

Abstract
The discovery of cancer dependencies has the potential to inform therapeutic strategies and to identify putative drug targets. Integrating data from comprehensive genomic profiling of cancer cell lines and from functional characterization of cancer cell dependencies, we discovered that loss of the enzyme methylthioadenosine phosphorylase (MTAP) confers a selective dependence on protein arginine methyltransferase 5 (PRMT5) and its binding partner WDR77. MTAP is frequently lost due to its proximity to the commonly deleted tumor suppressor gene, CDKN2A. We observed increased intracellular concentrations of methylthioadenosine (MTA; the metabolite cleaved by MTAP) in cells harboring MTAP deletions. Furthermore, MTA specifically inhibited PRMT5 enzymatic activity. Administration of either MTA or a small molecule PRMT5 inhibitor showed a modest preferential impairment of cell viability for MTAP-null cancer cell lines compared to isogenic MTAP-expressing counterparts. Together, our findings reveal PRMT5 as a potential vulnerability across multiple cancer lineages augmented by a common “passenger” genomic alteration.
Main Text
The gene encoding methylthioadenosine phosphorylase (MTAP) is ubiquitously expressed in normal tissues (fig. S1). However, homozygous deletion of MTAP occurs frequently in cancer due to its proximity to CDKN2A, one of the most commonly deleted tumor suppressor genes (Fig. 1A) (1-7). For example, MTAP is deleted in 40% of glioblastomas; 25% of melanomas, urothelial carcinomas, and pancreatic adenocarcinomas; and 15% of non-small cell lung carcinomas (NSCLC) (8). MTAP cleaves methylthioadenosine (MTA) to generate precursor substrates for methionine and adenine salvage pathways. Synthetic lethal strategies to exploit MTAP loss with methionine starvation or by inhibiting de novo purine synthesis have been proposed; however, clinical efficacy of such approaches has not been demonstrated (9-11).
Fig. 1. Cancer cell lines with homozygous MTAP loss are selectively sensitive to suppression of PRMT5 or WDR77.
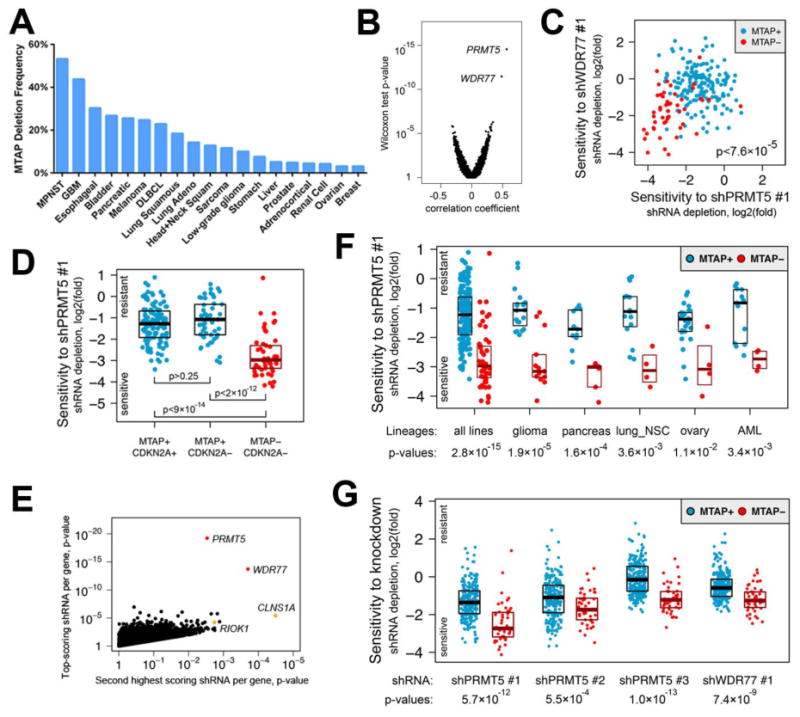
(A) Frequency of MTAP deletion for selected cancers is shown. Data was obtained from the cBioPortal for Cancer Genomics (http://www.cbioportal.org). MPNST, malignant peripheral nerve sheath tumor; GBM, glioblastoma; DLBCL, diffuse large B-cell lymphoma. (B) Point biserial correlation coefficients are plotted against Wilcoxon two-class comparison test p-values for 50,529 shRNAs. (C) Log2(fold) of depletion of shPRMT5 #1 and shWDR77 #1 are shown, demonstrating a correlation between sensitivity to these shRNAs for MTAP− lines. (D) Log2(fold) of shPRMT5 #1 depletion is plotted for cell lines with the indicated genotypes. Median with upper and lower 25th percentiles are shown. (E) Pearson correlation test p-values for the top-scoring shRNAs are plotted against p-values for the second best-scoring shRNAs targeting the same gene. Selective sensitivity of MTAP− lines to depletion of the methylosome is supported by at least two hairpins against four members of the complex including constitutive members of the complex (PRMT5 and WDR77, red) and mutually exclusive substrate adaptors (CLNS1A and RIOK1, orange). (F) Log2(fold) of shPRMT5 #1 depletion is plotted for all 216 cell lines (left) and for lines from the indicated lineages. lung_NSC, non-small cell lung cancer; AML, acute myeloid leukemia. (G) Log2(fold) depletion for the indicated shRNAs is shown for all 275 cell lines from the validation cohort.
