

Abstract
AIM
To investigate the anticancer effect of a recombinant adenovirus-mediated p53 (rAd-p53) combined with 5-fluorouracil (5-FU) in human colon cancer resistant to 5-FU in vivo and the mechanism of rAd-p53 in reversal of 5-FU resistance.
METHODS
Nude mice bearing human colon cancer SW480/5-FU (5-FU resistant) were randomly assigned to four groups (n = 25 each): control group, 5-FU group, rAd-p53 group, and rAd-p53 + 5-FU group. At 24 h, 48 h, 72 h, 120 h and 168 h after treatment, 5 mice were randomly selected from each group and sacrificed using an overdose of anesthetics. The tumors were removed and the protein expressions of p53, protein kinase C (PKC), permeability-glycoprotein (P-gp) and multidrug resistance-associated protein 1 (MRP1) (Western blot) and apoptosis (TUNEL) were determined.
RESULTS
The area ratios of tumor cell apoptosis were larger in the rAd/p53 + 5-FU group than that in the control, 5-FU and rAd/p53 groups (P < 0.05), and were larger in the rAd/p53 group than that of the control group (P < 0.05) and the 5-FU group at more than 48 h (P < 0.05). The p53 expression was higher in the rAd/p53 and the rAd/p53 + 5-FU groups than that of the control and 5-FU groups (P < 0.05), and were higher in the rAd/p53 + 5-FU group than that of the rAd/p53 group (P < 0.05). Overexpression of PKC, P-gp and MRP1 was observed in the 5-FU and control groups. In the rAd/p53 + 5-FU group, the expression of P-gp and MRP1 was lower that of the control and 5-FU groups (P < 0.05), and the expression of PKC was lower than that of the control, 5-FU and rAd/p53 groups at more than 48 h (P < 0.05). In the rAd/p53 group, the expression of P-gp and MRP1 was lower that of the control and 5-FU groups at more than 48 h (P < 0.05), and the expression of PKC was lower than that of the control and 5-FU groups at more than 120 h (P < 0.05).
CONCLUSION
5-FU combined with rAd-p53 has a synergistic anticancer effect in SW480/5-FU (5-FU resistance), which contributes to reversal of 5-FU resistance.
Keywords: Human colon cancer, Multidrug resistance, 5-Fluorouracil, Recombinant adenovirus-mediated p53, Xenografts in nude mice
Core tip: To observe anticancer action of a recombinant adenovirus-mediated p53 (rAd-p53) combined with 5-fluorouracil (5-FU) in human colon cancer with resistance to 5-FU in vivo to investigate the potential and mechanism of rAd-p53 in the reversal of resistance to 5-FU in human colon cancer. Our previous results revealed that exogenous wild-type p53 gene from rAd-p53 can decrease expression of PKC, P-gp and MRP1 in SW480/5-FU (5-FU resistance) and promote apoptosis of tumor cell, which contributes to reversing 5-FU resistance in vivo. 5-FU can increase the expression of exogenous wild-type p53, so 5-FU combined with rAd-p53 has a synergistic anticancer effect for colon cancer of 5-FU resistance in vivo.
INTRODUCTION
Colorectal cancer (CRC) is one of the most common gastrointestinal cancers. In 2013, there were 1.6 million incident cases of CRC worldwide, with 56% occurring in developing countries and 44% in developed countries, which caused 771000 deaths[1]. Most patients are usually at an advanced stage at the time of diagnosis.
To date, 5-fluorouracil (5-FU) remains a widely used chemotherapeutic drug in the treatment of advanced CRC; however, response rates are only 10% to 15%, due to severe side effects and resistance[2]. The anticancer efficacy of 5-FU is thought to be partly attributed to its ability to induce p53-dependent cell growth arrest and apoptosis; consequently, mutations or deletions of p53 can cause cells to become resistant to 5-FU[3-6]. Therefore, overcoming 5-FU resistance caused by mutations or deletions of p53 will be a key issue in the design of more effective individualized therapeutic strategies.
Gene replacement therapy for a mutated p53 gene using a recombinant adenovirus-mediated p53 (rAd-p53) gene reportedly increases apoptosis after administration[7-12]. Our previous results revealed that exogenous wild-type p53 (wt-p53) from rAd-p53 increased tumor necrosis in human colon cancer SW480 (5-FU responsive) harboring mutant p53, and 5-FU combined with rAd-p53 had a synergistic anticancer effect in vivo[13]. Therefore, rAd-p53 may contribute to the reversal of resistance to 5-FU in colon cancer.
The present study determined the early therapeutic effectiveness of rAd-p53 alone or in combination with 5-FU for the treatment of human colon cancer SW480/5-FU (5-FU resistant) in a nude mouse model. The potential and mechanism of rAd-p53 in the reversal of resistance to 5-FU in human colon cancer in vivo was also investigated.
MATERIALS AND METHODS
The present study strictly complied with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Sun Yat-sen University (2011-0702) and Guangzhou Medical University, Guangzhou, China.
Cell culture
The human colon cancer cell line SW480 was purchased from the Cell Bank of Sun Yat-sen University. The cells were cultured in RPMI 1640 with 10% fetal calf serum, 100 U/mL penicillin and 100 μg/mL streptomycin, and grown at 37 °C in a 5% CO2 humidified atmosphere. 5-FU resistant SW480 (SW480/5-FU) cells were generated by continuous exposure to increasing concentrations of 5-FU for more than 5 mo. SW480/5-FU cells were able to survive in 6 μg/mL of 5-FU. The IC50 of 5-FU, based on the results of a [3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT)] assay, was 23.593 μg/mL for parental cells (SW480) and 140.642 μg/mL for resistant cells (SW480/5-FU). The resistance index (RI) was 5.93. The IC50 of 5-FU in SW480 and SW480/5-FU cells was assayed with MTT.
