

Abstract
Staphylococcus aureus osteomyelitis is a common and debilitating invasive infection of bone. Treatment of osteomyelitis is confounded by widespread antimicrobial resistance and the propensity of bacteria to trigger pathological changes in bone remodeling that limit antimicrobial penetration to the infectious focus. Adjunctive therapies that limit pathogen-induced bone destruction could therefore limit morbidity and enhance traditional antimicrobial therapies. In this study, we evaluate the efficacy of the U.S. Food and Drug Administration-approved, nonsteroidal anti-inflammatory (NSAID) compound diflunisal in limiting S. aureus cytotoxicity toward skeletal cells and in preventing bone destruction during staphylococcal osteomyelitis. Diflunisal is known to inhibit S. aureus virulence factor production by the accessory gene regulator (agr) locus, and we have previously demonstrated that the Agr system plays a substantial role in pathological bone remodeling during staphylococcal osteomyelitis. Consistent with these observations, we find that diflunisal potently inhibits osteoblast cytotoxicity caused by S. aureus secreted toxins independently of effects on bacterial growth. Compared to commonly used NSAIDs, diflunisal is uniquely potent in the inhibition of skeletal cell death in vitro. Moreover, local delivery of diflunisal by means of a drug-eluting, bioresorbable foam significantly limits bone destruction during S. aureus osteomyelitis in vivo. Collectively, these data demonstrate that diflunisal potently inhibits skeletal cell death and bone destruction associated with S. aureus infection and may therefore be a useful adjunctive therapy for osteomyelitis.
INTRODUCTION
Osteomyelitis is an invasive infection of bone, most frequently caused by the Gram-positive pathogen Staphylococcus aureus (1). Treatment of osteomyelitis is confounded by an increasing prevalence of antimicrobial resistance, as well as the propensity for patients to develop pathological changes in bone remodeling that limit antimicrobial penetration to the infectious focus. For this reason, patients suffering from osteomyelitis often undergo multiple surgical debridements to remove infected and necrotic bone, while also receiving prolonged antimicrobial therapy (1, 2). Despite these aggressive measures, a subset of patients with acute osteomyelitis will progress to chronic infection, which is highly refractory to further intervention and may require months to years of antimicrobial treatment. Moreover, although adult osteomyelitis is often secondary to comorbidities such as trauma, surgery, diabetes, or vascular insufficiency, hematogenous osteomyelitis in previously healthy children is more prevalent in the current era of community-acquired staphylococcal infections (3). Children are inherently vulnerable to adverse outcomes during osteomyelitis given the presence of active epiphyseal plates and thus have the potential to develop limb-length discrepancy (4). Taken together, these facts illustrate the need for adjunctive therapies that lessen the morbidity of staphylococcal osteomyelitis and promote the efficacy of traditional antimicrobials through inhibition of destructive bone remodeling.
S. aureus is capable of producing numerous virulence factors, including host-binding proteins, secreted toxins and superantigens, degradative enzymes, and immune evasion factors. We recently defined the secreted alpha-type phenol soluble modulins (PSMs) as key contributors to the pathogenesis of staphylococcal osteomyelitis (5). Alpha PSMs were both necessary and sufficient to elicit destruction of osteoblasts, the skeletal cells responsible for synthesis of new bone and regulation of bone-resorbing osteoclasts. Moreover, inactivation of the alpha-type PSMs led to significantly decreased bone destruction in a murine model of S. aureus osteomyelitis, despite not having a significant effect on bacterial burdens. Production of PSMs in S. aureus is regulated by the accessory gene regulator protein AgrA, which is a component of the quorum-sensing agr regulatory locus. In contrast to most agr-regulated virulence factors, the PSMs are regulated directly by AgrA, rather than via the regulatory RNA molecule RNAIII (6). Accordingly, inactivation of the agr locus also leads to a significant reduction in bone destruction during osteomyelitis (5). These data suggest that the agr locus might be an attractive target for adjunctive therapies that lessen the morbidity of staphylococcal osteomyelitis.
To date, a number of studies have shown preclinical efficacy of agr inhibitory compounds in limiting S. aureus virulence (7 – 10). One such study identified the U.S. Food and Drug Administration (FDA)-approved nonsteroidal anti-inflammatory drug (NSAID) diflunisal as a potent inhibitor of AgrA-mediated transcriptional regulation (11). In an in silico screen, diflunisal was predicted to inhibit AgrC-mediated phosphorylation of AgrA. Consistent with this prediction, diflunisal treatment inhibited rabbit red blood cell lysis and significantly lowered transcript levels of hla, RNAIII, and psmα without affecting bacterial growth (7, 11). However, the efficacy of diflunisal in mitigating staphylococcal virulence in vivo has yet to be determined.
Based on the critical role of agr-regulated virulence factors in the pathogenesis of staphylococcal osteomyelitis, we hypothesized that diflunisal treatment would limit skeletal cell death and cortical bone destruction during S. aureus infection. Here, we test this hypothesis by evaluating the effects of diflunisal and other commonly used NSAIDs on staphylococcal growth, virulence factor production, and host cell cytotoxicity. We also test the preclinical efficacy of local diflunisal therapy during staphylococcal osteomyelitis by synthesizing drug-eluting polyurethane foams for direct delivery to infected bone. Collectively, our results suggest that diflunisal treatment is a promising strategy for adjunctive therapy of osteomyelitis given its osteoprotective and anti-virulence properties.
MATERIALS AND METHODS
Bacterial strains, reagents, and growth conditions.