We searched for genetic vulnerabilities associated with MTAP loss by leveraging genome-scale pooled short hairpin RNA (shRNA) screening data for 216 cancer cell lines from Project Achilles (12, 13). MTAP deletion status for each line was determined using profiles of MTAP copy number and mRNA expression from the Cancer Cell Line Encyclopedia (CCLE; Additional data table S1) (14). We correlated 50,529 shRNA sensitivity profiles with MTAP deletion status across these lines and identified two shRNAs that strongly correlated with reduced viability of MTAP-null (MTAP−) lines (n=50) but not MTAP-positive (MTAP+) lines (n=166; Fig. 1B; Additional data table S2). One shRNA targeted PRMT5 (shPRMT5 #1; two-sided Wilcoxon p <3 × 10-15) and the other targeted WDR77 (shWDR77 #1; p <4 × 10-12). We observed a correlation between sensitivity to these shRNAs (Fig. 1C), suggesting that MTAP− lines sensitive to suppression with either shRNA were generally also sensitive to suppression with the other shRNA. Cell lines with loss of CDKN2A but not MTAP were generally less sensitive to PRMT5 or WDR77 depletion than were lines with co-deletion of CDKN2A and MTAP, suggesting a correlation with MTAP (but not CDKN2A) loss (Fig. 1D; fig. S2). To provide further support for a possible dependency on PRMT5 or WDR77 in the setting of MTAP loss, we examined additional shRNAs against PRMT5 and WDR77 from the screening dataset. We identified a second shRNA against PRMT5 (shPRMT5 #2) and WDR77 (shWDR77 #2) that also demonstrated a strong correlation between impaired cell viability and MTAP loss (Fig. 1E; Additional data table S3).
False positive findings can occur from genome-scale shRNA analyses because of “off-target” microRNA-like effects attributable to partial sequence complementarity with the 5′ end of the shRNA (known as the “seed” region) (15, 16). To investigate this possibility, we identified shRNAs from the screening dataset that shared sequence identity in the seed region with each of the four shRNAs against PRMT5 or WDR77. None of the shRNAs with shared seed sequence identity demonstrated a correlation between cell viability and MTAP status comparable to that observed for the shRNAs against PMRT5 or WDR77, arguing that the differential viability was not caused by a seed effect (fig. S3; table S1, S2). We also confirmed on-target activity of all four shRNAs against PRMT5 or WDR77 by immunoblotting of lysates from shRNA-expressing cells (fig. S4).
PRMT5 and WDR77 encode critical components of the methylosome. PRMT5 forms a complex with WDR77 and catalyzes the transfer of methyl groups to arginine side-chains of target proteins including histones (involved in chromatin remodeling and gene expression) and Sm proteins (RNA-binding proteins involved in mRNA processing) (17-19). Genetic depletion of PRMT5 has previously been reported to impair cancer cell viability by promoting G1 cell cycle arrest and apoptosis (20-22). Interestingly, shRNAs targeting either PRMT5 or WDR77 reduced levels of both proteins (while demonstrating specific suppression of the target transcript), consistent with depletion of the methylosome complex using either shRNA (fig. S4). MTAP− cells were also sensitive to shRNA-mediated depletion of CLNS1A and RIOK1, which encode two additional components of the methylosome (Fig. 1E) (23). Finally, the correlation between MTAP loss and sensitivity to PRMT5 or WDR77 suppression was not confounded by cell lineage. Within individual lineages (including glioma, pancreatic adenocarcinoma, and NSCLC), MTAP− cell lines were generally (but not universally) more sensitive to depletion of PRMT5 and WDR77 than were MTAP+ lines (Fig. 1F; fig. S2).