Animal model
BALB/c nude mice were purchased from the Animal Center of Sun Yat-sen University. A total of 100 4-wk-old BALB/c nude mice, weighing 16-18 g regardless of sex, were subcutaneously implanted with SW480/5-FU tissues in the rear flank to generate xenograft models as described previously[13,14]. All surgical procedures were performed under anesthesia induced by chloral hydrate (4.5% chloral hydrate, 2 mL/100 g body weight, intraperitoneal injection), and all efforts were made to minimize suffering. Mice were fed in a specific pathogen-free (SPF) laboratory. One month after implantation, the mice were randomly assigned to four groups (25 per group): control group (medical-grade saline), 5-FU group, rAd-p53 group (Gendicine, SibionoGeneTech Co., Ltd, Shenzhen, China), and rAd-p53 + 5-FU group. The above-mentioned therapeutic agents were administered by intratumoral injection. The dose of rAd-p53 administered was 1 × 107 VIP/mm3 tumor for each group. The dose of 5-FU was 25 mg for tumors 0.5-0.9 cm, 50 mg for tumors 1.0-1.4 cm, and 75 mg for tumors more than 1.5 cm[13].
Assessment of tumor response
At 24, 48, 72, 120 and 168 h after treatment, 5 mice were randomly selected from each group and euthanized with an overdose of anesthetics. The tumors were removed and divided into equal halves. One half was immediately frozen at -30 °C for Western blot analysis. The other half was fixed in phosphate-buffered saline (pH 7.3) containing 4% formaldehyde and 0.2% glutaraldehyde, embedded in paraffin, and sectioned for TUNEL assay.
Measurement of apoptosis
Pathological sections were stained using the TUNEL apoptosis in situ detection reagent kit (Keygen, Nanjing, China) according to the manufacturer’s instructions. The area ratio of tumor cell apoptosis was calculated as the percentage of positively stained cell nuclei (dark brown) at magnification × 100. The average of the evaluations by the pathologists (Zhang & Xu) was used for analysis.
Western blot analysis
Western blot analysis was used to detect the protein expression of p53, protein kinase C (PKC), permeability-glycoprotein (P-gp) and multidrug resistance-associated protein 1 (MRP1) as described in the instruction manual (Phototope®-HRP Western blot kit; Cell Signaling Technology, United States). Cell extracts were obtained from frozen tumor tissues (-30 °C). Immunoblot analysis was performed using anti-p53 monoclonal antibodies (Santa Cruz Biotech, United States), anti-PKC and multidrug resistance protein 1 (MDR1) and MRP1 monoclonal antibodies (Santa Cruz Biotech). Subsequent protein detection was performed using an enhanced chemiluminescence (ECL) detection system (Hitachi, Japan).
The band intensities (IOD) of protein expression stated above were scanned into the computer and analyzed with Image-Pro Plus 6.0 software. The relative IOD (RIOD) of protein expression was calculated as the IOD of the protein in the control group and therapeutic groups at each time point divided by the corresponding IOD of GAPDH (internal control).
Statistical analysis
The SPSS 13.1 statistical package (SPSS Inc., United States) was used for all calculations. The tumor responses of the entire sample set (ANOVA repeated data) and between groups (SNK test) were compared, and the correlations between parameters were evaluated with Pearson’s correlation at a significance level of 0.05. Data are presented as mean ± SD of a representative of at least three independently performed experiments.
RESULTS
Tumor cell IC50
The IC50 of 5-FU based on the results of the MTT assay was 23.593 μg/mL for parental cells (SW480) and 140.642 μg/mL for resistant cells (SW480/5-FU). The resistance index (RI) was 5.93.
Tumor cell apoptosis
Tumor cell apoptosis was detected in sections from the 5-FU group and the control group at the observed time points (Figure 1); however, there were no significant differences in tumor cell apoptosis between the two groups (SNK test, P > 0.05; Table 1).
Figure 1.
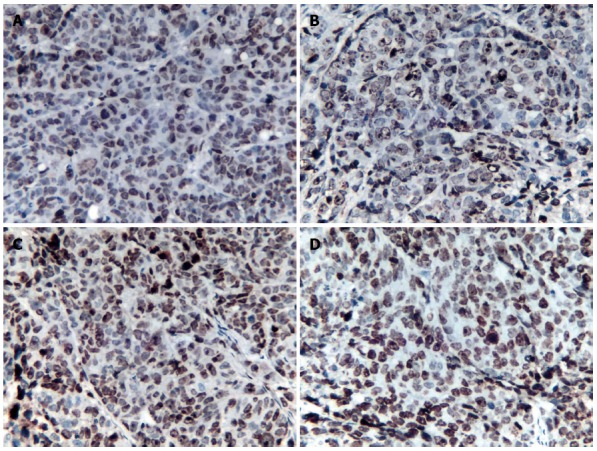
Tumor cell apoptosis. Tumor cell apoptosis at 72 h (magnification × 400) in the control group (A), 5-FU group (B), rAd-p53 group (C) and rAd-p53 + 5-FU group (D).
Table 1.
Tumor apoptosis ratios in experimental groups and control group (mean ± SD)
| Group | 24 h | 48 h | 72 h | 120 h | 168 h |
| Control | 0.25 ± 0.02 | 0.28 ± 0.01 | 0.27 ± 0.02 | 0.29 ± 0.02 | 0.24 ± 0.04 |
| 5-FU | 0.27 ± 0.03 | 0.31 ± 0.03 | 0.31 ± 0.05 | 0.30 ± 0.04 | 0.31 ± 0.02 |
| rAd-p53 | 0.29 ± 0.02 | 0.35 ± 0.03 | 0.43 ± 0.08 | 0.42 ± 0.06 | 0.46 ± 0.06 |
| 5-FU + rAd-p53 | 0.33 ± 0.03 | 0.44 ± 0.08 | 0.58 ± 0.07 | 0.59 ± 0.05 | 0.62 ± 0.07 |
| F | 13.235 | 41.487 | 53.812 | 61.676 | 80.755 |
| P value | 0 | 0 | 0 | 0 | 0 |
| P value | > 0.05 | > 0.05 | > 0.05 | > 0.05 | > 0.05 |
| 5-FU vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs rAd-p53 | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs 5-FU | |||||
| P value | >0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs 5-FU |
The area ratio of tumor cell apoptosis in the rAd-p53 + 5-FU group was significantly larger than that in the control group, 5-FU group and the rAd-p53 group (SNK test, P < 0.05; Table 1). The area ratio of tumor cell apoptosis in the rAd-p53 group was significantly larger than that in the control group (SNK test, P < 0.05). After > 48 h of treatment, the area ratio of tumor cell apoptosis in the rAd-p53 group was significantly larger than that in the 5-FU group (SNK test, P < 0.05).