An erythromycin-sensitive derivative of the USA300-lineage strain LAC was used for all experiments unless otherwise noted (12). Strain Newman was also used to test whether diflunisal could limit the cytotoxic potential of other clinical strains of S. aureus (13). For growth curve analysis, overnight cultures were back-diluted 1:1,000 into tryptic soy broth (TSB) supplemented with various NSAIDs or vehicle control. Cultures were grown in glass Erlenmeyer flasks at 37°C and 180 rpm, shaking at a flask-to-volume ratio of 5:1. The optical density at 600 nm (OD600) was measured at the indicated time points. Diflunisal, ibuprofen, ketorolac, piroxicam, and salicylic acid were purchased from Sigma and dissolved in either 100% ethanol or dimethyl sulfoxide (DMSO) at a final concentration of 10 mg/ml. PSMα2 was synthesized to >95% purity by AAPPTec (Louisville, KY) and resuspended in DMSO. Lysine triisocyanate (LTI) was purchased from Kyowa Hakko USA (New York, NY) and contained 42.2% NCO. For polyester triol synthesis, ε-caprolactone and stannous octoate were purchased from Sigma-Aldrich, and d,l-lactide and glycolide were purchased from Polysciences (Warrington, PA). Triethylene diamine (TEDA) catalyst was received from Goldschmidt (TEGOAMIN33, Hopewell, VA). All other reagents, including calcium stearate and turkey red oil, were purchased from Sigma-Aldrich.
Osteoblast cell culture.
Primary human osteoblasts were obtained from Lonza (Basel, Switzerland) and cultured according to the manufacturer's recommendations. To isolate primary murine osteoblasts, murine femurs were extracted, the bone marrow was flushed, and the diaphysis was cut into small pieces. The bone pieces were digested twice in 0.01% trypsin-EDTA and 2 mg/ml collagenase for 30 min at 37°C. After digestion, bone pieces were plated in α-MEM supplemented with 10% fetal bovine serum (FBS), and cells were allowed to migrate from the bone for 10 to 14 days prior to harvest and plating for cytotoxicity assays. Cell lines were obtained from the American Type Culture Collection (ATCC) and propagated at 37°C and 5% CO2 according to ATCC recommendations. The media were replaced every 2 to 3 days. All cell culture media were prepared with 1× penicillin-streptomycin and sterilized using a 0.22-μm-pore size filter prior to use. MC3T3 E-1 murine osteoblastic cells were cultured in α-MEM, supplemented with 10% FBS. Saos-2 human osteoblastic cells were grown in McCoy's 5A medium with 15% FBS. The following cell densities were used for cytotoxicity assays: MC3T3 E1 murine preosteoblastic cells and primary murine osteoblasts at 5,000 cells per well, Saos-2 human osteoblastic cells at 10,000 cells per well, and primary human osteoblasts at 3,500 cells per well.
Preparation of concentrated supernatants.
To prepare concentrated bacterial supernatants, three colonies were inoculated into triplicate 15-ml cultures in loosely capped 50-ml conical tubes and grown for 15 h in RPMI supplemented with 1% Casamino Acids and various NSAIDs or vehicle control. For some experiments, bacteria were alternatively grown in glass Erlenmeyer flasks that were tightly sealed with a rubber stopper. Bacteria were grown for 15 h at 37°C and 180 rpm, and then triplicate cultures were combined, an aliquot was taken for CFU enumeration, and supernatants were harvested by centrifugation. Supernatants were sterilized by passage through a 0.22-μm-pore size filter and then concentrated to a final volume of approximately 1.5 ml using Amicon Ultra 3-kDa nominal molecular mass columns. After concentration, supernatants were again filter sterilized and either used immediately or frozen at −80°C. For some experiments, synthetic PSMα2 (100 μg/ml) was added to concentrated supernatants prior to intoxication of the cells.
Osteoblast cytotoxicity assay.
The cytotoxic potential of concentrated supernatants toward MC3T3-E1 murine osteoblastic cells, Saos-2 human osteoblastic cells, primary murine osteoblasts, and primary human osteoblasts was determined as previously described (5, 14). Briefly, cells were seeded into 96-well tissue culture plates 12 to 24 h prior to intoxication and grown at 37°C and 5% CO2. The tissue culture medium was then replaced with fresh media containing various concentrations of concentrated supernatants. Cells were incubated an additional 22 h, at which time the cell viability was determined using CellTiter AQueous One solution (Promega, Madison, WI) according to the manufacturer's instructions. For some experiments, diflunisal was added to the cell culture medium at the time of intoxication at a final concentration of 10 μg/ml.
Synthesis of PUR foams for local drug delivery.
LTI, tertiary amine catalyst, polyester triol, and turkey red oil were all dried at 10 mm Hg for 3 h at 80°C to avoid excess amounts of water during polyurethane (PUR) foam synthesis. The polyol component (70% ε-caprolactone, 20% glycolide, and 10% lactide) of the PUR foam comprised a polyester triol of 900 g/mol that was synthesized using previously published techniques (15, 16). Appropriate amounts of ε-caprolactone, glycolide, d,l-lactide, dried glycerol, and stannous octoate were mixed in a 250-ml round-bottom flask and stirred under dry argon for 36 h at 140°C. The polyol was then washed with hexane and vacuum-dried at 80°C for 14 h. Foams were synthesized using a formulation that maintained constant ratios relative to the polyol component and premixed with diflunisal. Formulations of foams included 100 pphp (parts per hundred part polyol) polyol 7C2G1L900 mixed with 1.0 pphp water, 4.5 pphp tertiary catalyst, 1.5 pphp turkey red oil, 6.0 pphp calcium stearate, and the appropriate amount of LTI. Appropriate ratios of LTI and polyol were mixed to ensure foams contained targeted index values (100 × NCO equivalents/OH equivalents) of 115. Reactants were mixed in a 5-ml plastic container for 1.5 min using a Hauschild SpeedMixer DAC 150 FVZ-K vortex mixer (Flacktek) and left overnight to cure. The foams synthesized included blank foams and foams loaded with 10 mM diflunisal. Foams were cut into prisms (6 mm by 4.5 mm by 4.5 mm) for in vivo testing and sterilized by ethylene oxide treatment.