Based on these observations, we hypothesized that MTAP loss may confer enhanced sensitivity to genetic suppression of PRMT5 and WDR77. To validate this hypothesis, we examined effects of shRNAs targeting PRMT5 and WDR77 on cell viability in 275 additional cancer cell lines profiled through Project Achilles. This profiling data was generated using an expanded shRNA library with additional shRNAs not included in the initial study. Similar to findings from the initial screening dataset, we observed that MTAP− lines (n=47) were generally more sensitive to PRMT5 or WDR77 suppression than MTAP+ lines (n=228; Fig. 1G; Additional data table S4). Three of the four shRNAs used to establish our initial finding from the screening dataset again demonstrated a strong correlation between loss of cell viability and MTAP status, as did an additional shRNA targeting PRMT5 not included in the screening dataset (shPRMT5 #3). In total, the overall increased sensitivity of MTAP− cells to PRMT5 or WDR77 depletion was demonstrated with five shRNAs (three targeting PRMT5 and two targeting WDR77) from two independent functional datasets comprising 491 cancer cell lines (fig. S5).
To determine whether the effects of PRMT5 or WDR77 suppression on cell viability are affected by MTAP, we first introduced MTAP into four MTAP− cell lines [LU99 and H647 (NSCLC), SF-172 (glioma), and SU.86.86 (pancreatic ductal carcinoma)]. This resulted in robust MTAP protein expression in MTAP-reconstituted lines, whereas MTAP was absent from parental lines (Fig. 2A, fig. S6). We then performed colony formation assays to assess differences in cell viability following depletion of PRMT5 or WDR77 in the presence or absence of MTAP. We observed a reduction in cell viability for each MTAP− cell line with PRMT5 or WDR77 suppression, consistent with our screening and validation results (Fig. 2B, C; fig. S6). Overall, MTAP-reconstituted lines demonstrated reduced sensitivity to PRMT5 or WDR77 suppression compared to isogenic MTAP− counterparts, suggesting a functional link between MTAP loss and PRMT5 or WDR77 dependency (Fig. 2B, C; fig. S6).
Fig. 2. Cells with MTAP loss are more sensitive to suppression of PRMT5 and WDR77 than isogenic MTAP-reconstituted cells.
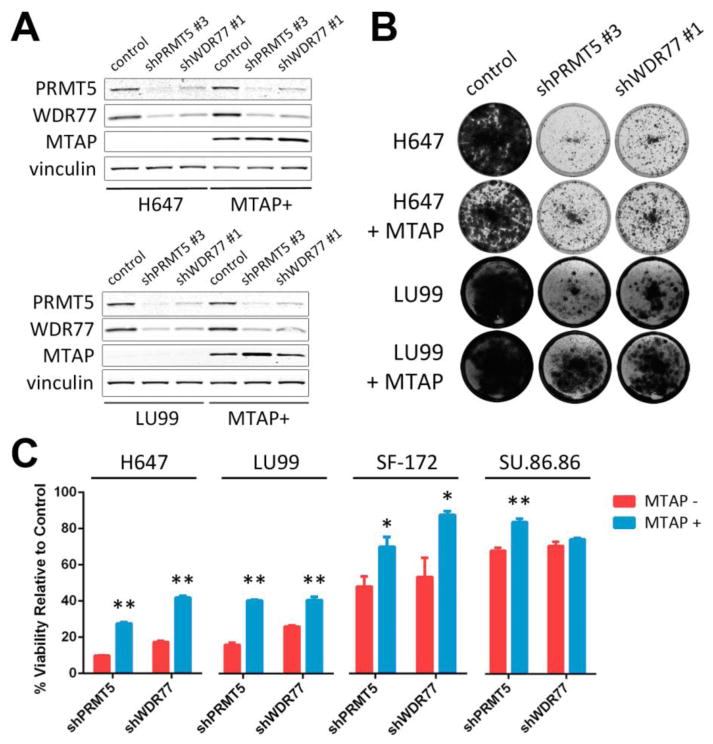
(A) Protein lysates were harvested from H647 (top) or LU99 (bottom) and from MTAP-reconstituted H647 or LU99 cells (MTAP+) 5 days after lentiviral transduction with the indicated shRNAs or control. Lysates were fractionated by SDS-PAGE, and immunoblotting was performed with the indicated antibodies. (B) H647 or LU99 cells and MTAP-reconstituted H647 or LU99 cells were transduced with lentivirus harboring the indicated shRNAs and stained with crystal violet after 10 to 18 days. Media change was performed every 3 days. (C) Quantitation of crystal violet uptake by cells transduced with shRNAs against PRMT5 or WDR77 (normalized to control shRNA for each cell line). Mean and standard error of 3-4 replicates are shown. The experiment was performed 2-3 times for each of the four cell line pairs. ** p < 0.01; * p < 0.05 by two-tailed Student's t-test.