The area ratio of tumor cell apoptosis in the rAd-p53 group and the rAd-p53 + 5-FU group tended to increase with time (ANOVA, P <0.05).
Protein expression
P53 expression: P53 protein showed weak expression in the 5-FU group and the control group at the observed time points (Figure 2A). There were no significant differences in the RIOD of p53 expression between the two groups (SNK test, P > 0.05; Table 2). P53 expression level in the rAd-p53 and the rAd-p53 + 5-FU group was higher than that in the control and 5-FU groups (SNK test, P < 0.05) and increased in a time-dependent manner (ANOVA, P < 0.05; Table 2), with peak expression at 120 h. P53 expression in the rAd-p53 + 5-FU group was significantly higher than that in the rAd-p53 group (SNK test, P < 0.05; Table 2).
Figure 2.
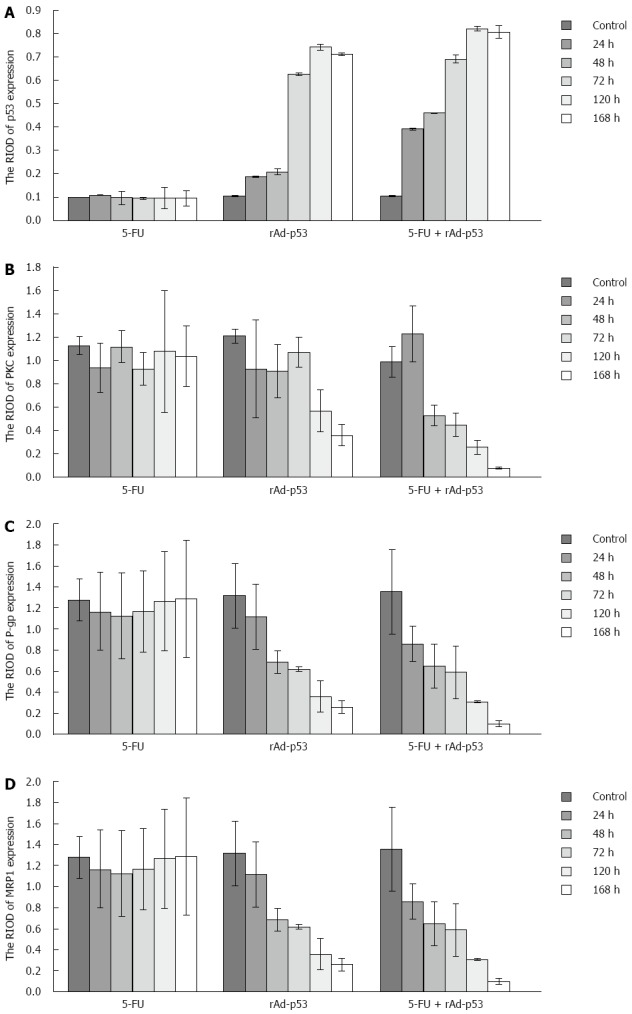
Relative band intensities of p53 expression (A), protein kinase C expression (B), permeability-glycoprotein expression (C) and MRP1 expression (D) in the three experimental groups.
Table 2.
Relative IOD of p53 expression in experimental groups and control group (mean ± SD)
| Group | 24 h | 48 h | 72 h | 120 h | 168 h |
| Control | 0.099 ± 0.001 | 0.105 ± 0.003 | 0.101 ± 0.007 | 0.105 ± 0.015 | 0.103 ± 0.001 |
| 5-FU | 0.108 ± 0.002 | 0.098 ± 0.028 | 0.096 ± 0.004 | 0.097 ± 0.047 | 0.096 ± 0.033 |
| rAd-p53 | 0.187 ± 0.003 | 0.209 ± 0.014 | 0.627 ± 0.005 | 0.743 ± 0.012 | 0.713 ± 0.005 |
| 5-FU + rAd-p53 | 0.393 ± 0.004 | 0.461 ± 0.002 | 0.691 ± 0.018 | 0.822 ± 0.009 | 0.808 ± 0.027 |
| F | 221.565 | 270.392 | 3983.173 | 9639.595 | 3867.703 |
| P value | 0 | 0 | 0 | 0 | 0 |
| P value | > 0.05 | > 0.05 | > 0.05 | > 0.05 | > 0.05 |
| 5-FU vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs rAd-p53 | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs 5-FU | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs 5-FU |
PKC, P-gp and MRP1 expression: Overexpression of PKC, P-gp and MRP1 was observed in the 5-FU group and the control group (Figure 2B-D). There were no significant differences in the RIOD of the expression of these proteins between the two groups (SNK test, P > 0.05; Tables 3, 4 and 5).
Table 3.
Relative IOD of protein kinase C expression in experimental groups and control group (mean ± SD)
| Group | 24 h | 48 h | 72 h | 120 h | 168 h |
| Control | 1.13 ± 0.08 | 1.21 ± 0.06 | 0.99 ± 0.13 | 1.26 ± 0.42 | 1.08 ± 0.20 |
| 5-FU | 0.94 ± 0.21 | 1.12 ± 0.14 | 0.93 ± 0.14 | 1.08 ± 0.52 | 1.04 ± 0.26 |
| rAd-p53 | 0.93 ± 0.42 | 0.91 ± 0.23 | 1.07 ± 0.13 | 0.57 ± 0.18 | 0.36 ± 0.09 |
| 5-FU + rAd-p53 | 1.23 ± 0.24 | 0.53 ± 0.09 | 0.45 ± 0.10 | 0.26 ± 0.06 | 0.08 ± 0.01 |
| F | 1.036 | 33.972 | 14.348 | 24.072 | 71.559 |
| P value | 0.427 | 0 | 0.01 | 0 | 0 |
| P value | > 0.05 | > 0.05 | > 0.05 | > 0.05 | > 0.05 |
| 5-FU vs Control | |||||
| P value | > 0.05 | < 0.05 | > 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs Control | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd/p53 + 5- FU vs Control | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs rAd-p53 | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs 5-FU | |||||
| P value | > 0.05 | < 0.05 | > 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs 5-FU |
Table 4.