In vitro release kinetics of diflunisal PUR foams.
Foams containing 10 mM (10.8 mg) of diflunisal were synthesized and cut into four equal slices. The foams were approximately 4.35 cm3 before being cut into slices at approximate sizes of 1.09 cm3. Each foam slice was then placed in a 65 ml vial and immersed in 60 ml of phosphate-buffered saline (PBS) solution. All vials were placed on a mixer and incubated at 37°C. Then, 50-ml samples were withdrawn at 4, 8, 24, 32, 48, 56, 72, and 144 h postincubation and replaced with 50 ml of fresh PBS. The samples were kept at 4°C until analyzed by high-pressure liquid chromatography using a system that contained a Waters 1525 binary pump and a 2487 Dual-Absorbance Detector at 255 nm. Samples were analyzed using published techniques (17). Diflunisal samples were eluted through an Xterra RP18 column containing 5-μm particle sizes and measured 4.6 mm in diameter by 250 mm in length. The mobile phase for this analysis contained a ratio of A40:B60 of acetonitrile (A) and 50 mM acetate buffer (pH 4.2) containing sodium acetate anhydrous and distilled water (B) flowing at 0.35 ml/min. The injection volume was 15 μl, and the column temperature was maintained at 30°C.
Murine model of osteomyelitis.
Osteomyelitis was induced in 7- to 8-week-old female C57BL/6J mice as previously described (5). An inoculum of 106 CFU in 2 μl of PBS was delivered into murine femurs. For the evaluation of local diflunisal therapy, PUR foams (6 mm × 4.5 mm × 4.5 mm) loaded with 10 mM diflunisal were fabricated, sterilized, and wrapped around the femur at the inoculation site and sutured into place. Empty PUR foams served as a mock treatment control. As a positive control for the feasibility of local therapy using PUR foams, 8% (wt/wt) vancomycin foams were synthesized and used in the osteomyelitis model. At various times postinfection, mice were euthanized, and the infected femurs were removed and either processed for CFU enumeration or imaged by micro-computed tomography (microCT). For CFU enumeration, femurs were homogenized in a Bullet Blender (Next Advance, Averill, NY) using the Navy Bead Lysis kit and plated at limiting dilution on tryptic soy agar. Analysis of cortical bone destruction was determined by microCT imaging as previously described (5). Specifically, axial images of each femur were acquired with 5.0-μm voxels at 70 kV, 200 μA, 2,000 projections per rotation, and an integration time of 350 ms in a 10.24-mm field of view. Each imaging scan comprised 1,635 slices (8.125 mm) of the length of the femur, centered on the inoculation site as visualized in the scout-view radiographs. A volume of interest including only the original cortical bone and any destruction was selected by drawing inclusive contours on the periosteal surface and excluding contours on the endosteal surface. New bone formed during the course of infection was excluded from this analysis. This process was repeated iteratively for all of the 1,635 slices of the microCT scan (comprising the entire diaphysis) using the manufacturer's analytical software. Volume of cortical bone destruction was determined by segmenting the image with a lower threshold of 0 and an upper threshold of 595 mg HA/ccm (milligrams of hydroxyapatite per cubic centimeter), sigma 1.3, and support 1 to exclude bone in the analysis. The direct voxel counting method was used for all reported calculations in each analysis.
Statistical evaluation.
Differences in cytotoxicity, cortical bone destruction, and CFU counts were analyzed by Student t test. A P value of ≤0.05 was considered significant.
RESULTS
Diflunisal inhibits S. aureus cytotoxicity toward human and murine osteoblasts without affecting bacterial growth.
In silico analyses identified the NSAID diflunisal as a potent inhibitor of AgrA-mediated gene expression (11). We previously demonstrated that Agr-dependent virulence factor production, and in particular synthesis of alpha-type PSMs, significantly contributes to skeletal cell death and bone destruction associated with S. aureus osteomyelitis (5, 14, 18). Accordingly, we hypothesized that diflunisal treatment would limit PSM production, and therefore abrogate osteoblast cytotoxicity. To test this hypothesis, a USA300-lineage strain of S. aureus was grown in the presence of various concentrations of diflunisal or vehicle control, after which concentrated supernatants were prepared and tested for cytotoxicity toward murine and human osteoblasts. Treatment with diflunisal significantly limited the cytotoxicity of supernatant from strain LAC toward murine osteoblastic MC3T3 cells in a dose-dependent manner (Fig. 1A). Moreover, this effect was not strain dependent, since diflunisal also significantly limited the cytotoxicity of supernatant from strain Newman (see Fig. S1A in the supplemental material). Diflunisal treatment also potently inhibited staphylococcal killing of human Saos-2 osteoblastic cells, primary murine osteoblasts, and primary human osteoblasts (Fig. 1B). The diminution of cytotoxicity in the presence of diflunisal was not a result of decreased bacterial density, since diflunisal treatment did not significantly affect staphylococcal growth kinetics (Fig. 1C). Collectively, these data indicate that diflunisal treatment significantly limits the ability of S. aureus to destroy osteoblasts.
FIG 1.

