumor-initiating properties (Filmore & Kuperwasser, 2008), similar to Wnt1-Tg mammary tumors, we show that serum-derived Kyn, but not 2-OHG, a metabolite also associated with poor breast cancer prognosis (Terunuma et al., 2014), can recapitulate the HFDO serum effects on reducing apoptosis and increasing mammosphere-formation. Increased levels of Kyn relative to Trp have been recently reported to correlate with disease progression in lung cancer through its immunosuppressive effects (Suzuki et al., 2010) and to avert immune surveillance in several cancer types including breast cancer (Heng et al., 2015). Our collective results suggest a novel mechanism (altered Trp catabolism) and a specific target population (basal stem cell-like subpopulation) that are subject to maternal metabolic dysregulations which can influence early chemotherapeutic response of mammary tumors of progeny.
The contribution of the tryptophan catabolic pathway and associated metabolites in bridging maternal metabolic status with breast cancer incidence and drug response in progeny, is currently unknown but is important to consider further given that Kyn has been implicated in regulating stem cell biology (Jones et al., 2013) by virtue of its presumed role in inflammation, a known driver of stem cell expansion and dysregulation (Iliopoulos et al., 2011). Moreover, Kyn levels are highly elevated in the more aggressive estrogen receptor-negative relative to estrogen receptor-positive breast cancers (Tang X et al., 2014). Since tumors generated in Wnt1-Tg mice are reported to be refractory to both ovariectomy and the estrogen receptor antagonist tamoxifen (Zhang et al., 2005), we speculate that the higher levels of Kyn observed with early exposure to maternal high-fat diet may constitute early indicators of the evolution of estrogen-refractory tumors.
Our Dox-treatment paradigm employed mice one week after initial tumor detection and a short-term regimen of two weeks; the limited treatment duration and dose were adopted to eliminate confounding effects of cardiac dysfunctions that may arise in tumor-bearing mice with prolonged Dox treatment. The higher heart rate and stroke volume in HFDO than in those of CDO before Dox treatment concur with HFD promotion of metabolic stress. While we did not expect the 16 mg/kg cumulative dose of Dox to significantly elicit cardiac pathology, Dox treatment decreased heart rates and stroke volumes, resulting in the lower cardiac output of HFDO; these effects are anticipated to be more apparent (and hence, significant) with chronic Dox treatment. Interestingly, while individual tumor volume responses to Dox were comparable between CDO and HFDO, the more sensitive parameters of gene expression, epithelial subpopulation frequencies and self-renewal capacity provided support for the significantly reduced Dox-responsiveness of HFDO than CDO tumors. Future studies will address whether with protracted Dox treatments, tumor volume response and the molecular parameters evaluated here will be more stringently correlated to conclusively establish a causal relationship between early-life exposure to maternal dysfunction and tumor drug resistance.
The higher levels of EGR1 transcription factor in HFDO when compared to CDO tumors, while counter to its previously reported tumor suppressor function (Huang et al., 1997), is consistent with its recently reported role as an oncogene (Li et al., 2013) and in enhancing drug-resistance of breast cancer cells (Tao et al., 2013). Moreover, the lack of Dox treatment effects on the frequencies of basal (CD29hiCD24+), luminal (CD29loCD24+) and tumor-initiating (CD29hiCD24+Thy1+) epithelial subpopulations in HFDO, and the higher transcript levels of stem cell markers Notch 1 and Aldh1 in HFDO than in CDO tumors are congruent with the reported positive association between drug insensitivity and expansion of breast cancer stem cells. Notably, the increased resistance to tamoxifen, an anti-estrogen receptor-positive breast cancer drug, was accompanied by the selection of breast cancer cells with mammosphere-forming capacity (Raffo et al., 2013). Further, in mouse xenograft models of breast cancer, the tyrosine kinase inhibitor sunitinib was found to reduce tumor volume of triple-negative MDA-MB-468 cells but increased the frequency of breast cancer stem cells (Chinchar et al., 2014). Importantly, inhibitors of the metabolic pathway-associated mTOR were found to counteract tamoxifen-induced activation of breast cancer cells (Karthik et al., 2015), suggesting that resistance to temoxifen is associated with metabolic dysregulation. The enhanced expression of PTEN in Dox-treated HFDO tumors appears counter-intuitive to its tumor suppressor role and the recent report that decreased PTEN function is associated with increased resistance of breast cancer cells to doxorubicin (Steelman et al., 2008). Nevertheless, our finding is consistent with PTEN-dependence of trastuzumab resistance in HER-2 gene-amplified breast cancer cells (Deb et al., 2015). The lower levels of Notch2 transcripts in HFDO vs. CDO tumors are aligned with the reported tumor-suppressive role of NOTCH 2 in human breast cancers (Parr et al., 2004). Our collective results highlight the context-dependent signaling pathways of traditionally-considered tumor suppressors and emphasize the need for epidemiological studies addressing the link between maternal metabolic status and drug response in daughters with different breast cancer clinical subtypes.