Prior studies suggest the activity of PRMT proteins may be inhibited by MTA (the substrate of MTAP) (24, 25). MTA is an analog of S-adenosyl methionine (SAM; the donor substrate for PRMT-mediated methylation) (26). We hypothesized that somatic MTAP loss may lead to increased intracellular MTA concentrations, which in turn confers a partial inhibition of PRMT5 activity. Together, these effects may heighten cell sensitivity to further reductions in PRMT5 activity (e.g., through genetic suppression). To test this hypothesis, we first determined whether MTAP− cells contain elevated MTA levels. We used liquid chromatography tandem mass spectrometry (LC-MS) to quantify levels of 56 metabolites (including MTA) from LU99, H647, SF-172, and SU.86.86 cells and their isogenic MTAP-reconstituted counterpart lines. The abundance of most measured metabolites was not significantly altered by ectopic MTAP expression (Fig. 3A). However, intracellular MTA abundance was reduced by 1.5 to 6-fold with MTAP reconstitution in each isogenic cell line pair, consistent with increased intracellular MTA in the absence of MTAP (Fig. 3A-C).
Fig. 3. Intracellular MTA is increased in MTAP− cells and correlates with sensitivity to PRMT5 suppression.
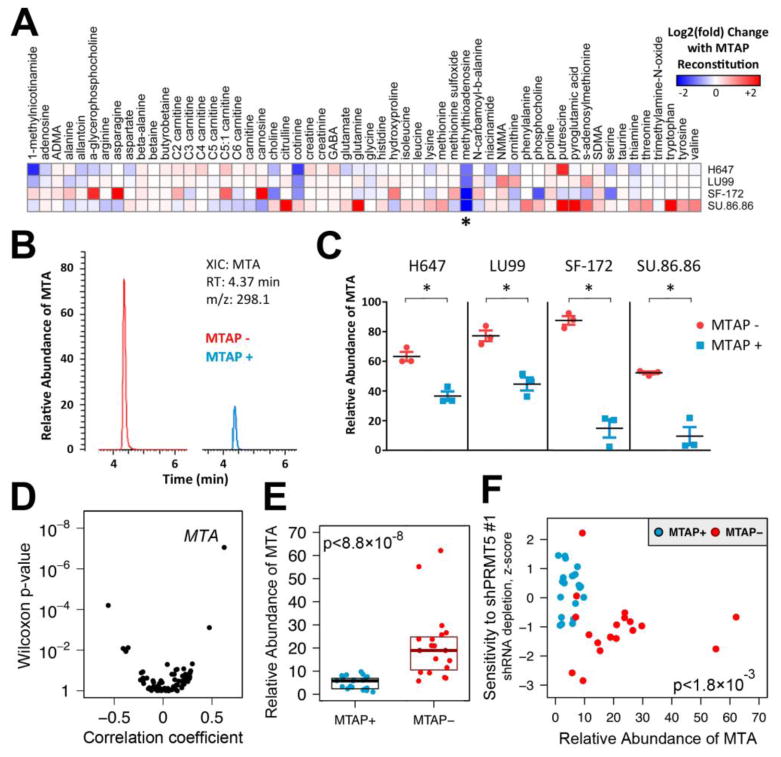
(A) Relative abundance of 56 profiled metabolites was compared for cell extracts from 4 isogenic cell line pairs. Fold-change in relative abundance of each metabolite with MTAP reconstitution is shown for each isogenic pair. Results represent the mean of 2 independent experiments with 3 replicates per cell line. Findings for MTA (methylthioadenosine) are indicated with an asterisk. (B) Representative extracted ion chromatograms (XICs) from LC-MS analysis of SF-172 (left) or MTAP-reconstituted SF-172 (right) cell extracts demonstrating a peak corresponding to MTA. RT, retention time; m/z, mass-to-charge ratio. (C) Relative abundance of MTA from cell extracts is displayed. Mean and standard error of 3 biological replicates are shown. The experiment was performed twice with similar findings. * p < 0.01 by Student's t-test. (D) Correlation of metabolite levels with MTAP status is shown. Point biserial correlation coefficients are plotted against Wilcoxon two-class comparison test p-values for 73 metabolites profiled across 40 MTAP+ and MTAP−cell lines. (E) Relative abundance of MTA from MTAP+ (n=21) and MTAP− (n=19) cell lines from various lineages is shown. For each cell line, mean of 3 biological replicates is displayed. Median with upper and lower 25th percentiles are shown for MTAP− and MTAP+ lines. (F) Correlation of intracellular MTA levels with sensitivity to PRMT5 depletion is shown. shPRMT5 sensitivity data from the screening and validation studies was normalized and combined using modified z-scores. Z-scores are plotted against relative intracellular abundance of MTA for the 40 assayed cell lines. Spearman rank correlation p-value is shown.