Diflunisal inhibits S. aureus cytotoxicity toward human and murine osteoblasts without affecting bacterial growth. (A) MC3T3 murine osteoblastic cells were intoxicated with 20 or 30% (vol/vol) of the concentrated supernatant (SN) from S. aureus grown in the presence of vehicle control (DMSO) or the indicated concentrations of diflunisal. Percent cell viability is depicted relative to mock intoxication with sterile RPMI. Error bars represent the standard deviation (SD). n = 5 per group, and data are representative of three independent trials. ***, P < 0.001 relative to the vehicle control. (B) Same as panel A, except that Saos-2 human osteoblastic cells, primary murine osteoblasts, or primary human osteoblasts were intoxicated with 30% (vol/vol) concentrated SN. n = 10, and the data are representative of at least two independent trials. Error bars represent the SD. ***, P < 0.001 relative to the vehicle control. (C) Growth analysis (i.e., the OD600) of S. aureus cultured in glass Erlenmeyer flasks with a 1:5 volume-to-flask ratio and in the presence of two different vehicle controls (100% ethanol or DMSO) or 10 μg/ml diflunisal. Error bars represent the SD. (D) Same as panel B, except that 100 μg/ml of synthetic PSMα2 was added to concentrated supernatant prior to intoxication of primary human osteoblasts or MC3T3 cells. Cells were treated with vehicle (DMSO) or 10 μg/ml diflunisal. n = 10, and the data are representative of two independent trials. Error bars represent the SD. ***, P < 0.001 relative to vehicle control; ###, P < 0.001 relative to diflunisal-treated cells without PSMα2.
During the completion of these experiments, we discovered that oxygenation significantly impacts the cytotoxic potential of S. aureus cultures (14). Specifically, the growth of S. aureus in tightly capped Erlenmeyer flasks, and thus lower oxygenation, led to enhanced cytotoxicity relative to culture in flasks loosely covered by foil. We therefore conducted additional assays to test whether diflunisal could inhibit the cytotoxic potential of staphylococcal cultures grown under altered oxygenation. In staphylococcal cultures grown in tightly capped flasks, diflunisal still significantly inhibited the cytotoxicity of concentrated supernatants toward MC3T3 cells (see Fig. S1B in the supplemental material). This result suggests that diflunisal can inhibit staphylococcal virulence factor production under conditions of lower oxygenation and therefore may have efficacy in the hypoxic environment of infected bone.
Replenishment of alpha-type PSMs restores cytotoxicity to diflunisal-treated S. aureus supernatants.
Structural modeling predicted that diflunisal binds to AgrA and inhibits phosphorylation by AgrC, thereby halting quorum sensing and virulence factor production (11). Given the critical role of AgrA in transcription of the alpha-type PSMs (6), we reasoned that supplementation of diflunisal-treated staphylococcal supernatants with alpha-type PSMs would restore cytotoxicity toward osteoblasts. Three of the alpha-type PSMs were previously noted to be dose-dependently cytotoxic toward osteoblasts, with PSMα2 being most potent (5). To test whether PSM supplementation could restore cytotoxicity to diflunisal-treated S. aureus, synthetic PSMα2 was added to concentrated supernatants harvested from diflunisal-treated cultures. Supplementation with PSMα2 restored the cytotoxicity of diflunisal-treated supernatants toward both MC3T3 cells and primary human osteoblasts (Fig. 1D), providing further evidence that the anti-virulence effects of diflunisal reflect AgrA inhibition.
Treatment of host cells with diflunisal fails to prevent staphylococcal osteoblast cytotoxicity.
Diflunisal treatment significantly inhibits S. aureus-induced osteoblast cell death. However, given that this drug is an anti-inflammatory compound, it is possible that diflunisal treatment modulates host cell death in a manner that is partially independent of bacterial factors. To investigate this possibility, supernatants were prepared from S. aureus cultures grown in the absence of diflunisal. The supernatants were subsequently combined with fresh cell culture medium containing diflunisal or vehicle control and then applied to osteoblast cell monolayers to assess cytotoxicity. Supplementation of cell culture media with diflunisal failed to inhibit the cytotoxicity of diflunisal-naive S. aureus supernatants (Fig. 2). Thus, inhibition of staphylococcal cytotoxicity by diflunisal is largely independent of anti-inflammatory effects on host cells.
FIG 2.
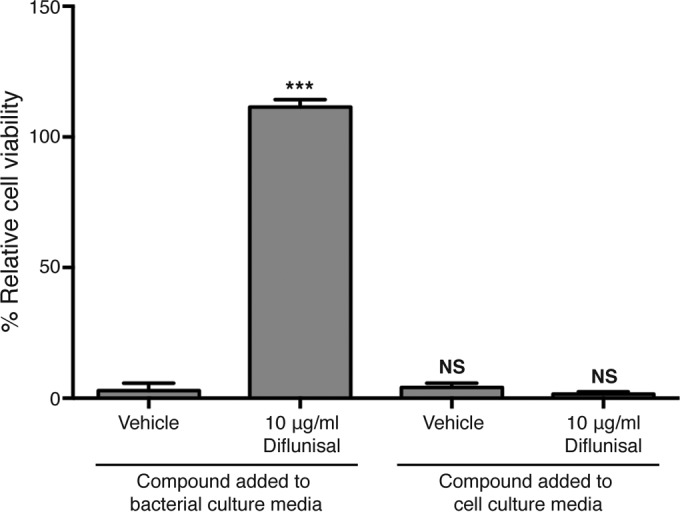
Supplementation of host cells with diflunisal does not inhibit cytotoxicity of untreated S. aureus supernatant. MC3T3 cells were intoxicated with concentrated supernatant from S. aureus grown in the presence (left) or absence (right) of vehicle control (DMSO) or 10 μg/ml diflunisal. Host cells were then cultured in the absence (left) or presence (right) of vehicle control (DMSO) or 10 μg/ml diflunisal. More specifically, fresh diflunisal or DMSO-supplemented cell culture medium was added back to the cell monolayers at the time of intoxication with concentrated supernatant. n = 5 per group, and the data are representative of two independent trials. Error bars represent the SD. ***, P < 0.001 relative to vehicle control. NS, not significant.