Relative IOD of P-gp expression in experimental groups and control group (mean ± SD)
| Group | 24 h | 48 h | 72 h | 120 h | 168 h |
| Control | 1.28 ± 0.20 | 1.32 ± 0.31 | 1.36 ± 0.40 | 1.29 ± 0.43 | 1.30 ± 0.56 |
| 5-FU | 1.17 ± 0.37 | 1.13 ± 0.41 | 1.17 ± 0.39 | 1.27 ± 0.47 | 1.29 ± 0.56 |
| rAd-p53 | 1.12 ± 0.31 | 0.69 ± 0.11 | 0.62 ± 0.02 | 0.36 ± 0.15 | 0.26 ± 0.06 |
| 5-FU + rAd-p53 | 0.86 ± 0.17 | 0.65 ± 0.21 | 0.59 ± 0.25 | 0.31 ± 0.01 | 0.10 ± 0.03 |
| F | 5.670 | 7.301 | 14.645 | 9.844 | 7.971 |
| P value | 0.022 | 0.011 | 0.001 | 0.005 | 0.009 |
| P value | > 0.05 | > 0.05 | > 0.05 | > 0.05 | > 0.05 |
| 5-FU vs Control | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs Control | |||||
| P value | < 0.05 | > 0.05 | > 0.05 | > 0.05 | > 0.05 |
| rAd-p53 + 5-FU vs rAd-p53 | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs 5-FU | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs 5-FU |
Table 5.
Relative IOD of MRP1 expression in experimental groups and control group (mean ± SD)
| Group | 24 h | 48 h | 72 h | 120 h | 168 h |
| Control | 1.31 ± 0.36 | 1.33 ± 0.08 | 1.09 ± 0.24 | 1.22 ± 0.35 | 1.07 ± 0.22 |
| 5-FU | 1.27 ± 0.10 | 1.31 ± 0.11 | 1.14 ± 0.16 | 1.20 ± 0.31 | 1.13 ± 0.14 |
| rAd-p53 | 0.98 ± 0.09 | 0.74 ± 0.11 | 0.58 ± 0.18 | 0.87 ± 0.31 | 0.31 ± 0.08 |
| 5-FU + rAd-p53 | 0.71 ± 0.17 | 0.62 ± 0.02 | 0.51 ± 0.10 | 0.42 ± 0.08 | 0.13 ± 0.10 |
| F | 6.438 | 123.754 | 20.567 | 15.512 | 36.073 |
| P value | 0.016 | 0 | 0 | 0.001 | 0 |
| P value | > 0.05 | > 0.05 | > 0.05 | > 0.05 | > 0.05 |
| 5-FU vs Control | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs Control | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs Control | |||||
| P value | > 0.05 | < 0.05 | > 0.05 | < 0.05 | > 0.05 |
| rAd-p53 + 5-FU vs rAd-p53 | |||||
| P value | < 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 + 5-FU vs 5-FU | |||||
| P value | > 0.05 | < 0.05 | < 0.05 | < 0.05 | < 0.05 |
| rAd-p53 vs 5-FU |
At the observed time points, the expression of PKC, P-gp and MRP1 in the rAd/p53 + 5-FU group gradually decreased in a time-dependent manner (ANOVA, P < 0.05; Tables 3-5). The expression of P-gp and MRP1 was significantly lower than that in the control group and the 5-FU group (SNK test, P < 0.05). More than 48 h after treatment, the expression of PKC in the rAd-p53 + 5-FU group was significantly lower than that in the control, 5-FU and rAd-p53 groups.
In the rAd-p53 group, the expression of P-gp and MRP1 was significantly lower than that in the control group and the 5-FU group at > 48 h of treatment (SNK test, P < 0.05). The expression of PKC was significantly lower than that in the control group and the 5-FU group at > 120 h of treatment (SNK test, P < 0.05).
Pearson’s correlation test
The RIOD of p53 expression was positively correlated with the area ratio of tumor cell apoptosis (the correlation coefficient and P value were 0.545 and 0.000, respectively).
The RIOD of PKC, P-gp and MRP1 expression was negatively correlated with the area ratio of tumor cell apoptosis (correlation coefficients were -0.322, 0.012 and -0.335 and P values were 0.009, -0.541 and 0.000, respectively).
The RIOD of p53 expression and the RIOD of PKC, P-gp and MRP1 expression showed a negative correlation (correlation coefficients were -0.366, 0.004 and -0.406 and P values were 0.001, -0.488 and 0.000, respectively).
DISCUSSION
5-FU is still widely used as a major anticancer drug in the treatment of colon cancer[3]. However, a major impediment to the success of colon cancer chemotherapy is the development of cancer variants exhibiting multidrug resistance (MDR)[2-6,15,16].
MDR usually presents as cross-resistance to multiple chemotherapeutic drugs with different structures[16,17]. Anti-cancer drug resistance in colon cancer cells can be caused by various factors, including alterations in drug influx and efflux, enhancement of drug inactivation and mutation of the drug target induced by various proteins[18-21]. To date, multiple factors have been reported to lead to resistance to chemotherapeutic drugs[16-23]. P-gp, PKC and the multidrug resistance-associated proteins (MRPs) contribute to chemotherapy resistance[17-23]. In our previous studies, overexpression of P-gp, PKC and MRP1 was observed in human colon cancer SW480/5-FU cells (5-FU resistant) and weak expression of these proteins was seen in parental human colon cancer SW480 cells (5-FU responsive)[14].
Various mechanisms contribute to MDR, including the overexpression of drug efflux pumps (pump resistance) and the up-regulation of cellular antiapoptotic defense systems (non-pump resistance)[23,24]. P-gp encoded by the MDR1 gene and MRPs belong to the ATP-binding cassette (ABC) superfamily. These transporter proteins (responsible for pump resistance) mediate the efflux of drugs in the MDR spectrum, such as anthracyclines, out of cells, thus reducing drug efficacy[24]. Generally, there are two approaches used to reverse ABC superfamily-mediated MDR: blocking its drug-pump function and inhibiting its expression[25-27].