To determine whether MTA levels are generally higher in MTAP− cell lines compared to MTAP+ lines, we quantified intracellular levels of 73 metabolites from MTAP− (n=19) and MTAP+ (n=21) cancer cell lines from various lineages including NSCLC, melanoma, and breast. Among profiled metabolites, the abundance of MTA was most strongly correlated with MTAP loss (Fig. 3D; Additional data table S5, S6). We observed an approximately 3.3-fold increase in median MTA levels in MTAP− lines compared to MTAP+, consistent with the hypothesis that MTAP loss leads to increased intracellular MTA (Fig. 3E). In contrast, intracellular levels of the methyl donor SAM were not significantly different between MTAP− and MTAP+ lines (fig. S7). Using shRNA sensitivity data from Project Achilles, we also observed a significant correlation between MTA levels and PRMT5 dependency across profiled cell lines (Fig. 3F; fig. S7, Additional data table S7).
Next, we assessed whether elevated MTA might inhibit PRMT5 activity. PRMT5 catalyzes the formation of symmetric dimethyl arginine (sDMA), while most other PRMTs generate asymmetric dimethyl arginine (aDMA) (17, 27, 28). Using an antibody previously shown to recognize sDMAs generated by PRMT5 (29), we observed decreased sDMA levels in MTAP− cells compared to isogenic, MTAP-reconstituted lines (Fig. 4A). In addition, reduced sDMA was observed in MTAP-reconstituted cells exposed to exogenous MTA, consistent with inhibition of PRMT5 enzymatic activity (Fig. 4A). Similar findings were observed with an antibody recognizing symmetric methylation of histone H4 arginine 3 (H4R3), an established substrate of PRMT5 (Fig. 4A) (30). In contrast, we observed only modest effects of MTAP status or exogenous MTA on levels of aDMA (Fig. 4A).
Fig. 4. Pharmacological inhibition of PRMT5.
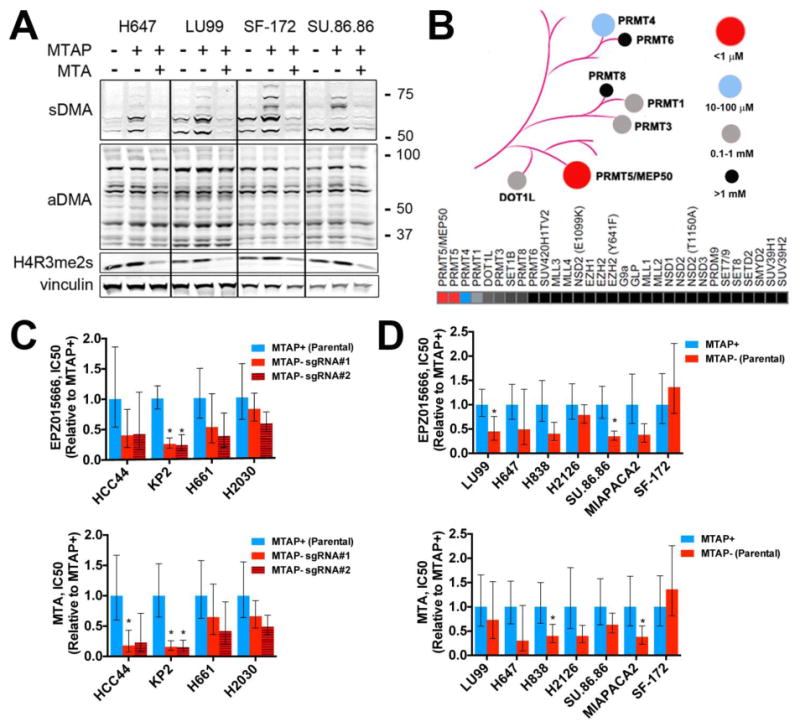
(A) Cells were exposed to DMSO or 200 μM MTA for 48 hours. Lysates were harvested and fractionated by SDS-PAGE. Immunoblotting was performed with the indicated antibodies [recognizing symmetric or asymmetric di-methyl arginine motifs (sDMA and aDMA, respectively), symmetric di-methyl histone H4 arginine 3 (H4R3me2s), or vinculin (loading control)]. Molecular weight is indicated on the right in kDa. (B) Dendrogram and heat map indicating relative sensitivity of 31 histone methyltransferases to inhibition by MTA as determined by radioisotope filter binding assay. (C, D) Relative cell viability IC50 (normalized to MTAP+ cells for each line) for cells treated with EPZ015666 (top) or MTA (bottom), for isogenic cell lines derived from (C) MTAP-expressing parental cell lines or (D) MTAP− parental cell lines. Mean IC50 and 95% confidence interval of 6 replicates are shown for each cell line. Each experiment was performed twice for each cell line. * = p < 0.05 by two-tailed Student's t-test (vs. MTAP+ cells).