Potent inhibition of S. aureus cytotoxicity toward osteoblasts is unique to diflunisal.
Diflunisal is a member of the salicylate class of nonsteroidal anti-inflammatory compounds. It was therefore postulated that other NSAIDs with similar chemical properties might also inhibit staphylococcal cytotoxicity toward osteoblasts. To test this, a panel of commonly used anti-inflammatory drugs representing the major classes of NSAIDs was tested for the ability to limit S. aureus cytotoxicity. Aspirin (salicylate class), its active metabolite salicylic acid, ibuprofen (propionic acid derivative class), ketorolac (acetic acid derivative class), and piroxicam (enolic acid derivative class) were included in the analysis. All compounds except piroxicam were tested using two different solvents, DMSO and 100% ethanol. Piroxicam was only soluble in DMSO at 10 mg/ml. None of the compounds significantly inhibited S. aureus growth at 10 μg/ml (see Fig. S2 in the supplemental material). Interestingly, although these compounds share similar mechanisms of action, only diflunisal potently inhibited staphylococcal cytotoxicity toward osteoblastic cells (Fig. 3A and B). Piroxicam partially inhibited cytotoxicity at 10 μg/ml (Fig. 3B) but also caused the most pronounced lag in exponential growth (see Fig. S2 in the supplemental material). Salicylic acid modestly inhibited cytotoxicity when dissolved in ethanol at a concentration of 50 μg/ml (Fig. 3C), which is consistent with previous reports showing efficacy of this compound in limiting staphylococcal virulence factor production (19 – 21). To further investigate the relative potency of diflunisal, piroxicam, and salicylic acid in preventing cytotoxicity, dose-response studies were performed using 10, 25, 50, or 100 μg/ml of each compound dissolved in DMSO. Diflunisal potently inhibited cytotoxicity toward MC3T3 cells at all of the concentrations tested (Fig. 3D). Salicylic acid did not inhibit cytotoxicity at any of the tested concentrations, suggesting that its effects on staphylococcal virulence may be solvent dependent. Piroxicam again inhibited cytotoxicity at higher concentrations (Fig. 3D), but growth curve analysis indicated that this diminution of cytotoxicity was likely related to inhibition of bacterial growth (Fig. 3E). Diflunisal also inhibited bacterial growth at concentrations greater than 50 μg/ml. Collectively, these data indicate that diflunisal potently inhibits staphylococcal cytotoxicity toward osteoblasts, likely reflecting the important role of AgrA and alpha-type PSMs in inducing cell death of these host cells.
FIG 3.

Potent inhibition of S. aureus cytotoxicity toward osteoblasts is unique to diflunisal. (A) MC3T3 cells were intoxicated with 20 or 30% (vol/vol) of concentrated SN from S. aureus grown in the presence of vehicle control (ethanol) or 10 μg/ml of the indicated NSAIDs. (B) Same as panel A except that S. aureus was grown in the presence of vehicle control (DMSO) or 10 μg/ml of piroxicam or diflunisal, since piroxicam was not soluble in ethanol. (C) Same as panel A except that S. aureus was grown in the presence of vehicle control (ethanol), 10 μg/ml diflunisal, or 50 μg/ml salicylic acid. For panels A to C, the percent cell viability is depicted relative to mock intoxication with sterile RPMI. Error bars represent the SD. n = 10 per group, and the data are representative of two independent trials. ***, P < 0.001; **, P < 0.01 (relative to the vehicle control). (D) Dose-response curves were prepared with concentrated SNs grown in the presence of increasing amounts of diflunisal, piroxicam, and salicylic acid (in DMSO). Then, 30% (vol/vol) concentrated SNs were used to intoxicate MC3T3 monolayers. The percent cell viability relative to mock intoxication with sterile RPMI containing equivalent concentrations of each NSAID was determined. n = 10 per group. Error bars represent the SD. (E) Growth analysis (i.e., the OD600) of S. aureus cultured in glass Erlenmeyer flasks with a 1:5 volume-to-flask ratio and in the presence of the indicated concentrations of diflunisal, piroxicam, or vehicle control (DMSO). Error bars represent the SD. n = 3 per group, and the data represent the average of two independent trials.
PUR drug delivery foams are effective as local therapies for experimental osteomyelitis.
Given the potent blockade of PSM-induced osteoblast cell death by diflunisal in vitro, we sought to test whether diflunisal would limit the pathogenesis of S. aureus osteomyelitis in vivo in a murine model. We reasoned that a local drug delivery system would achieve clinical efficacy while avoiding the potential untoward effects of NSAIDs on the gastrointestinal, renal, and cardiovascular systems. PUR drug delivery foams were therefore fabricated to deliver diflunisal directly to the infectious focus. To test the feasibility of PUR as a drug delivery system in the murine model of S. aureus osteomyelitis, we first synthesized foams containing 8% (wt/wt) of vancomycin. Groups of mice were subjected to experimental S. aureus osteomyelitis, and either an empty foam or a vancomycin-loaded foam was sutured into place around the affected femur, centered on the inoculation site, at the time of wound closure. Mice received no additional antibiotic therapy throughout the experiment. At 14 days postinfection, mice were euthanized, and the infected femur was harvested and processed for CFU enumeration. Local vancomycin therapy dramatically reduced the bacterial burdens in affected femurs relative to mock-treated animals (Fig. 4). In fact, four of five vancomycin-treated animals had bacterial burdens that were below the limit of detection, suggesting that these mice may have cleared the infection completely. Therefore, PUR drug delivery foams have therapeutic efficacy in a murine model of S. aureus osteomyelitis.
FIG 4.