PKC is one of the signaling enzymes that is positively regulated by reactive oxygen species (ROS) and plays a crucial role in a variety of pathophysiological states, including tumor progression. PKC contains multiple cysteine residues that can be activated by ROS oxidatively[28,29]. PKC represents a family of serine/threonine kinases that are involved in the regulation of cell growth, cell death and stress responsiveness[30]. Generally, the PKCs are classified into three subfamilies based on their structural and activation characteristics: the conventional or classic (α, βI, βII, and γ), the novel or non-classic (δ, ε, η and θ), and the atypical PKC isoenzymes (ζ, ι and λ)[30]. Different PKC isoenzymes may exert similar or opposite cellular effects by differential coupling of signaling pathways[31]. Cancer cells survive by evading apoptosis or promoting proliferation, invasion and metastasis. PKC may act as a downstream effector of the signaling protein phosphatidylinositol 3-kinase (PI3K). The PI3K-mediated signaling cascade regulates cell proliferation, cell survival, differentiation and apoptosis[32-34]. Phosphorylation of the regulatory subunit p85a is linked to increased survival of cancer cells. The p85 subunit regulates the catalytic subunit p110 by stabilization and inactivation of its kinase activity in the basal state as well as by recruitment of PI3K to phospho-tyrosine residues of the activated receptors[34,35].
PKCα and PKCβ may promote ABCB1 function by phosphorylation[18,33,34]. Notably, the effects of PKC signaling on ABCB1 phosphorylation and function appear to be cell type-dependent. In ovarian carcinoma cells, antisense oligomers directed against PKCα and PKCβ reversed ABCB1-mediated drug resistance[36]. In contrast, PKCβ was not detectable in some reports, and siRNAs targeting PKCα interfered with PKC signaling, but not with ABCB1 function[18]. Moreover, p53 was shown to suppress PKCα-mediated ABCB1 activation in leiomyosarcoma, fibrosarcoma, and osteosarcoma cells[18,34].
Mutations or deletions of suppressor gene p53 are the most common genetic abnormalities that occur during cancerogenesis in the majority of human neoplasms. The p53 gene, localized on the short arm of chromosome 17 (17p13), encodes nucleic phosphoproteins, and affects several cell functions (induction of many genes, regulation of the cell cycle and apoptosis control)[37]. Under the condition of p53 gene mutation, cancer cells remain intact and survive[38].
Evasion from chemotherapy-induced apoptosis due to p53 loss strongly contributes to drug resistance[3-6,16,38]. Wild-type p53 is a key tumor suppressor in preventing tumorigenesis and cancer progression; however, mutant p53, detected in over 50% of all human tumors and in approximately 70% of colorectal cancers[39-43], promotes tumor progression and resistance to therapies[3-6,38,44], and such mutants have become the most common prognostic indicators for both tumor recurrence and cancer death[40,43,45,46]. Prevention of p53 mutation to restore wild-type p53 activity is an attractive anticancer therapy to reverse 5-FU resistance in colon cancer.
Infection with Ad-p53 can significantly down-regulate MDR1 transcription and P-gp expression in breast cancer cell lines and reverse resistance to adriamycin[47]. Treatment with rAd-p53 alone, oxaliplatin alone or combined treatment led to a decrease in Bcl-2 expression and an increase in Bax expression in gastric cancer cells, and induced apoptosis of gastric cancer cells, which was accompanied by increased expression of caspase-3[12]. Therefore, rAd-p53 may enhance the sensitivity of gastric cancer cells to chemotherapy by promoting apoptosis. In the present study, exogenous wild-type p53 gene from rAd-p53 decreased the expression of PKC, P-gp and MRP1 and promoted apoptosis of colon carcinoma cells in nude mice implanted with human colon carcinoma SW480/5-FU (5-FU resistant), which contributed to the reversal of 5-FU resistance.
The antimetabolite, 5-FU, is an analogue of uracil with a fluorine atom at the C5 position of the pyrimidine ring. 5-FU is converted in cells to different active metabolites, including fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP) and fluorouridine triphosphate (FUTP). These metabolites have been implicated in both global RNA metabolism due to incorporation of the ribonucleotide FUMP into RNA, and DNA metabolism due to thymidylate synthase (TS) inhibition or direct incorporation of FdUMP into DNA, leading to a wide range of biological effects which can act as triggers for apoptotic cell death[3,4,15,16]. Therefore, 5-FU can be regarded as a genotoxic agent.
P53 protein in the production of wild-type p53 expression plays a key role in cell cycle regulation and in the cellular response to cytotoxic stress and DNA damage[48-51]. P53 protein is maintained at low levels by MDM2, an E3-ligase that binds p53 and promotes its degradation[52-54]. DNA damage and other stresses, including gamma and UV irradiation, chemotherapeutic agents, hypoxia, heat or alterations in intracellular nucleotide pools, disrupt p53-MDM2 binding, causing p53 levels to increase[49-51,55]. Wild-type p53 is induced in response to a host of genotoxic and environmental stresses, a host of target genes are then transcriptionally activated, including p21, GADD45, Bax and Bcl-2. Induction of p21, in turn, leads to cell cycle arrest at both G1 and G2 checkpoints. This function is thought to be essential in preserving the integrity of the cellular genome in response to treatment with cytotoxic agents. In addition to mediating cell cycle arrest, p53 is a potent inducer of apoptosis and programmed cell death[49-51].