This finding raised the possibility that among PRMT family members, PRMT5 may exhibit heightened sensitivity to MTA intracellular concentrations. To test this, we measured the ability of MTA to inhibit the catalytic function of 31 histone methyltransferases (including PRMT5 and the PRMT5/WDR77 complex) using a radioisotope filter binding assay (Additional Data Table S8) (31). We observed more than 100-fold selectivity for MTA against both PRMT5 and PRMT5/WDR77 activity compared to all other profiled methyltransferases, consistent with the hypothesis that PRMT5 function is selectively vulnerable to elevated MTA concentrations (Fig. 4B). Furthermore, we demonstrate that MTA is a SAM-competitive inhibitor of PRMT5 (fig. S8).
Next, we sought to determine whether MTAP− cell lines might exhibit increased sensitivity to pharmacologic inhibition of PRMT5 compared to MTAP+ lines. We identified two inhibitors with distinct PRMT5 binding sites: the metabolite MTA itself and EPZ015666, a potent peptide-competitive and SAM-cooperative inhibitor with >10,000-fold specificity against PRMT5 relative to other methyltransferases (32). We tested the ability of these inhibitors to selectively impair viability of parental MTAP− cell lines compared to isogenic lines expressing MTAP, as well as parental MTAP+ cell lines compared to isogenic CRISPR-mediated MTAP knockout lines (fig. S9). Among the 11 isogenic cell line pairs assayed, the IC50 values (concentrations of inhibitor that led to a 50% reduction in viability) for MTAP− cell lines treated with MTA or EPZ015666 were generally lower than IC50 values for isogenic MTAP+ lines, consistent with our findings from genetic depletion of PRMT5 (although with a smaller effect size) (Fig. 4C, D). While the results for any given cell line pair were consistent using either PRMT5 inhibitor (fig. S9), the differences between each isogenic cell line pair were generally modest and more pronounced for some pairs than others (the differential sensitivity was absent altogether in SF-172). Furthermore, we did not observe significant differences in mean IC50 values between MTAP+ and MTAP− cell lines for either compound (fig. S9).
The discrepancy in effect size that we observed between genetic depletion and enzymatic inhibition of PRMT5 may be caused by several factors. For example, it is possible that the reported SAM-cooperative mechanism of action of EPZ015666 limits inhibition of PRMT5 in the setting of excess MTA and reduced SAM binding (32). Consistent with this, assays of PRMT5 activity in the setting of excess MTA are reported to increase the IC50 of EPZ015666 by an order of magnitude (33). In addition, we cannot exclude the possibility that a non-catalytic PRMT5 function also contributes to the dependency. In this case, therapeutic approaches to exploit this type of vulnerability may require strategies that deplete protein levels of either PRMT5 itself or the larger methylosome complex. Further work will be necessary to explore these and other mechanistic possibilities.
Collectively, our findings suggest that MTAP loss leads to increased intracellular MTA, which in turn inhibits PRMT5 activity and confers heightened susceptibility to further depletion of PRMT5 (fig. S10). While PRMT5 has recently emerged as a possible therapeutic target in some cancers (26), genetic alterations correlated with sensitivity to PRMT5 inhibition have not previously been identified. Our data suggest that many MTAP− tumors are more sensitive to depletion of the methylosome, although there is an overlapping distribution of sensitivities to PRMT5 or WDR77 suppression between MTAP− and MTAP+ cell lines (Fig. 1D, F, G). Thus, MTAP status alone is not sufficient to distinguish cell lines that are sensitive to PRMT5 inhibition. These observations suggest the presence of other modifiers of sensitivity to methylosome depletion that function in a manner independent of MTAP status. Nevertheless, our results endorse the unexpected notion that MTAP loss confers sensitivity to PRMT5 depletion. More generally, these findings highlight the value of comprehensive functional and molecular characterization of large cancer cell line collections to promote identification of potentially targetable dependencies conferred by common genetic lesions.