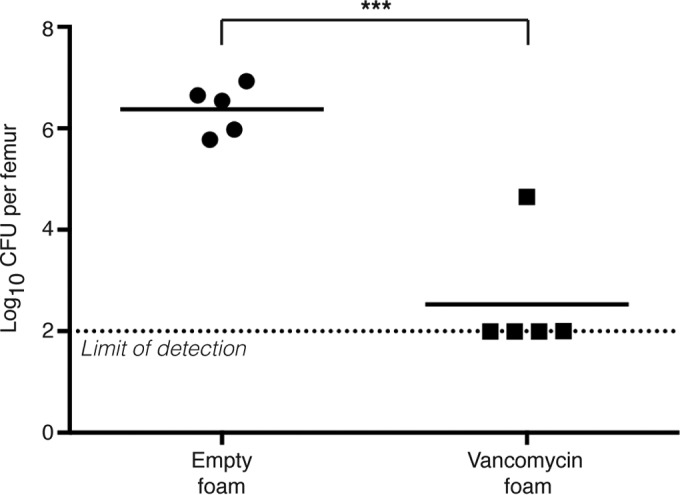
PUR drug delivery foams are effective as local therapies for experimental osteomyelitis. Groups (n = 5) of 7- to 8-week-old female C57BL/6J mice were subjected to experimental osteomyelitis by inoculation with S. aureus. After inoculation, either an empty PUR foam or a foam containing 8% (wt/wt) vancomycin was sutured into place around the inoculation site. At 14 days postinoculation, femurs were harvested and processed for CFU enumeration. The log10 CFU per femur is depicted. The horizontal bar represents the mean. The dotted line depicts the limit of detection for bacterial burdens. ***, P < 0.001 relative to mice administered an empty foam.
Local diflunisal therapy significantly decreases S. aureus-induced bone destruction during osteomyelitis.
Having established the feasibility of local drug delivery in a murine model of osteomyelitis, we next sought to determine whether local diflunisal therapy could limit bone destruction during S. aureus infection. PUR foams were synthesized with a final concentration of approximately 10 mM diflunisal. To ensure that the diflunisal-loaded foams would elute the drug at a sufficient concentration to inhibit skeletal cell death without impacting bacterial growth, the foams were first tested in vitro for the ability to block staphylococcal osteoblast cytotoxicity. Cultures grown in the presence of diflunisal-loaded foams achieved similar bacterial densities compared to cultures incubated with empty foams (9.93 ± 0.1008 log10 CFU/ml for diflunisal treatment versus 9.81 ± 0.1307 log10 CFU/ml for mock treatment; n = 3). However, supernatants prepared from cultures incubated in the presence of diflunisal-loaded foams had substantially less cytotoxicity toward osteoblastic cells in comparison to cultures incubated with empty foams (Fig. 5A). Of note, the potency of concentrated S. aureus supernatant was lower in the presence of empty foams relative to experiments with other vehicle controls, which may reflect nonspecific binding or inhibition of the amphipathic phenol soluble modulins by the foams. Nevertheless, diflunisal-loaded foams still showed significant inhibition of cytotoxicity relative to empty foams. In addition, the in vitro release kinetics of diflunisal indicated an initial burst release from the foam in the first 24 to 48 h, with approximately 50% of the loaded diflunisal having released within the first day followed by a sustained release until day 7 (see Fig. S3 in the supplemental material). From these results we concluded that diflunisal-loaded PUR foams elute the drug within the range of therapeutic efficacy.
FIG 5.

Local diflunisal therapy significantly decreases S. aureus-induced bone destruction during osteomyelitis. (A) MC3T3 cells were intoxicated with 20 or 30% (vol/vol) concentrated S. aureus supernatant prepared from cultures grown in the presence of either an empty PUR foam or a foam containing 10 mM diflunisal. n = 10 per group, and the data are representative of three independent trials. Error bars represent the SD. The percent cell viability is depicted relative to mock intoxication with sterile RPMI. ***, P < 0.001 relative to empty foam treatment. (B to D) Groups of 7- to 8-week-old female C57BL/6J mice were subjected to experimental osteomyelitis by inoculation with S. aureus. After inoculation, either an empty PUR foam or a foam containing 10 mM diflunisal was sutured into place around the inoculation site. At 14 days postinoculation, femurs were harvested and either processed for CFU enumeration (B) or microCT analysis (C and D). For CFU enumeration, n = 5 per group, and the log10 CFU per femur is depicted. The horizontal bar represents the mean. The dotted line depicts the limit of detection for bacterial burdens. For microCT analysis, n = 9 or 10 per group (one mouse in the control group suffered a pathological fracture), and the data are the average of two independent trials. (C) Anteroposterior microCT images of femurs subjected to either mock treatment (empty foam) or local diflunisal therapy. (D) Quantification of cortical bone destruction. Error bars represent the SD. **, P < 0.01.
To test the ability of diflunisal to inhibit pathogen-induced bone destruction, groups of mice were subjected to staphylococcal osteomyelitis and administered a diflunisal-loaded or empty foam. At 14 days postinfection, infected femurs were removed and either imaged by microCT or processed for CFU enumeration. There were no significant differences in bacterial burdens in the femurs of mice receiving local diflunisal therapy versus those receiving mock treatment (Fig. 5B). However, mice treated with diflunisal experienced significantly less cortical bone destruction than mock-treated mice, with an average reduction in bone destruction of 36% in two independent trials (Fig. 5C and D). In sum, these data show that diflunisal therapy ameliorates the pathogenesis of staphylococcal osteomyelitis by blocking osteoblast cell cytotoxicity and limiting cortical bone destruction. We therefore propose that diflunisal could be a promising adjunctive therapy for invasive staphylococcal disease, and particularly musculoskeletal infections.