Our identification of poor metabolic history elicited by maternal HFD exposure as a potential (and significant) contributor to early response to chemotherapeutic drug in progeny may inform clinical procedures to increase drug efficacy in early-stage breast cancer. Further, our results documenting the higher frequency of an epithelial subpopulation with self-renewing capacity in mice exposed to maternal HFD, suggest that strategies for breast cancer prevention starting during early development, is highly relevant to the current make-up of reproductive-aged women with high incidence of obesity and metabolic dysregulation. Our studies supporting the remarkable ability of maternal metabolic status to potentially influence breast cancer risk and treatment in progeny may uncover insights underlying the heterogeneity of response to chemotherapeutic drugs of women with breast cancer.
Supplementary Material
Acknowledgments
The authors thank Olekssandra Pavliv and Mahdav Shroff for technical assistance and Andrea Harris (UAMS Flow Core) for assistance with flow cytometry analyses.
Funding
This work was supported by the Arkansas Breast Cancer Research Program, the University of Arkansas for Medical Sciences Translational Research Institute (CTSA Grant Award UL1TR000039), and the National Institutes of Health/National Cancer Institute (R01CA136493).
Footnotes
Declaration of Interest
The authors declare no conflict of interest.
Author’s Contributions
Conception and Design: RCM Simmen, FA Simmen
Acquisition of data: MTE Montales, SB Melnyk, SJ Liu, YL Liu
Analyses and interpretation of data: MTE Montales, SB Melnyk, SJ Liu, YL Liu, FA Simmen, RCM Simmen
Writing, review and/or revision of manuscript: MTE Montales, SB Melnyk, SJ Liu, YL Liu, FA Simmen, RCM Simmen
References
- Al-Dwairi A, Pabona JM, Simmen RC, Simmen FA. Cytosolic malic enzyme 1 (ME1) mediates high fat diet-induced adiposity, endocrine profile, and gastrointestinal tract proliferation-associated biomarkers in male mice. PLoS One. 2012;7:e46716. doi: 10.1371/journal.pone.0046716. 10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WF, Rosenberg PS, Prat A, Perou CM, Sherman ME. How many etiological subtypes of breast cancer: two, three, four, or more? J Natl Cancer Inst. 2014;106:165. doi: 10.1093/jnci/dju165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. The fetal origins of hypertension. J Hypertens Suppl. 1996;14:S117–1120. [PubMed] [Google Scholar]
- Blackmore HL, Niu Y, Fernandez-Twinn DS, Tarry-Adkins JL, Giussani DA, Ozanne SE. Maternal diet-induced obesity programs cardiovascular dysfunction in adult male mouse offspring independent of current body weight. Endocrinology. 2014;155:3970–3980. doi: 10.1210/en.2014-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks MD, Burness ML, Wicha MS. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell. 2015;17:260–271. doi: 10.1016/j.stem.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchar E, Makey KL, Gibson J, Chen F, Cole SA, Megason GC, Vijayakumar S, Miele L, Gu JW. Sunitinib significantly suppresses the proliferation, migration, apoptosis resistance, tumor angiogenesis and growth of triple-negative breast cancers but increases breast cancer stem cells. Vasc Cell. 2014;6:12. doi: 10.1186/2045-824X-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Wang X, Diehn M, Shedden K, Chen GY, Sherlock G, Gurney A, Lewicki J, Clarke MF. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells. 2008;26:364–371. doi: 10.1634/stemcells.2007-0440. [DOI] [PubMed] [Google Scholar]