Supplementary Material
Acknowledgments
Data from Project Achilles and the Cancer Cell Line Encyclopedia can be accessed at https://www.broadinstitute.org/achilles and http://www.broadinstitute.org/ccle/home, respectively. We thank members of the Garraway lab for helpful discussion. We thank Desiree Hernandez for assistance with seeding cell lines for metabolomics studies. We gratefully acknowledge the Carlos Slim Foundation for providing access to shRNA screening data (funded in part by the SIGMA project) for validation studies. Funding for this work was provided by the Novartis Institutes for BioMedical Research, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Melanoma Research Alliance, a NIH PO1 Research Program Project grant (each to L.A.G.), an ICBP grant (U54 CA112962 to T.R.G.), and an NIH U01 CA176058 (to W.C.H.). F.H.W. is supported by a Conquer Cancer Foundation of ASCO Young Investigator Award, the 2014 AACR-Bristol-Myers Squibb Oncology Fellowship in Clinical Cancer Research (Grant 14-40-15-WILS), a grant from the Karin Grunebaum Cancer Research Foundation, and a KL2/Catalyst Medical Research Investigator Training award (an appointed KL2 award) from Harvard Catalyst / The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH Award KL2 TR001100). L.A.G. is an equity holder and consultant in Foundation Medicine. L.A.G. is a consultant to Novartis, Millenium/Takeda, and Boehringer Ingelheim and a recipient of a grant from Novartis. W.C.H. is a consultant for Novartis and recipient of a grant from Novartis. M.T. is a visiting scientist from Mitsubishi Tanabe Pharma Corporation (MTPC) and a recipient of non-research support from MTPC. G.V.K., A.T., F.V., B.A.W., and C.B. performed computational analysis of datasets generated from Project Achilles and CCLE under the supervision of J.S.B., T.R.G., W.C.H., and L.A.G. G.S.C., D.E.R., and W.C.H. supervised the generation of Project Achilles screening data. F.H.W. and S.E.M. performed experiments to validate the computational shRNA screening findings. G.V.K., F.H.W., S.E.M., J.M.S., C.D., and C.B.C. performed metabolomic studies and analyzed data. Isogenic cell line pairs were generated and characterized by F.H.W. and J.R.R. J.R.R. conducted cell viability IC50 experiments and the MTA methyltransferase selectivity assay. J.P. performed the MTA mechanism of action study under the supervision of J.E.B. M.E.F. and M.T. synthesized EPZ015666 under the supervision of J.E.B. F.H.W., G.V.K., J.R.R., and L.A.G. wrote the manuscript.
References and Notes
- 1.Nobori T, Takabayashi K, Tran P, Orvis L, Batova A, Yu AL, Carson DA. Genomic cloning of methylthioadenosine phosphorylase: a purine metabolic enzyme deficient in multiple different cancers. Proc Natl Acad Sci U S A. 1996;93:6203–6208. doi: 10.1073/pnas.93.12.6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen ZH, Zhang H, Savarese TM. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the alpha- and beta-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res. 1996;56:1083–1090. [PubMed] [Google Scholar]
- 3.Illei PB, Rusch VW, Zakowski MF, Ladanyi M. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin Cancer Res. 2003;9:2108–2113. [PubMed] [Google Scholar]
- 4.Hustinx SR, Hruban RH, Leoni LM, Iacobuzio-Donahue C, Cameron JL, Yeo CJ, Brown PN, Argani P, Ashfaq R, Fukushima N, Goggins M, Kern SE, Maitra A. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: a potential new target for therapy. Cancer Biol Ther. 2005;4:83–86. doi: 10.4161/cbt.4.1.1380. [DOI] [PubMed] [Google Scholar]
- 5.Powell EL, Leoni LM, Canto MI, Forastiere AA, Iocobuzio-Donahue CA, Wang JS, Maitra A, Montgomery E. Concordant loss of MTAP and p16/CDKN2A expression in gastroesophageal carcinogenesis: evidence of homozygous deletion in esophageal noninvasive precursor lesions and therapeutic implications. Am J Surg Pathol. 2005;29:1497–1504. doi: 10.1097/01.pas.0000170349.47680.e8. [DOI] [PubMed] [Google Scholar]
- 6.Karikari CA, Mullendore M, Eshleman JR, Argani P, Leoni LM, Chattopadhyay S, Hidalgo M, Maitra A. Homozygous deletions of methylthioadenosine phosphorylase in human biliary tract cancers. Mol Cancer Ther. 2005;4:1860–1866. doi: 10.1158/1535-7163.MCT-05-0103. [DOI] [PubMed] [Google Scholar]
- 7.Christopher SA, Diegelman P, Porter CW, Kruger WD. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer Res. 2002;62:6639–6644. [PubMed] [Google Scholar]
- 8.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertino JR, Waud WR, Parker WB, Lubin M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: current strategies. Cancer Biol Ther. 2011;11:627–632. doi: 10.4161/cbt.11.7.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kindler HL, Burris HA, 3rd, Sandler AB, Oliff IA. A phase II multicenter study of L-alanosine, a potent inhibitor of adenine biosynthesis, in patients with MTAP-deficient cancer. Invest New Drugs. 2009;27:75–81. doi: 10.1007/s10637-008-9160-1. [DOI] [PubMed] [Google Scholar]