DISCUSSION
Invasive S. aureus infections are a considerable source of global morbidity and mortality. Treatment of staphylococcal infections is hindered by intrinsic antimicrobial resistance, as well as by extrinsic factors that lessen the clinical efficacy of antimicrobial agents with proven activity in vitro. Staphylococcal osteomyelitis is perhaps most demonstrative of these barriers to treatment, as many musculoskeletal infections in the United States are caused by antibiotic-resistant S. aureus (3), and pathogen-induced bone destruction limits antimicrobial penetration to the infectious focus. Osteomyelitis therefore requires prolonged antibiotic therapy and invasive debridement procedures, after which a proportion of patients still progress to chronic infection.
Given that S. aureus accounts for nearly half of all deaths caused by antibiotic-resistant pathogens in the United States, there is an urgent focus on the development of new antimicrobial therapies. This includes new and improved antibiotics for the treatment of resistant pathogens, as well as complementary therapies aimed at boosting host immune responses or limiting the extrinsic factors that promote treatment failure. In regard to staphylococcal osteomyelitis, therapies that limit pathological bone destruction and improve antibiotic delivery to the infectious focus could significantly enhance traditional antimicrobial therapy. Such complementary therapies could shorten the duration of therapy for osteomyelitis while also limiting progression to chronic infection, thereby lessening the likelihood of a pathogen developing intrinsic antimicrobial resistance. In this study, we tested the FDA-approved NSAID diflunisal as a potential complementary therapy for S. aureus osteomyelitis. Diflunisal potently inhibited staphylococcal cytotoxicity toward murine and human osteoblasts in vitro and significantly reduced bone destruction during experimental osteomyelitis. Because the agr system is necessary for expression of critical virulence factors by S. aureus, blockade of AgrA activity by diflunisal may also have therapeutic efficacy in other invasive staphylococcal diseases. Our in vivo studies likely underestimate the efficacy of diflunisal as an osteoprotective agent, as the release kinetics of diflunisal-loaded foams indicated the majority of the compound is released in the first week of the 2-week experiment. In future studies, we will tune the foams to deliver the most efficacious release pattern in vivo.
Since surgical debridement of infected bone is a standard practice in the treatment of acute osteomyelitis, a unique opportunity exists for administration of local therapies. In order to create a preclinical model to evaluate local therapies for staphylococcal infection, we utilized drug-eluting PUR foams in a murine model of osteomyelitis. PUR foams effectively delivered both vancomycin and diflunisal to the infectious focus, resulting in efficient bacterial killing and amelioration of bone destruction, respectively. Since PUR foams can be synthesized in a variety of shapes and sizes, are readily compressible, and can be loaded with a variety of antimicrobial and tissue regenerative compounds, they may be particularly useful for the delivery of local therapies to infected tissues (22). The murine model of osteomyelitis did not allow for testing the efficacy of diflunisal in limiting the pathogenesis of established infections, since this would require stabilization of the induced fracture with a fixator device to facilitate multiple surgical procedures. Instead, we are currently developing alternative delivery vehicles for diflunisal to facilitate percutaneous administration to the infectious focus.
Future studies will determine whether local diflunisal therapy enhances systemic antimicrobial therapy, in terms of reducing the necessary duration of therapy, limiting the outgrowth of resistant pathogens, or by decreasing the progression to chronic infection. One limitation of the present study is the inability to parse out the antivirulence effects of diflunisal from its anti-inflammatory properties. NSAIDs are known to affect bone remodeling, and they specifically impact osteoclast biology via inhibition of the synthesis of proresorptive, inflammatory mediators such as prostaglandins (23). Additional studies are required to further investigate the cellular mechanisms underlying the osteoprotective effects of diflunisal in the context of osteomyelitis and whether osteoprotection is mediated by osteoblasts, osteoclasts, or both. Nevertheless, since NSAIDs are a mainstay of therapy for patients recovering from osteomyelitis, any antivirulence effects that can be added by diflunisal will be valuable. Another potential limitation of diflunisal therapy is that AgrA inhibition may have the unintended consequence of increasing biofilm formation. Inactivation of the agr system leads to enhanced in vitro biofilm formation in a number of S. aureus strains, and Agr-defective strains have been isolated from patients with chronic musculoskeletal infection (24 – 27). It may therefore be prudent to only use diflunisal in combination with traditional antibiotics, and perhaps to focus on patients with acute osteomyelitis, rather than those with implant-associated infection or established chronic disease. Despite these caveats, our work suggests that diflunisal may be a promising adjunctive therapy for osteomyelitis with antivirulence and osteoprotective effects.
Supplementary Material
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00834-16.