- Cox J, Williams S, Grove K, Lane RH, Aagaard-Tillery KM. A maternal high-fat diet is accompanied by alterations in the fetal primate metabolome. Am J Obstet Gynecol. 2009;201:281 e1–9. doi: 10.1016/j.ajog.2009.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb TB, Zuo AH, Barndt RJ, Sengupta S, Jankovic R, Johnson MD. Pnck overexpression in HER-2 gene-amplified breast cancer causes Trastuzumab resistance through a paradoxical PTEN-mediated process. Breast Cancer Res Treat. 2015;150:347–361. doi: 10.1007/s10549-015-3337-z. [DOI] [PubMed] [Google Scholar]
- Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. Br J Cancer. 2000;83:1688–1695. doi: 10.1054/bjoc.2000.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis C, Ma J, Bryan L, Jemal ACA. Breast cancer statistics, 2013. Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos CO, Rebbeck C, Rozhkova E, Valentine A, Samuels A, Kadiri LR, Osten P, Harris EY, Uren PJ, Smith AD, Hannon GJ. Molecular hierarchy of mammary differentiation yields refined markers of mammary stem cells. Proc Natl Acad Sci USA. 2013;110:7123–7130. doi: 10.1073/pnas.1303919110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer JS, Rosenfeld CR. Metabolic imprinting by prenatal, perinatal, and postnatal overnutrition: a review. Semin Reprod Med. 2011;29:266–276. doi: 10.1055/s-0031-1275521. [DOI] [PubMed] [Google Scholar]
- Filmore C, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson KM, Ten Brink HJ, Schor DSM, Kok RM, Bootsma AH, Hoffmann GF, Jakobs C. Stable-isotope dilution analysis of D- and L-2-hydroxyglutaric acid: application to the detection and prenatal diagnosis of D- and L-2-hydroxyglutaric acidemias. Pediatric Res. 1993;34:277–280. doi: 10.1203/00006450-199309000-00007. [DOI] [PubMed] [Google Scholar]
- Gostner JM, Becker K, Überall F, Fuchs D. The potential of targeting indoleamine 2,3-dioxygenase for cancer treatment. Expert Opin Ther Targets. 2015;19:605–615. doi: 10.1517/14728222.2014.995092. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hauner D, Hauner H. Metabolic syndrome and breast cancer: is there a link? Breast Care. 2014;9:277–281. doi: 10.1159/000365951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard ME, Simmons CD, Simmen FA, Simmen RC. Krüppel-like factor 9 deficiency in uterine endometrial cells promotes ectopic lesion establishment associated with activated notch and hedgehog signaling in a mouse model of endometriosis. Endocrinology. 2014;155:1532–1546. doi: 10.1210/en.2013-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng B, Lim CK, Lovejoy DB, Bessede A, Gluch L, Guillemin GJ. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget. 2015:6506–6520. doi: 10.18632/oncotarget.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton LC, Ester WA, Lumey LH, Michels KB, Wei Y, Cohn BA, Susser E, Terry MB. Maternal weight gain in excess of pregnancy guidelines is related to daughters being overweight 40 years later. Am J Obstet Gynecol. 2016 doi: 10.1016/j.ajog.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer. 1997;72:102–109. doi: 10.1002/(sici)1097-0215(19970703)72:1<102::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SP, Guillemin GJ, Brew BJ. The kynurenine pathway in stem cell biology. Int J Tryptophan Res. 2013;6:57–66. doi: 10.4137/IJTR.S12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthik GM, Ma R, Lövrot J, Kis LL, Lindh C, Blomquist L, Fredriksson I, Bergh J, Hartman J. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015;367:76–87. doi: 10.1016/j.canlet.2015.07.017. [DOI] [PubMed] [Google Scholar]