- 11.Munshi PN, Lubin M, Bertino JR. 6-thioguanine: a drug with unrealized potential for cancer therapy. Oncologist. 2014;19:760–765. doi: 10.1634/theoncologist.2014-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, Jiang G, Hsiao J, Mermel CH, Getz G, Barretina J, Gopal S, Tamayo P, Gould J, Tsherniak A, Stransky N, Luo B, Ren Y, Drapkin R, Bhatia SN, Mesirov JP, Garraway LA, Meyerson M, Lander ES, Root DE, Hahn WC. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WFJ, Pantel SE, Lizotte PH, Jiang G, Hsiao J, Tsherniak A, Dwinell E, Aoyama S, Okamoto M, Harrington W, Gelfand E, Green TM, Tomko MJ, Gopal S, Wong TC, Li H, Howell S, Stransky N, Liefeld T, Jang D, Bistline J, Meyers B Hill, Armstrong SA, Anderson KC, Stegmaier K, Reich M, Pellman D, Boehm JS, Mesirov JP, Golub TR, Root DE, Hahn WC. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Scientific Data. 2014;1 doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. Rna. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012;9:363–366. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci. 2011;36:633–641. doi: 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, Rappsilber J, Mann M, Dreyfuss G. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol. 2001;21:8289–8300. doi: 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friesen WJ, Wyce A, Paushkin S, Abel L, Rappsilber J, Mann M, Dreyfuss G. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem. 2002;277:8243–8247. doi: 10.1074/jbc.M109984200. [DOI] [PubMed] [Google Scholar]
- 20.Scoumanne A, Zhang J, Chen X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009;37:4965–4976. doi: 10.1093/nar/gkp516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim JH, Lee YM, Lee G, Choi YJ, Lim BO, Kim YJ, Choi DK, Park JW. PRMT5 is essential for the eIF4E-mediated 5′-cap dependent translation. Biochem Biophys Res Commun. 2014;452:1016–1021. doi: 10.1016/j.bbrc.2014.09.033. [DOI] [PubMed] [Google Scholar]
- 22.Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabo A, Amati B, Wee KB, Guccione E. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96–100. doi: 10.1038/nature14351. [DOI] [PubMed] [Google Scholar]
- 23.Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U, Grimmler M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J Biol Chem. 2011;286:1976–1986. doi: 10.1074/jbc.M110.148486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Williams-Ashman HG, Seidenfeld J, Galletti P. Trends in the biochemical pharmacology of 5′-deoxy-5′-methylthioadenosine. Biochem Pharmacol. 1982;31:277–288. doi: 10.1016/0006-2952(82)90171-x. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 27.Branscombe TL, Frankel A, Lee JH, Cook JR, Yang Z, Pestka S, Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem. 2001;276:32971–32976. doi: 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- 28.Pollack BP, Kotenko SV, He W, Izotova LS, Barnoski BL, Pestka S. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J Biol Chem. 1999;274:31531–31542. doi: 10.1074/jbc.274.44.31531. [DOI] [PubMed] [Google Scholar]
- 29.Liu F, Cheng G, Hamard PJ, Greenblatt S, Wang L, Man N, Perna F, Xu H, Tadi M, Luciani L, Nimer SD. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest. 2015;125:3532–3544. doi: 10.1172/JCI81749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, Cunningham JM, Jane SM. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16:304–311. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horiuchi KY, Eason MM, Ferry JJ, Planck JL, Walsh CP, Smith RF, Howitz KT, Ma H. Assay development for histone methyltransferases. Assay Drug Dev Technol. 2013;11:227–236. doi: 10.1089/adt.2012.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, West KA, Lingaraj T, Stickland K, Ribich SA, Raimondi A, Scott MP, Waters NJ, Pollock RM, Smith JJ, Barbash O, Pappalardi M, Ho TF, Nurse K, Oza KP, Gallagher KT, Kruger R, Moyer MP, Copeland RA, Chesworth R, Duncan KW. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol, ( 2015 doi: 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- 33.Mavrakis KJ, McDonald ER, III, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, Yue Y, Ho S, Liu Y, Yan-Neale Y, Yang G, Lin F, Yin H, Gao H, Kipp DR, Zhao S, McNamara JT, Sprague ER, Zheng B, Lin Y, Cho YS, Gu J, Crawford K, Ciccone D, Vitari A, Lai A, Capka V, Hurov K, Porter JA, Tallarico J, Mickanin C, Lees E, Pagliarini R, Keen N, Schmelzle T, Hofmann F, Stegmeier F, Sellers WR. Disordered methionine metabolism in MTAP/CDKN2A deleted cancers leads to dependence on PRMT5. Science. doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.