REFERENCES
- 1.Lew DP, Waldvogel FA. 2004. Osteomyelitis. Lancet 364:369–379. [DOI] [PubMed] [Google Scholar]
- 2.Copley LA. 2009. Pediatric musculoskeletal infection: trends and antibiotic recommendations. J Am Acad Orthop Surg 17:618–626. doi: 10.5435/00124635-200910000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Gerber JS, Coffin SE, Smathers SA, Zaoutis TE. 2009. Trends in the incidence of methicillin-resistant Staphylococcus aureus infection in children's hospitals in the United States. Clin Infect Dis 49:65–71. doi: 10.1086/599348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu T, Zhang X, Li Z, Peng D. 2011. Management of combined bone defect and limb-length discrepancy after tibial chronic osteomyelitis. Orthopedics 34:e363–367. [DOI] [PubMed] [Google Scholar]
- 5.Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP. 2013. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe 13:759–772. doi: 10.1016/j.chom.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M. 2008. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell 32:150–158. doi: 10.1016/j.molcel.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, Sklar LA, Horswill AR, Hall PR, Gresham HD. 2014. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog 10:e1004174. doi: 10.1371/journal.ppat.1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray EJ, Crowley RC, Truman A, Clarke SR, Cottam JA, Jadhav GP, Steele VR, O'Shea P, Lindholm C, Cockayne A, Chhabra SR, Chan WC, Williams P. 2014. Targeting Staphylococcus aureus quorum sensing with nonpeptidic small molecule inhibitors. J Med Chem 57:2813–2819. doi: 10.1021/jm500215s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.George EA, Novick RP, Muir TW. 2008. Cyclic peptide inhibitors of staphylococcal virulence prepared by Fmoc-based thiolactone peptide synthesis. J Am Chem Soc 130:4914–4924. doi: 10.1021/ja711126e. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen A, Mansson M, Bojer MS, Gram L, Larsen TO, Novick RP, Frees D, Frokiaer H, Ingmer H. 2014. Solonamide B inhibits quorum sensing and reduces Staphylococcus aureus mediated killing of human neutrophils. PLoS One 9:e84992. doi: 10.1371/journal.pone.0084992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khodaverdian V, Pesho M, Truitt B, Bollinger L, Patel P, Nithianantham S, Yu G, Delaney E, Jankowsky E, Shoham M. 2013. Discovery of antivirulence agents against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:3645–3652. doi: 10.1128/AAC.00269-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duthie ES, Lorenz LL. 1952. Staphylococcal coagulase; mode of action and antigenicity. J Gen Microbiol 6:95–107. [DOI] [PubMed] [Google Scholar]
- 14.Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, Hinger SA, Aysanoa EE, Blanchard C, Dunman PM, Wasserman GA, Chen J, Shopsin B, Gilmore MS, Skaar EP, Cassat JE. 2015. Bacterial hypoxic responses revealed as critical determinants of the host-pathogen outcome by TnSeq analysis of Staphylococcus aureus invasive infection. PLoS Pathog 11:e1005341. doi: 10.1371/journal.ppat.1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guelcher SA, Patel V, Gallagher KM, Connolly S, Didier JE, Doctor JS, Hollinger JO. 2006. Synthesis and in vitro biocompatibility of injectable polyurethane foam scaffolds. Tissue Eng 12:1247–1259. doi: 10.1089/ten.2006.12.1247. [DOI] [PubMed] [Google Scholar]
- 16.Sawhney AS, Hubbell JA. 1990. Rapidly degraded terpolymers of dl-lactide, glycolide, and epsilon-caprolactone with increased hydrophilicity by copolymerization with polyethers. J Biomed Mater Res 24:1397–1411. doi: 10.1002/jbm.820241011. [DOI] [PubMed] [Google Scholar]
- 17.Wahbi AA, Mabrouk MM, Moneeb MS, Kamal AH. 2009. Simultaneous determination of the two non-steroidal anti-inflammatory drugs; diflunisal and naproxen in their tablets by chemometric spectrophotometry and HPLC. Pak J Pharm Sci 22:8–17. [PubMed] [Google Scholar]
- 18.Hammer ND, Cassat JE, Noto MJ, Lojek LJ, Chadha AD, Schmitz JE, Creech CB, Skaar EP. 2014. Inter- and intraspecies metabolite exchange promotes virulence of antibiotic-resistant Staphylococcus aureus. Cell Host Microbe 16:531–537. doi: 10.1016/j.chom.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kupferwasser LI, Yeaman MR, Nast CC, Kupferwasser D, Xiong YQ, Palma M, Cheung AL, Bayer AS. 2003. Salicylic acid attenuates virulence in endovascular infections by targeting global regulatory pathways in Staphylococcus aureus. J Clin Invest 112:222–233. doi: 10.1172/JCI200316876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palma M, Bayer A, Kupferwasser LI, Joska T, Yeaman MR, Cheung A. 2006. Salicylic acid activates sigma factor B by rsbU-dependent and -independent mechanisms. J Bacteriol 188:5896–5903. doi: 10.1128/JB.01960-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kupferwasser LI, Yeaman MR, Shapiro SM, Nast CC, Sullam PM, Filler SG, Bayer AS. 1999. Acetylsalicylic acid reduces vegetation bacterial density, hematogenous bacterial dissemination, and frequency of embolic events in experimental Staphylococcus aureus endocarditis through antiplatelet and antibacterial effects. Circulation 99:2791–2797. doi: 10.1161/01.CIR.99.21.2791. [DOI] [PubMed] [Google Scholar]
- 22.Guelcher SA, Brown KV, Li B, Guda T, Lee BH, Wenke JC. 2011. Dual-purpose bone grafts improve healing and reduce infection. J Orthop Trauma 25:477–482. doi: 10.1097/BOT.0b013e31821f624c. [DOI] [PubMed] [Google Scholar]
- 23.Harder AT, An YH. 2003. The mechanisms of the inhibitory effects of nonsteroidal anti-inflammatory drugs on bone healing: a concise review. J Clin Pharmacol 43:807–815. doi: 10.1177/0091270003256061. [DOI] [PubMed] [Google Scholar]
- 24.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valour F, Rasigade JP, Trouillet-Assant S, Gagnaire J, Bouaziz A, Karsenty J, Lacour C, Bes M, Lustig S, Benet T, Chidiac C, Etienne J, Vandenesch F, Ferry T, Laurent F, Lyon BJISG. 2015. Delta-toxin production deficiency in Staphylococcus aureus: a diagnostic marker of bone and joint infection chronicity linked with osteoblast invasion and biofilm formation. Clin Microbiol Infect 21:568.e561-11. doi: 10.1016/j.cmi.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 26.Beenken KE, Blevins JS, Smeltzer MS. 2003. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun 71:4206–4211. doi: 10.1128/IAI.71.7.4206-4211.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beenken KE, Mrak LN, Griffin LM, Zielinska AK, Shaw LN, Rice KC, Horswill AR, Bayles KW, Smeltzer MS. 2010. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS One 5:e10790. doi: 10.1371/journal.pone.0010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.