Increased p53 protein in response to genotoxic stress also occurs in cancer cells[56-59]. Treatment of human colon cancer RKO cells and tetraploid cancer cells with 5-FU resulted in a significant increase in the levels of the endogenous p53 protein family in vitro and enhanced tumor suppression[59]. The p53 protein family forms an interacting network of proteins[60]. Cancer cell responses to 5-FU treatment are determined by the total activity of the entire p53 family rather than p53 alone[60]. Suppressor p53 is one of the molecular targets of 5-FU. With regard to 5-FU, translational regulation is an important process for controlling endogenous p53 expression[59-61]. Our previous study demonstrated that the expression of exogenous wild-type p53 gene in colon cancer cells in nude mice bearing human colon carcinoma SW480 (5-FU responsive) treated with rAd-p53 + 5-FU was significantly higher than that with rAd-p53 alone, and tumor necrosis was positively correlated with p53 expression in vivo[13]. 5-FU also increased the anticancer effect of rAd/p53 in vivo.
In the present study, p53 expression in colon cancer SW480/5-FU in the rAd/p53 group and rAd-p53 + 5-FU group was higher than in the control and 5-FU groups and increased in a time-dependent manner. P53 expression in the rAd-p53 + 5-FU group was significantly higher than in the rAd-p53 group. These results suggest that 5-FU increased the expression of exogenous wild-type p53 gene in colon cancer (resistant to 5-FU) in vivo. Exogenous wild-type p53 is also induced in response to genotoxic stress in chemotherapy-resistant cancer cells.
In summary, exogenous wild-type p53 gene from rAd-p53 can decrease the expression of PKC, P-gp and MRP1 in SW480/5-FU (5-FU resistant) and promote apoptosis of tumor cells, which contributes to the reversal of 5-FU resistance in vivo. 5-FU can increase the expression of exogenous wild-type p53, thus 5-FU combined with rAd-p53 has a synergistic anticancer effect in colon cancer resistant to 5-FU in vivo. Therefore, the DNA-damaging agent 5-FU combined with exogenous wild-type p53 provides a potential therapeutic strategy and can enhance the sensitivity and reduce the toxicity of chemotherapy and improve the clinical efficacy of colon cancer chemotherapy.
ACKNOWLEDGMENTS
A statistical review of the study was performed by Gui-Qin Wang (a biomedical statistician at Nansha Central Hospital, Guangzhou First People’s Hospital, Guangzhou Medical University, Guangzhou, China).
COMMENTS
Background
5-fluorouracil (5-FU) is a widely used chemotherapeutic drug in the treatment of advanced colorectal cancer (CRC); however, response rates are only 10% to 15%, due to severe side effects and resistance. Mutations or deletions of p53 can cause cells to become resistant to 5-FU. Therefore, gene replacement therapy for a mutated p53 gene using recombinant adenovirus-mediated p53 (rAd-p53) may overcome 5-FU resistance caused by mutations or deletions of p53. Therefore, rAd-p53 may contribute to the reversal of 5-FU resistance in colon cancer.
Research frontiers
Exogenous wild-type p53 from rAd-p53 increased tumor necrosis in human colon cancer SW480 (5-FU responsive) harboring mutant p53, and 5-FU combined with rAd-p53 had a synergistic anticancer effect in vivo.
Innovations and breakthroughs
This is the first study to evaluate 5-FU combined with rAd-p53 in the treatment of colon cancer SW480/5-FU (5-FU resistant) compared with controls in vivo.
Applications
This study first showed that exogenous wild-type p53 gene from rAd-p53 can decrease the expression of PKC, P-gp and MRP1 in colon cancer SW480/5-FU (5-FU resistant) and promote apoptosis of tumor cells, which contributes to the reversal of 5-FU resistance in vivo. 5-FU can increase the expression of exogenous wild-type p53; thus, 5-FU combined with rAd-p53 had a synergistic anticancer effect in 5-FU resistant colon cancer in vivo.
Terminology
5-FU increased the expression of exogenous wild-type p53 gene in colon cancer (5-FU resistant) in vivo. Exogenous wild-type p53 is also induced in response to genotoxic stress in chemotherapy-resistant cancer cells.
Peer-review
The authors demonstrated that 5-FU increased the expression of exogenous wild-type p53 gene, and exogenous wild-type p53 is induced in response to genotoxic stress by 5-FU in colon cancer (resistant to 5-FU) in vivo. These results are interesting. Previous studies have established that increased p53 protein in response to genotoxic stress occurs in cancer cells in vitro. In this study, the authors showed for the first time that 5-FU combined with rAd-p53 has a synergistic anticancer effect for colon cancer resistant to 5-FU in vivo.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Experimental Animal Center of Sun Yat-sen University and Guangzhou Medical University respectively.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University and Guangzhou Medical University.
Conflict-of-interest statement: To the best of our knowledge, no conflict of interest exists.