- Li D, Ilnytskyy Y, Kovalchuk A, Khachigian LM, Bronson RT, Wang B, Kovalchuk O. Crucial role for early growth response-1 in the transcriptional regulation of miR-20b in breast cancer. Oncotarget. 2013;4:1373–1387. doi: 10.18632/oncotarget.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying for studying the genetic basis of breast cancer. Oncogene. 2000;19:1002–1009. doi: 10.1038/sj.onc.1203273. [DOI] [PubMed] [Google Scholar]
- Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ. Ultrasound biomicroscopic study of arteries in detection of doxorubicin-induced disorders. Artery Res. 2014;8:173. [Google Scholar]
- Martelotto LG, Ng CK, Piscuoglio S, Weigelt B, Reis-Filho JS. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014;16:210–215. doi: 10.1186/bcr3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Romero A, Cheang MC, López García-Asenjo JA, García-Saenz JA, Oliva B, Román JM, He X, Casado A, de la Torre J, Furio V, Puente J, et al. Genomic predictors of response to doxorubicin versus docetaxel in primary breast cancer. Breast Cancer Res Treat. 2011;128:127–136. doi: 10.1007/s10549-011-1461-y. [DOI] [PubMed] [Google Scholar]
- Medana IM, Day NP, Salahifar-Sabet H, Stocker R, Smythe G, Bwanaisa L, Njobvu A, Kayira K, Turner GD, Taylor TE, Hunt NH. Metabolites of the kynurenine pathway of tryptophan metabolism in the cerebrospinal fluid of Malawian children with malaria. J Infect Dis. 2003;188:844–849. doi: 10.1086/377583. [DOI] [PubMed] [Google Scholar]
- Melynk S, Pogribna M, Pogribny I, Hine RJ, James SJ. A new HPLC method for the simultaneous determination of oxidized and reduced plasma aminothiols using coulometric electrochemical detection. J Nutr Biochem. 1999;10:490–497. doi: 10.1016/s0955-2863(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Montales MT, Rahal OM, Kang J, Rogers TJ, Prior RL, Wu X, Simmen RC. Repression of mammosphere formation of human breast cancer cells by soy isoflavone genistein and blueberry polyphenolic acids suggests diet-mediated targeting of cancer stem-like/progenitor cells. Carcinogenesis. 2012;33:652–660. doi: 10.1093/carcin/bgr317. [DOI] [PubMed] [Google Scholar]
- Montales MT, Melnyk SB, Simmen FA, Simmen RC. Maternal metabolic perturbations elicited by high-fat diet promote Wnt-1-induced mammary tumor risk in adult female offspring via long-term effects on mammary and systemic phenotypes. Carcinogenesis. 2014;35:2102–2112. doi: 10.1093/carcin/bgu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montales MT, Simmen RC, Ferreira ES, Neves VA, Simmen FA. Metformin and soybean-derived bioactive molecules attenuate the expansion of stem cell-like epithelial subpopulation and confer apoptotic sensitivity in human colon cancer cells. Genes Nutr. 2015;10:49. doi: 10.1007/s12263-015-0499-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narod SA. Breast cancer prevention in the era of precision medicine. J Natl Cancer Inst. 2015;107(5) doi: 10.1093/jnci/djv078. pii: djv078. [DOI] [PubMed] [Google Scholar]
- Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14:779–786. doi: 10.3892/ijmm.14.5.779. [DOI] [PubMed] [Google Scholar]
- Pfefferle AD, Spike BT, Wahl GM, Perou CM. Luminal progenitor and fetal mammary stem cell expression features predict breast tumor response to neoadjuvant chemotherapy. Breast Cancer Res Treat. 2015;149:425–437. doi: 10.1007/s10549-014-3262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffo D, Berardi DE, Pontiggia O, Todaro L, de Kier Joffé EB, Simian M. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast Cancer Res Treat. 2013;142:537–548. doi: 10.1007/s10549-013-2760-2. [DOI] [PubMed] [Google Scholar]