Data sharing statement: Technical appendix, statistical code, and dataset available from the corresponding author at xieqi8@yeah.net.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 23, 2016
First decision: May 27, 2016
Article in press: July 6, 2016
P- Reviewer: Sam MR S- Editor: Ma YJ L- Editor: Filipodia E- Editor: Ma S
References
- 1.Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, Wolfe C, et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1:505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardini B, Kumar R, Naccarati A, Novotny J, Prasad RB, Forsti A, Hemminki K, Vodicka P, Lorenzo Bermejo J. 5-Fluorouracil-based chemotherapy for colorectal cancer and MTHFR/MTRR genotypes. Br J Clin Pharmacol. 2011;72:162–163. doi: 10.1111/j.1365-2125.2010.03892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y, Chen W, Wang K, Li D, Jin W, et al. JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Sci Rep. 2014;4:4694. doi: 10.1038/srep04694. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Sun XX, Dai MS, Lu H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J Biol Chem. 2007;282:8052–8059. doi: 10.1074/jbc.M610621200. [DOI] [PubMed] [Google Scholar]
- 5.Subbarayan PR, Sarkar M, Nelson G, Benitez E, Singhal S, Ardalan B. Chronic exposure of colorectal cancer cells in culture to fluoropyrimidine analogs induces thymidylate synthase and suppresses p53. A molecular explanation for the mechanism of 5-FU resistance. Anticancer Res. 2010;30:1149–1156. [PubMed] [Google Scholar]
- 6.Huang C, Zhang XM, Tavaluc RT, Hart LS, Dicker DT, Wang W, El-Deiry WS. The combination of 5-fluorouracil plus p53 pathway restoration is associated with depletion of p53-deficient or mutant p53-expressing putative colon cancer stem cells. Cancer Biol Ther. 2009;8:2186–2193. doi: 10.4161/cbt.8.22.10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baek JH, Agarwal ML, Tubbs RR, Vladisavljevic A, Tomita H, Bukowski RM, Milsom JW, Kim JM, Kwak JY. In vivo recombinant adenovirus-mediated p53 gene therapy in a syngeneic rat model for colorectal cancer. J Korean Med Sci. 2004;19:834–841. doi: 10.3346/jkms.2004.19.6.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guan YS, La Z, Yang L, He Q, Li P. p53 gene in treatment of hepatic carcinoma: status quo. World J Gastroenterol. 2007;13:985–992. doi: 10.3748/wjg.v13.i7.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue H, Shiraki K, Murata K, Sugimoto K, Kawakita T, Yamaguchi Y, Saitou Y, Enokimura N, Yamamoto N, Yamanaka Y, et al. Adenoviral-mediated transfer of p53 gene enhances TRAIL-induced apoptosis in human hepatocellular carcinoma cells. Int J Mol Med. 2004;14:271–275. [PubMed] [Google Scholar]
- 10.Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther. 2006;6:55–61. doi: 10.1517/14712598.6.1.55. [DOI] [PubMed] [Google Scholar]
- 11.Shimada H, Shimizu T, Ochiai T, Liu TL, Sashiyama H, Nakamura A, Matsubara H, Gunji Y, Kobayashi S, Tagawa M, et al. Preclinical study of adenoviral p53 gene therapy for esophageal cancer. Surg Today. 2001;31:597–604. doi: 10.1007/s005950170093. [DOI] [PubMed] [Google Scholar]
- 12.Chen GX, Zheng LH, Liu SY, He XH. rAd-p53 enhances the sensitivity of human gastric cancer cells to chemotherapy. World J Gastroenterol. 2011;17:4289–4297. doi: 10.3748/wjg.v17.i38.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie Q, Liang BL, Wu YH, Zhang J, Chen MW, Liu HY, Gu XF, Xu J. Synergistic anticancer effect of rAd/P53 combined with 5-fluorouracil or iodized oil in the early therapeutic response of human colon cancer in vivo. Gene. 2012;499:303–308. doi: 10.1016/j.gene.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Xie Q, Yang YM, Wu MYi, Zhang DX, Lei ZX, Zhang J, Xu J, Zhong WD. Detection of the drug resistance of human colon cancer in mice with magnetic resonance diffusion imaging. ZhongguoLinchuangYixueYingxiangZazhi. 2016;17:355–358, 366. [Google Scholar]
- 15.Ahn JY, Lee JS, Min HY, Lee HY. Acquired resistance to 5-fluorouracil via HSP90/Src-mediated increase in thymidylate synthase expression in colon cancer. Oncotarget. 2015;6:32622–32633. doi: 10.18632/oncotarget.5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang N, Yin Y, Xu SJ, Chen WS. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules. 2008;13:1551–1569. doi: 10.3390/molecules13081551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kathawala RJ, Gupta P, Ashby CR, Chen ZS. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Michaelis M, Rothweiler F, Löschmann N, Sharifi M, Ghafourian T, Cinatl J. Enzastaurin inhibits ABCB1-mediated drug efflux independently of effects on protein kinase C signalling and the cellular p53 status. Oncotarget. 2015;6:17605–17620. doi: 10.18632/oncotarget.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balcerczak E, Panczyk M, Piaskowski S, Pasz-Walczak G, Sałagacka A, Mirowski M. ABCB1/MDR1 gene polymorphisms as a prognostic factor in colorectal cancer. Int J Colorectal Dis. 2010;25:1167–1176. doi: 10.1007/s00384-010-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He T, Mo A, Zhang K, Liu L. ABCB1/MDR1 gene polymorphism and colorectal cancer risk: a meta-analysis of case-control studies. Colorectal Dis. 2013;15:12–18. doi: 10.1111/j.1463-1318.2012.02919.x. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Wang C, Qin YW, Yan SK, Gao YR. Simultaneous suppression of multidrug resistance and antiapoptotic cellular defense induces apoptosis in chemoresistant human acute myeloid leukemia cells. Leuk Res. 2007;31:989–994. doi: 10.1016/j.leukres.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 22.O’Brian CA, Ward NE, Stewart JR, Chu F. Prospects for targeting protein kinase C isozymes in the therapy of drug-resistant cancer--an evolving story. Cancer Metastasis Rev. 2001;20:95–100. doi: 10.1023/a:1013186430906. [DOI] [PubMed] [Google Scholar]
- 23.Zhan M, Yu D, Liu J, Glazer RI, Hannay J, Pollock RE. Transcriptional repression of protein kinase Calpha via Sp1 by wild type p53 is involved in inhibition of multidrug resistance 1 P-glycoprotein phosphorylation. J Biol Chem. 2005;280:4825–4833. doi: 10.1074/jbc.M407450200. [DOI] [PubMed] [Google Scholar]