- Rahal OM, Pabona JM, Kelly T, Huang Y, Hennings LJ, Prior RL, Al-Dwairi A, Simmen FA, Simmen RC. Suppression of Wnt1-induced mammary tumor growth and lower serum insulin in offspring exposed to maternal blueberry diet suggest early dietary influence on developmental programming. Carcinogenesis. 2013a;34:464–474. doi: 10.1093/carcin/bgs353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahal OM, Machado HL, Montales MT, Pabona JM, Heard ME, Nagarajan S, Simmen RC. Dietary suppression of the mammary CD29hiCD24+ epithelial subpopulation and its cytokine/chemokine transcriptional signatures modifies mammary tumor risk in MMTV-Wnt1 transgenic mice. Stem Cell Res. 2013b;11:1149–1162. doi: 10.1016/j.scr.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette L, Guenancia C, Gudjoncik A, Hachet O, Zeller M, Cottin Y, Vergely C. Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol Sci. 2015;36:326–348. doi: 10.1016/j.tips.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Ruager-Martin R, Hyde MJ, Modi N. Maternal obesity and infant outcomes. Early Hum Dev. 2010;86:715–722. doi: 10.1016/j.earlhumdev.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Simmen FA, Simmen RC. The maternal womb: a novel target for cancer prevention in the era of the obesity pandemic? Eur J Cancer Prev. 2011;20:539–548. doi: 10.1097/CEJ.0b013e328348fc21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike BT, Engle DD, Lin JC, Cheung SK, La J, Wahl GM. A mammary stem cell population identified and characterized in late embryogenesis reveals similarities to human breast cancer. Cell Stem Cell. 2012;10:183–197. doi: 10.1016/j.stem.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab. 2006;291:E792–E799. doi: 10.1152/ajpendo.00078.2006. [DOI] [PubMed] [Google Scholar]
- Steelman LS, Navolanic PM, Sokolosky ML, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman KM, Terrian DM, et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008;27:4086–4095. doi: 10.1038/onc.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Suda T, Furuhashi K, Suzuki M, Fujie M, Hahimoto D, Nakamura Y, Inui N, Nakamura H, Chida K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer. 2010;67:361–365. doi: 10.1016/j.lungcan.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Tang X, Lin CC, Spasojevic I, Iversen ES, Chi JT, Marks JR. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014;16:415. doi: 10.1186/s13058-014-0415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Shi JF, Zhang Q, Xue B, Sun YJ, Li CJ. Egr-1 enhances drug resistance of breast cancer by modulating MDR1 expression in a GGPPS-independent manner. Biomed Pharmacother. 2013;67:197–202. doi: 10.1016/j.biopha.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Terunuma A, Putluri N, Mishra P, Mathé EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, Neupert S, Nicholls HT, Mauer J, Hausen AC, Predel R, Kloppenburg P, Horvath TL, Brüning JC. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell. 2014;156:495–509. doi: 10.1016/j.cell.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CR, Tran MK, Salazar KL, Young ME, Taegtmeyer H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem J. 2007;406:457–467. doi: 10.1042/BJ20070392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Podsypanina K, Huang S, Mohsin SK, Chamness GC, Hatsell S, Cowin P, Schiff R, Li Y. Estrogen receptor positivity in mammary tumors of Wnt-1 transgenic mice is influenced by collaborating oncogenic mutations. Oncogene. 2005;24:4220–4231. doi: 10.1038/sj.onc.1208597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.