- 24.Lo YL, Liu Y. Reversing multidrug resistance in Caco-2 by silencing MDR1, MRP1, MRP2, and BCL-2/BCL-xL using liposomal antisense oligonucleotides. PLoS One. 2014;9:e90180. doi: 10.1371/journal.pone.0090180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue C, Wang C, Liu Q, Meng Q, Sun H, Huo X, Ma X, Liu Z, Ma X, Peng J, et al. Targeting P-glycoprotein expression and cancer cell energy metabolism: combination of metformin and 2-deoxyglucose reverses the multidrug resistance of K562/Dox cells to doxorubicin. Tumour Biol. 2016 doi: 10.1007/s13277-015-4478-8. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 26.Abdallah HM, Al-Abd AM, El-Dine RS, El-Halawany AM. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J Adv Res. 2015;6:45–62. doi: 10.1016/j.jare.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobori T, Harada S, Nakamoto K, Tokuyama S. Mechanisms of P-glycoprotein alteration during anticancer treatment: role in the pharmacokinetic and pharmacological effects of various substrate drugs. J Pharmacol Sci. 2014;125:242–254. doi: 10.1254/jphs.14r01cr. [DOI] [PubMed] [Google Scholar]
- 28.Gopalakrishna R, Gundimeda U. Antioxidant regulation of protein kinase C in cancer prevention. J Nutr. 2002;132:3819S–3823S. doi: 10.1093/jn/132.12.3819S. [DOI] [PubMed] [Google Scholar]
- 29.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, Missiroli S, Patergnani S, Poletti F, Rimessi A, et al. Redox control of protein kinase C: cell- and disease-specific aspects. Antioxid Redox Signal. 2010;13:1051–1085. doi: 10.1089/ars.2009.2825. [DOI] [PubMed] [Google Scholar]
- 31.Caino MC, Meshki J, Kazanietz MG. Hallmarks for senescence in carcinogenesis: novel signaling players. Apoptosis. 2009;14:392–408. doi: 10.1007/s10495-009-0316-z. [DOI] [PubMed] [Google Scholar]
- 32.Maurya AK, Vinayak M. Quercetin regresses Dalton’s lymphoma growth via suppression of PI3K/AKT signaling leading to upregulation of p53 and decrease in energy metabolism. Nutr Cancer. 2015;67:354–363. doi: 10.1080/01635581.2015.990574. [DOI] [PubMed] [Google Scholar]
- 33.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 34.Maurya AK, Vinayak M. Anticarcinogenic action of quercetin by downregulation of phosphatidylinositol 3-kinase (PI3K) and protein kinase C (PKC) via induction of p53 in hepatocellular carcinoma (HepG2) cell line. Mol Biol Rep. 2015;42:1419–1429. doi: 10.1007/s11033-015-3921-7. [DOI] [PubMed] [Google Scholar]
- 35.Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci USA. 2007;104:7809–7814. doi: 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masanek U, Stammler G, Volm M. Modulation of multidrug resistance in human ovarian cancer cell lines by inhibition of P-glycoprotein 170 and PKC isoenzymes with antisense oligonucleotides. J Exp Ther Oncol. 2002;2:37–41. doi: 10.1046/j.1359-4117.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- 37.Carson DA, Lois A. Cancer progression and p53. Lancet. 1995;346:1009–1011. doi: 10.1016/s0140-6736(95)91693-8. [DOI] [PubMed] [Google Scholar]
- 38.Hodorová I, Rybárová S, Vecanová J, Solár P, Plank L, Mihalik J. Relation between expression pattern of wild-type p53 and multidrug resistance proteins in human nephroblastomas. Acta Histochem. 2013;115:273–278. doi: 10.1016/j.acthis.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Bookstein R, Demers W, Gregory R, Maneval D, Park J, Wills K. p53 gene therapy in vivo of herpatocellular and liver metastatic colorectal cancer. Semin Oncol. 1996;23:66–77. [PubMed] [Google Scholar]
- 40.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 41.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 42.Spitz FR, Nguyen D, Skibber JM, Cusack J, Roth JA, Cristiano RJ. In vivo adenovirus-mediated p53 tumor suppressor gene therapy for colorectal cancer. Anticancer Res. 1996;16:3415–3422. [PubMed] [Google Scholar]
- 43.Liu YY, Hill RA, Li YT. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv Cancer Res. 2013;117:59–89. doi: 10.1016/B978-0-12-394274-6.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grassilli E, Narloch R, Federzoni E, Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE, Bonin S, et al. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res. 2013;19:3820–3831. doi: 10.1158/1078-0432.CCR-12-3289. [DOI] [PubMed] [Google Scholar]
- 45.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 46.Olivier M, Langerød A, Carrieri P, Bergh J, Klaar S, Eyfjord J, Theillet C, Rodriguez C, Lidereau R, Bièche I, et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006;12:1157–1167. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 47.Qi X, Chang Z, Song J, Gao G, Shen Z. Adenovirus-mediated p53 gene therapy reverses resistance of breast cancer cells to adriamycin. Anticancer Drugs. 2011;22:556–562. doi: 10.1097/CAD.0b013e328345b4e7. [DOI] [PubMed] [Google Scholar]
- 48.García MA, Carrasco E, Aguilera M, Alvarez P, Rivas C, Campos JM, Prados JC, Calleja MA, Esteban M, Marchal JA, et al. The chemotherapeutic drug 5-fluorouracil promotes PKR-mediated apoptosis in a p53-independent manner in colon and breast cancer cells. PLoS One. 2011;6:e23887. doi: 10.1371/journal.pone.0023887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 50.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 51.Vogelstein B, Kinzler KW. p53 function and dysfunction. Cell. 1992;70:523–526. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- 52.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 53.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 54.Davaadelger B, Shen H, Maki CG. Novel roles for p53 in the genesis and targeting of tetraploid cancer cells. PLoS One. 2014;9:e110844. doi: 10.1371/journal.pone.0110844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev. 1996;10:934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- 56.Mowat MR. p53 in tumor progression: life, death, and everything. Adv Cancer Res. 1998;74:25–48. doi: 10.1016/s0065-230x(08)60764-2. [DOI] [PubMed] [Google Scholar]
- 57.Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA. 1992;89:4495–4499. doi: 10.1073/pnas.89.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 59.Ju J, Schmitz JC, Song B, Kudo K, Chu E. Regulation of p53 expression in response to 5-fluorouracil in human cancer RKO cells. Clin Cancer Res. 2007;13:4245–4251. doi: 10.1158/1078-0432.CCR-06-2890. [DOI] [PubMed] [Google Scholar]
- 60.Das R, Bhattacharya K, Sarkar S, Samanta SK, Pal BC, Mandal C. Mahanine synergistically enhances cytotoxicity of 5-fluorouracil through ROS-mediated activation of PTEN and p53/p73 in colon carcinoma. Apoptosis. 2014;19:149–164. doi: 10.1007/s10495-013-0907-6. [DOI] [PubMed] [Google Scholar]
- 61.Vilgelm AE, Washington MK, Wei J, Chen H, Prassolov VS, Zaika AI. Interactions of the p53 protein family in cellular stress response in gastrointestinal tumors. Mol Cancer Ther. 2010;9:693–705. doi: 10.1158/1535-7163.MCT-09-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]